含硼氮咔唑茚类化合物及其应用的制作方法
1.本发明涉及电致发光材料领域,尤其涉及一种含硼氮咔唑茚类化合物及其应用。
背景技术:
2.由于有机半导体材料在合成上具有多样性,制造成本相对较低以及优良的光学与电学性能,其在光电器件,特别是oled器件方面的应用具有很大的潜力。
3.oled发光器件犹如三明治的结构,包括两个电极,以及夹在两个电极膜层之间的有机功能材料,各种不同功能材料根据用途相互叠加在一起共同组成oled发光器件。作为电致发光器件,当对oled发光器件的两端电极施加电压,并通过电场作用有机层功能材料膜层中的正负电荷,正负电荷进一步在发光层中复合,即产生oled电致发光。
4.为了提高有机发光二极管的发光效率,各种基于荧光和磷光的发光材料体系已被开发出来,使用荧光材料的有机发光二极管具有可靠性高的特点,但其在电气激发下其内部电致发光量子效率被限制为25%,这是因为激子的单重激发态和三重激发态的分支比为1:3。
5.与此相反,使用磷光材料的有机发光二极管已经取得了几乎100%的内部电致发光量子效率。但磷光oled有一显著的问题,就是roll-off效应,即发光效率随电流或亮度的增加而迅速降低,这对高亮度的应用尤为不利。
技术实现要素:
6.鉴于上述现有技术的不足,本发明的目的在于提供一种含硼氮咔唑茚类化合物及其应用,作为一类新型的有机光电功能材料,可提高器件的效率和寿命,降低roll-off效应,同时降低制造成本。
7.本发明的技术方案如下:
8.一种含硼氮咔唑茚类化合物,其结构通式如式(1)所示:
[0009][0010]
其中:
[0011]
ar
1-ar4每次出现时,独立选自取代或未取代的含有6至60个c原子的芳香基团,或取代或未取代的含有5至60个环原子的杂芳香基团,或取代或未取代含有3-30个环原子的非芳香环;
[0012]
x每次出现分别独立表示cr1r2、nr1、sir1r2、o、s、se、s=o、s(=o)2、或pr1;
[0013]r1-r2每次出现时,独立选自h,d,或具有1至20个c原子的直链烷基,具有1至20个c原子的直链烷氧基或直链硫代烷氧基,或具有3至20个c原子的支链烷基或环状的烷基,具有3至20个c原子的支链烷氧基或支链硫代烷氧基,具有3至20个c原子的环状的烷氧基或环状的硫代烷氧基,或甲硅烷基,或具有1至20个c原子的酮基,或具有2至20个c原子的烷氧基羰基,或具有7至20个c原子的芳氧基羰基,氰基,氨基甲酰基,卤甲酰基,甲酰基,异氰基,异氰酸酯基,硫氰酸酯基或异硫氰酸酯基,羟基,硝基,cf3,cl,br,f,i,可交联的基团,或者取代或未取代的具有5至60个环原子的芳香基团或杂芳香基团,或具有5至60个环原子的芳氧基或杂芳氧基基团,或这些基团的组合。
[0014]
本发明还提供一种混合物,包含一种上述的含硼氮咔唑茚类化合物,及至少一种有机功能材料,所述有机功能材料选自空穴注入材料、空穴传输材料、电子传输材料、电子注入材料、电子阻挡材料、空穴阻挡材料、发光体、主体材料和有机染料中的至少一种。
[0015]
本发明还提供一种组合物,包括一种上述的含硼氮咔唑茚类化合物或上述的混合物,及至少一种有机溶剂。
[0016]
本发明还提供一种有机电子器件,所述电子器件的制备原料至少包括一种上所述的含硼氮咔唑茚类化合物,或上述的混合物,或上述的组合物。
[0017]
与现有技术相比,本发明具有如下有益效果:
[0018]
本发明提供的的含硼氮咔唑茚类化合物,便于提高材料分子的刚性,提高材料的稳定性,以其制备发光器件,延长器件寿命。本发明的有机化合物可作为蓝光客体材料,通过与合适的主体材料配合,能提高其作为电致发光器件的发光效率及寿命,提供了一种制造成本低、效率高、寿命长、低滚降的发光器件的解决方案。
具体实施方式
[0019]
本发明提供一种含硼氮咔唑茚类化合物、混合物、组合物及其应用。以下结合具体实施例对本发明作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
[0020]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0021]
在本发明中,组合物和印刷油墨,或油墨具有相同的含义,它们之间可以互换。
[0022]
在本发明中,主体材料,基质材料,host或matrix材料具有相同的含义,它们之间可以互换。
[0023]
在本发明中,“取代”表示被取代基中的氢原子被取代基所取代。
[0024]
在本发明中,同一取代基多次出现时,可独立选自不同基团。如通式含有多个r1、r4,则r1、r4可独立选自不同基团。
[0025]
本发明中,“取代或未取代”表示所定义的基团可以被取代,也可以不被取代。当所定义的基团被取代时,应理解为任选被本领域可接受的基团所取代,包括但不限于:c
1-30
烷基、含有3-20个环原子的杂环基、含有5-20个环原子的芳基、含有5-20个环原子的杂芳基、
硅烷基、羰基、烷氧基羰基、芳氧基羰基、氨基甲酰基、卤甲酰基、甲酰基、-nrr
′
、氰基、异氰基、异氰酸酯基、硫氰酸酯基、异硫氰酸酯基、羟基、三氟甲基、硝基或卤素,且上述基团也可以进一步被本领域可接受取代基取代;可理解的,-nrr
′
中的r和r
′
各自独立地为本领域可接受的基团所取代,包括但不限于h、c
1-6
烷基、含有3-8个环原子的环烷基、含有3-8个环原子的杂环基、含有5-20个环原子的芳基或含有5-10个环原子的杂芳基;所述c
1-6
烷基、含有3-8个环原子的环烷基、含有3-8个环原子的杂环基、含有5-20个环原子的芳基或含有5-10个环原子的杂芳基任选进一步被一个或多个以下基团取代:c
1-6
烷基、含有3-8个环原子的环烷基、含有3-8个环原子的杂环基、卤素、羟基、硝基或氨基。
[0026]
在本发明中,“环原子数”表示原子键合成环状而得到的结构化合物(例如,单环化合物、稠环化合物、交联化合物、碳环化合物、杂环化合物)的构成该环自身的原子之中的原子数。该环被取代基所取代时,取代基所包含的原子不包括在成环原子内。关于以下所述的“环原子数”,在没有特别说明的条件下也是同样的。例如,苯环的环原子数为6,萘环的环原子数为10,噻吩基的环原子数为5。
[0027]
在本发明中,“烷基”可以表示直链、支链和/或环状烷基。烷基的碳数可以为1至50、1至30、1至20、1至10或1至6。包含该术语的短语,例如,“c
1-9
烷基”是指包含1~9个碳原子的烷基,每次出现时,可以互相独立地为c1烷基、c2烷基、c3烷基、c4烷基、c5烷基、c6烷基、c7烷基、c8烷基或c9烷基。烷基的非限制性实例包括甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、异丁基、2-乙基丁基、3,3-二甲基丁基、正戊基、异戊基、新戊基、叔戊基、环戊基、1-甲基戊基、3-甲基戊基、2-乙基戊基、4-甲基-2-戊基、正己基、1-甲基己基、2-乙基己基、2-丁基己基、环己基、4-甲基环己基、4-叔丁基环己基、正庚基、1-甲基庚基、2,2-二甲基庚基、2-乙基庚基、2-丁基庚基、正辛基、叔辛基、2-乙基辛基、2-丁基辛基、2-己基辛基、3,7-二甲基辛基、环辛基、正壬基、正癸基、金刚烷基、2-乙基癸基、2-丁基癸基、2-己基癸基、2-辛基癸基、正十一烷基、正十二烷基、2-乙基十二烷基、2-丁基十二烷基、2-己基十二烷基、2-辛基十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、2-乙基十六烷基、2-丁基十六烷基、2-己基十六烷基、2-辛基十六烷基、正十七烷基、正十八烷基、正十九烷基、正二十烷基、2-乙基二十烷基、2-丁基二十烷基、2-己基二十烷基、2-辛基二十烷基、正二十一烷基、正二十二烷基、正二十三烷基、正二十四烷基、正二十五烷基、正二十六烷基、正二十七烷基、正二十八烷基、正二十九烷基、正三十烷基、金刚烷等。
[0028]
术语“烷氧基”是指具有-o-烷基的基团,即如上所定义的烷基经由氧原子连接至母核结构。包含该术语的短语,合适的实例包括但不限于:甲氧基(-o-ch3或-ome)、乙氧基(-o-ch2ch3或-oet)和叔丁氧基(-o-c(ch3)3或-otbu)。
[0029]“芳基或芳香基团”是指在芳香环化合物的基础上除去一个氢原子衍生的芳族烃基,可以为单环芳基、或稠环芳基、或多环芳基,对于多环的环种,至少一个是芳族环系。例如,“具有5至60个环原子的取代或未取代的芳基”是指包含5至60个环原子的芳基,且芳基上任选进一步被取代;合适的实例包括但不限于:苯、联苯、萘、蒽、菲、二萘嵌苯、三亚苯及其衍生物。可以理解地,多个芳基也可以被短的非芳族单元间断(例如《10%的非h原子,比如c、n或o原子),具体如苊、芴,或者9,9-二芳基芴、三芳胺、二芳基醚体系也应该包含在芳基的定义中。
[0030]“杂芳基或杂芳香基团”是指在芳基的基础上至少一个碳原子被非碳原子所替代,
非碳原子可以为n原子、o原子、s原子等。例如,“具有5至60个环原子的取代或未取代的杂芳基”是指具有5至60个环原子的杂芳基,且杂芳基任选进一步被取代,合适的实例包括但不限于:呋喃、苯并呋喃、噻吩、苯并噻吩、吡咯、吡唑、三唑、咪唑、噁唑、噁二唑、噻唑、四唑、吲哚、咔唑、吡咯并咪唑、吡咯并吡咯、噻吩并吡咯、噻吩并噻吩、呋喃并吡咯、呋喃并呋喃、噻吩并呋喃、苯并异噁唑、苯并异噻唑、苯并咪唑、吡啶、吡嗪、哒嗪、嘧啶、三嗪、喹啉、异喹啉、邻二氮萘、喹喔啉、菲啶、伯啶、喹唑啉和喹唑啉酮、二苯并噻吩、二苯并呋喃、咔唑及其衍生物。
[0031]
在本发明中,“环原子数”表示原子键合成环状而得到的结构化合物(例如,单环化合物、稠环化合物、交联化合物、碳环化合物、杂环化合物)的构成该环自身的原子之中的原子数。该环被取代基所取代时,取代基所包含的原子不包括在成环原子内。关于以下所述的“环原子数”,在没有特别说明的条件下也是同样的。例如,苯环的环原子数为6,萘环的环原子数为10,噻吩基的环原子数为5。
[0032]“胺基”是指氨的衍生物,具有式-n(x)2的结构特征,其中每个“x”独立地是h、取代的或未被取代的烷基、取代的或未被取代的环烷基、取代的或未被取代的杂环基等。胺基的非限制性类型包括-nh2、-n(烷基)2、-nh(烷基)、-n(环烷基)2、-nh(环烷基)、-n(杂环基)2、-nh(杂环基)、-n(芳基)2、-nh(芳基)、-n(烷基)(芳基)、-n(烷基)(杂环基)、-n(环烷基)(杂环基)、-n(芳基)(杂芳基)、-n(烷基)(杂芳基)等。
[0033]“卤素”或“卤基”是指f、cl、br或i。
[0034]“烷基氨基”是指被至少一个烷基取代的氨基。合适的实例包括但不限于:-nh2、-nh(ch3)、-n(ch3)2、-nh(ch2ch3)、-n(ch2ch3)2。
[0035]“芳基烷基”是指烷基上至少一个键合至碳原子的氢原子被芳基代替衍生形成的烃基。其中芳基部分可以包括5~20个碳原子,烷基部分可以包括1~9个碳原子。合适的实例包括但不限于:苄基、2-苯基乙-1-基、萘基甲基、2-萘基乙-1-基、萘并苄基和2-萘并苯基乙-1-基。
[0036]
本发明中,与单键相连的“*”表示连接位点;本发明中,取代基相连的单键贯穿相应的环,表述该取代基可与环的任选位置连接,例如中r2与苯环的任一可取代位点相连,如表示中y1和y2与苯环的任选的两个相邻c原子形成并环,同理等。
[0037]
本发明中,当同一基团上含有多个相同符号的取代基时,各取代基可以彼此相同或不同,例如苯环上6个r1可以彼此相同或不同。
[0038]
在本发明中,取代基缩写对应为:n-正,sec-仲,i-异,t-叔,o-邻,m-间,p-对,me甲基,et乙基,pr丙基,bu丁基,am正戊基,hx己基,cy环己基。
[0039]
t-am表示2-(2-甲基)丁基;t-bu表示叔丁基。
[0040]
本发明提供一种含硼氮咔唑茚类化合物,其结构通式如式(1)所示:
[0041][0042]
b为硼原子;
[0043]
ar
1-ar4每次出现时,独立选自取代或未取代的含有6至60个c原子的芳香基团,或取代或未取代的含有5至60个环原子的杂芳香基团,或取代或未取代含有3-30个环原子的非芳香环;
[0044]
x每次出现分别独立表示cr1r2、nr1、sir1r2、o、s、se、s=o、s(=o)2、或pr1;
[0045]r1-r2每次出现时,独立选自h,d,或具有1至20个c原子的直链烷基,具有1至20个c原子的直链烷氧基或直链硫代烷氧基,或具有3至20个c原子的支链烷基或环状的烷基,具有3至20个c原子的支链烷氧基或支链硫代烷氧基,具有3至20个c原子的环状的烷氧基或环状的硫代烷氧基,或甲硅烷基,或具有1至20个c原子的酮基,或具有2至20个c原子的烷氧基羰基,或具有7至20个c原子的芳氧基羰基,氰基,氨基甲酰基,卤甲酰基,甲酰基,异氰基,异氰酸酯基,硫氰酸酯基或异硫氰酸酯基,羟基,硝基,cf3,cl,br,f,i,可交联的基团,或者取代或未取代的具有5至60个环原子的芳香基团或杂芳香基团,或具有5至60个环原子的芳氧基或杂芳氧基基团,或这些基团的组合。
[0046]
在本发明中,所述的取代是指被r取代,r含义同r1。
[0047]
在一实施例中,所述的x选自cr1r2、nr1、o、s或pr1。优选地,所述的x选自nr1、o、s或pr1。
[0048]
进一步地,r1选自具有6至30个c原子的芳香基团或被c
1-10
的烷基取代的具有6至30个c原子的芳香基团芳香基团;
[0049]
在一实施例中,r1选自苯基或被c
1-10
的烷基取代的苯基。
[0050]
在一实施例中,x多次出现时,选自相同的基团;
[0051]
在另一实施例中,x多次出现时,选自不同的基团。
[0052]
在一实施例中,所述的ar
1-ar4每次出现时,分别独立地选自取代或未取代的含有5至40个环原子的芳香基团或杂芳香基团。
[0053]
在一实施例中,所述的ar
1-ar4每次出现时,分别独立地选自取代或未取代的含有6个c原子的芳香基团,或取代或未取代的含有5至6个环原子的杂芳香基团。
[0054]
在另一实施例中,所述的ar
1-ar4中至少有一个选自取代或未取代的具有9至30个环原子的稠环芳香基团,或取代或未取代的具有9至30个环原子的稠环杂芳香基团。
[0055]
进一步地,ar
1-ar4中至少有两个选自取代或未取代的具有9至30个环原子的稠环芳香基团或稠环杂芳香基团。
[0056]
在一实施例中,ar
1-ar4中至少有三个选自取代或未取代的具有9至30个环原子的
稠环芳香基团或稠环杂芳香基团。
[0057]
在一实施例中,ar
1-ar4均选自取代或未取代的具有9至30个环原子的稠环芳香基团或稠环杂芳香基团。
[0058]
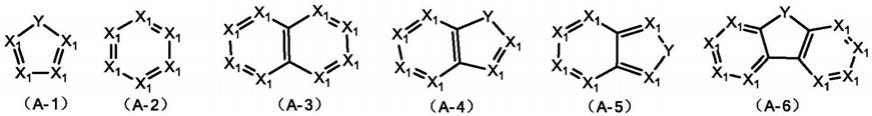
在一实施例中,所述的ar
1-ar4分别独立地选自(a-1)-(a-6)中的任意一种:
[0059][0060]
其中:
[0061]
x1每次出现时,独立地选自cr3或n;
[0062]
y每次出现时,独立地选自cr4r5、sir4r5、nr4、pr4、c(=o)、s、或o;
[0063]r3-r5每次出现时,独立选自h,d,或具有1至20个c原子的直链烷基,具有1至20个c原子的直链烷氧基或直链硫代烷氧基,或具有3至20个c原子的支链烷基或环状的烷基,具有3至20个c原子的支链烷氧基或支链硫代烷氧基,具有3至20个c原子的环状的烷氧基或环状的硫代烷氧基,或甲硅烷基,或具有1至20个c原子的酮基,或具有2至20个c原子的烷氧基羰基,或具有7至20个c原子的芳氧基羰基,氰基,氨基甲酰基,卤甲酰基,甲酰基,异氰基,异氰酸酯基,硫氰酸酯基或异硫氰酸酯基,羟基,硝基,cf3,cl,br,f,i,可交联的基团,或者取代或未取代的具有5至40个环原子的芳香环或杂芳香环,或具有5至40个环原子的芳氧基或杂芳氧基基团,或这些基团的组合;
[0064]
其中一个或多个r3可以彼此形成单环或多环的脂族环,或形成单环或多环的芳族环,或形成单环或多环的杂环。
[0065]
优选地,上述取代或未取代的具有9至30个环原子稠环芳香基团或稠环杂芳香基团选自(a-3)或(a-6)。
[0066]
当x1为连接位点时,x1选自c。
[0067]
在一实施例中,所述的ar
1-ar4均选自(a-2)、(a-3)或(a-6)。
[0068]
在一实施例中,所述的ar
1-ar4均选自(a-2)。所述有机化合物的结构通式选自式(2):
[0069][0070]
在一实施例中,通式(2)中的x1均选自cr3。
[0071]
在一实施例中,所述的ar
1-ar4中至少有一个选自(a-3)或(a-6)。
[0072]
在一实施例中,进一步,ar1选自(a-3);进一步,ar4选自(a-2)、(a-3)或(a-6);优选地,ar4选自(a-2)或(a-3)。
[0073]
在一实施例中,ar2选自(a-3)或(a-6)。
[0074]
在一实施例中,ar2选自(a-3),ar3选自(a-2)、(a-3)或(a-6);进一步地,ar3选自(a-2)或(a-3)。
[0075]
在另一实施例中,ar2选自(a-6),ar4选自(a-2)或(a-6);进一步地,ar4选自(a-2)。
[0076]
在一实施例中,ar
1-ar4中至少有一个选自(a-6);优选地,ar2或ar3选自选自(a-6)。
[0077]
更进一步地,通式(1)选自通式(3-1)-(3-14)中的任意一种:
[0078][0079][0080]
在一实施例中,x1每次出现时,独立地选自cr3;
[0081]
在一实施例中,r3每次出现时,独立选自h,d,或具有1至10个c原子的直链烷基,或具有3至10个c原子的支链烷基或环状的烷基,或者取代或未取代的具有5至20个环原子的
芳香基团或杂芳香基团;进一步地,r3每次出现时,独立选自h,d,或具有1至8个c原子的直链烷基,或具有3至8个c原子的支链烷基。
[0082]
优选地,至少一个r3选自具有1至20个c原子的直链烷基,或者具有3至20个c原子的支链或环状的烷基。
[0083]
进一步地,至少一个r3包含有一个甲基或基团,n为0、1、2、3或4,*表示取代位点。
[0084]
在一实施例中,至少一个r3选自甲基、t-am或t-bu。
[0085]
在一实施例中,按照本发明的有机化合物,通式(3-1)-(3-14)中的y选自nr4、pr4、o、s。
[0086]
下面列出按照本发明所述的有机化合物的例子,但不限于:
[0087]
[0088]
[0089]
[0090]
[0091][0092]
按照发明所述的的含硼氮咔唑茚类化合物,可以作为功能材料应用于电子器件,特别是oled器件中。有机功能材料可分为空穴注入材料(him),空穴传输材料(htm),电子传输材料(etm),电子注入材料(eim),电子阻挡材料(ebm),空穴阻挡材料(hbm),发光材料(emitter),主体材料(host)和有机染料中的至少一种。
[0093]
在一实施例中,按照本发明的含硼氮咔唑茚类化合物用于发光层中,优选地,可作为发光层客体材料用于发光层中;更优选地,可作为蓝光发光层客体材料用于发光层中。
[0094]
本发明还涉及一种混合物,包括如一种上述的含硼氮咔唑茚类化合物,以及至少一种有机功能材料。所述的有机功能材料,包括空穴注入材料,空穴传输材料,电子传输材料,电子注入材料,电子阻挡材料,空穴阻挡材料,发光体,或主体材料。发光体选自单重态发光体(荧光发光体)、三重态发光体(磷光发光体)级有机热激发延迟荧光材料(tadf材料)。例如在wo2010135519a1、us20090134784a1和wo 2011110277a1中对各种有机功能材料有详细的描述,特此将此3专利文件中的全部内容并入本文作为参考。
[0095]
在一实施例中,所述另一种有机功能材料选自主体材料;进一步地,所述另一种有机功能材料选自蓝光主体材料。
[0096]
本发明还涉及一种组合物,包含至少一种如上所述的含硼氮咔唑茚类化合物或混合物,及至少一种有机溶剂;所述的至少一种的有机溶剂选自芳族或杂芳族、酯、芳族酮或芳族醚、脂肪族酮或脂肪族醚、脂环族或烯烃类化合物,或硼酸酯或磷酸酯类化合物,或两种及两种以上溶剂的混合物。
[0097]
在一个优选的实施例中,按照本发明的一种组合物,其特征在于,所述的至少的一种有机溶剂选自基于芳族或杂芳族的溶剂。
[0098]
适合本发明的基于芳族或杂芳族溶剂的例子有,但不限制于:对二异丙基苯、戊苯、四氢萘、环己基苯、氯萘、1,4-二甲基萘、3-异丙基联苯、对甲基异丙苯、二戊苯、三戊苯、戊基甲苯、邻二乙苯、间二乙苯、对二乙苯、1,2,3,4-四甲苯、1,2,3,5-四甲苯、1,2,4,5-四甲苯、丁苯、十二烷基苯、二己基苯、二丁基苯、对二异丙基苯、环己基苯、苄基丁基苯、二甲基萘、3-异丙基联苯、对甲基异丙苯、1-甲基萘、1,2,4-三氯苯、4,4-二氟二苯甲烷、1,2-二甲氧基-4-(1-丙烯基)苯、二苯甲烷、2-苯基吡啶、3-苯基吡啶、n-甲基二苯胺、4-异丙基联苯、α,α-二氯二苯甲烷、4-(3-苯基丙基)吡啶、苯甲酸苄酯、1,1-双(3,4-二甲基苯基)乙烷、2-异丙基萘、喹啉、异喹啉、2-呋喃甲酸甲酯、2-呋喃甲酸乙酯等;
[0099]
适合本发明的基于芳族酮溶剂的例子有,但不限制于:1-四氢萘酮,2-四氢萘酮,2-(苯基环氧)四氢萘酮,6-(甲氧基)四氢萘酮,苯乙酮、苯丙酮、二苯甲酮、及它们的衍生物,如4-甲基苯乙酮、3-甲基苯乙酮、2-甲基苯乙酮、4-甲基苯丙酮、3-甲基苯丙酮、2-甲基苯丙酮等;
[0100]
适合本发明的基于芳族醚溶剂的例子有,但不限制于:3-苯氧基甲苯、丁氧基苯、对茴香醛二甲基乙缩醛、四氢-2-苯氧基-2h-吡喃、1,2-二甲氧基-4-(1-丙烯基)苯、1,4-苯并二噁烷、1,3-二丙基苯、2,5-二甲氧基甲苯、4-乙基本乙醚、1,3-二丙氧基苯、1,2,4-三甲氧基苯、4-(1-丙烯基)-1,2-二甲氧基苯、1,3-二甲氧基苯、缩水甘油基苯基醚、二苄基醚、4-叔丁基茴香醚、反式-对丙烯基茴香醚、1,2-二甲氧基苯、1-甲氧基萘、二苯醚、2-苯氧基甲醚、2-苯氧基四氢呋喃、乙基-2-萘基醚;
[0101]
在一些优选的实施例中,按照本发明的组合物,所述的至少一种的有溶剂可选自:脂肪族酮,例如,2-壬酮、3-壬酮、5-壬酮、2-癸酮、2,5-己二酮、2,6,8-三甲基-4-壬酮、葑酮、佛尔酮、异佛尔酮、二正戊基酮等;或脂肪族醚,例如,戊醚、己醚、二辛醚、乙二醇二丁醚、二乙二醇二乙醚、二乙二醇丁基甲醚、二乙二醇二丁醚、三乙二醇二甲醚、三乙二醇乙基甲醚、三乙二醇丁基甲醚、三丙二醇二甲醚、四乙二醇二甲醚等。
[0102]
在另一些优选的实施例中,按照本发明的组合物,所述的至少一种的有溶剂可选自基于酯的溶剂:辛酸烷酯、癸二酸烷酯、硬脂酸烷酯、苯甲酸烷酯、苯乙酸烷酯、肉桂酸烷酯、草酸烷酯、马来酸烷酯、烷内酯、油酸烷酯等。特别优选辛酸辛酯、癸二酸二乙酯、邻苯二甲酸二烯丙酯、异壬酸异壬酯。
[0103]
所述的溶剂可以是单独使用,也可以是作为两种或多种有机溶剂的混合物使用。
[0104]
在某些优选的实施例中,按照本发明的一种组合物,其特征在于,包含至少一种如上所述的有机化合物或高聚物或混合物及至少一种有机溶剂,还可进一步包含另一种有机溶剂。另一种有机溶剂的例子包括(但不限于):甲醇、乙醇、2-甲氧基乙醇、二氯甲烷、三氯甲烷、氯苯、邻二氯苯、四氢呋喃、苯甲醚、吗啉、甲苯、邻二甲苯、间二甲苯、对二甲苯、1,4二氧杂环己烷、丙酮、甲基乙基酮、1,2二氯乙烷、3-苯氧基甲苯、1,1,1-三氯乙烷、1,1,2,2-四氯乙烷、醋酸乙酯、醋酸丁酯、二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、四氢萘、萘烷、茚和/或它们的混合物。
[0105]
一些优选的实施例中,特别适合本发明的溶剂是汉森(hansen)溶解度参数在以下范围内的溶剂:
[0106]
δd(色散力)在17.0~23.2mpa1/2的范围,尤其是在18.5~21.0mpa1/2的范围;
[0107]
δp(极性力)在0.2~12.5mpa1/2的范围,尤其是在2.0~6.0mpa1/2的范围;
[0108]
δh(氢键力)在0.9~14.2mpa1/2的范围,尤其是在2.0~6.0mpa1/2的范围。
[0109]
按照本发明的组合物,其中有机溶剂在选取时需考虑其沸点参数。本发明中,所述的有机溶剂的沸点≥150℃;优选为≥180℃;较优选为≥200℃;更优为≥250℃;最优为≥300℃。这些范围内的沸点对防止喷墨印刷头的喷嘴堵塞是有益的。所述的有机溶剂可从溶剂体系中蒸发,以形成包含功能材料薄膜。
[0110]
在一个优选的实施方案中,按照本发明的组合物是一溶液。
[0111]
在另一个优选的实施方案中,按照本发明的组合物是一悬浮液。
[0112]
本发明实施例中的组合物中可以包括0.01至10wt%的按照本发明的化合物或混
合物,较好的是0.1至15wt%,更好的是0.2至5wt%,最好的是0.25至3wt%。
[0113]
本发明还涉及所述组合物作为涂料或印刷油墨在制备有机电子器件时的用途,特别优选的是通过打印或涂布的制备方法。
[0114]
其中,适合的打印或涂布技术包括(但不限于)喷墨打印,喷印(nozzle printing),活版印刷,丝网印刷,浸涂,旋转涂布,刮刀涂布,辊筒印花,扭转辊印刷,平版印刷,柔版印刷,轮转印刷,喷涂,刷涂或移印,狭缝型挤压式涂布等。首选的是凹版印刷,喷印及喷墨印刷。溶液或悬浮液可以另外包括一个或多个组份例如表面活性化合物,润滑剂,润湿剂,分散剂,疏水剂,粘接剂等,用于调节粘度,成膜性能,提高附着性等。有关打印技术,及其对有关溶液的相关要求,如溶剂及浓度,粘度等。
[0115]
本发明还提供一种如上所述的含硼氮咔唑茚类化合物、混合物或组合物在有机电子器件中的应用,所述的有机电子器件可选于,但不限于,有机发光二极管(oled),有机光伏电池(opv),有机发光电池(oleec),有机场效应管(ofet),有机发光场效应管,有机激光器,有机自旋电子器件,有机传感器及有机等离激元发射二极管(organic plasmon emitting diode)等,特别优选为oled。本发明实施例中,优选将所述有机化合物用于oled器件的发光层。
[0116]
本发明进一步涉及一种有机电子器件,包含至少一功能层,所述功能层包含一种如上所述的含硼氮咔唑茚类化合物、混合物或由上述的组合物制备而成。进一步地,所述有机电子器件,包含阴极、阳极和至少一功能层,所述功能层包含一种如上所述的多环化合物或混合物或由上述的组合物制备而成。所述功能层选自空穴注入层(hil)、空穴传输层(htl)、发光层(eml)、电子阻挡层(ebl)、电子注入层(eil)、电子传输层(etl)、空穴阻挡层(hbl);优选地,所述功能层选自发光层。
[0117]
所述的有机电子器件可选于,但不限于,有机发光二极管(oled),有机光伏电池(opv),有机发光电池(oleec),有机场效应管(ofet),有机发光场效应管,有机激光器,有机自旋电子器件,有机传感器及有机等离激元发射二极管(organic plasmon emitting diode)等,特别优选的是有机电致发光器件,如oled,oleec,有机发光场效应管。
[0118]
在以上所述的电致发光器件,特别是oled中,包括一基片,一阳极,至少一发光层,一阴极。
[0119]
基片可以是不透明或透明。一个透明的基板可以用来制造一个透明的发光元器件。例如可参见,bulovic等nature 1996,380,p29,和gu等,appl.phys.lett.1996,68,p2606。基片可以是刚性的或弹性的。基片可以是塑料,金属,半导体晶片或玻璃。最好是基片有一个平滑的表面。无表面缺陷的基板是特别理想的选择。在一个优选的实施例中,基片是柔性的,可选于聚合物薄膜或塑料,其玻璃化温度tg为150℃以上,较好是超过200℃,更好是超过250℃,最好是超过300℃。合适的柔性基板的例子有聚(对苯二甲酸乙二醇酯)(pet)和聚乙二醇(2,6-萘)(pen)。
[0120]
阳极可包括一导电金属或金属氧化物,或导电聚合物。阳极可以容易地注入空穴到空穴注入层(hil)或空穴传输层(htl)或发光层中。在一个的实施例中,阳极的功函数和发光层中的发光体或作为hil或htl或电子阻挡层(ebl)的p型半导体材料的homo能级或价带能级的差的绝对值小于0.5ev,较好是小于0.3ev,最好是小于0.2ev。阳极材料的例子包括但不限于:al、cu、au、ag、mg、fe、co、ni、mn、pd、pt、ito、铝掺杂氧化锌(azo)等。其他合适
的阳极材料是已知的,本领域普通技术人员可容易地选择使用。阳极材料可以使用任何合适的技术沉积,如一合适的物理气相沉积法,包括射频磁控溅射,真空热蒸发,电子束(e-beam)等。在某些实施例中,阳极是图案结构化的。图案化的ito导电基板可在市场上买到,并且可以用来制备根据本发明的器件。
[0121]
阴极可包括一导电金属或金属氧化物。阴极可以容易地注入电子到eil或etl或直接到发光层中。在一个的实施例中,阴极的功函数和发光层中发光体或作为电子注入层(eil)或电子传输层(etl)或空穴阻挡层(hbl)的n型半导体材料的lumo能级或导带能级的差的绝对值小于0.5ev,较好是小于0.3ev,最好是小于0.2ev。原则上,所有可用作oled的阴极的材料都可能作为本发明器件的阴极材料。阴极材料的例子包括但不限于:al、au、ag、ca、ba、mg、lif/al、mgag合金、baf2/al、cu、fe、co、ni、mn、pd、pt、ito等。阴极材料可以使用任何合适的技术沉积,如一合适的物理气相沉积法,包括射频磁控溅射,真空热蒸发,电子束(e-beam)等。
[0122]
oled还可以包含其他功能层,如空穴注入层(hil)、空穴传输层(htl)、电子阻挡层(ebl)、电子注入层(eil)、电子传输层(etl)、空穴阻挡层(hbl)。适合用于这些功能层中的材料在上面及在wo2010135519a1、us20090134784a1和wo2011110277a1中有详细的描述,特此将此3篇专利文件中的全部内容并入本文作为参考。
[0123]
在一个优选的实施例中,按照本发明的发光器件中,其发光层是通过按照本发明的组合物制备而成。
[0124]
按照本发明的发光器件,其发光波长在300到1000nm之间,较好的是在350到900nm之间,更好的是在400到800nm之间。
[0125]
本发明还涉及按照本发明的有机电子器件在各种电子设备中的应用,包括,但不限于,显示设备,照明设备,光源,传感器等等。
[0126]
本发明还涉及包含有按照本发明的有机电子器件的电子设备,包括,但不限于,显示设备,照明设备,光源,传感器等等。
[0127]
具体实施例
[0128]
1、化合物的合成
[0129][0130]
实施例1
[0131]
化合物(1)的合成路线如下所示:
[0132][0133]
中间体1-3的合成:
[0134]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体1-1与10mmol中间体1-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷多次萃取有机相,合并有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体1-3摩尔量为8.42mmol,产率:84.2%。ms(asap)=277.1。
[0135]
中间体1-5的合成:
[0136]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体1-3与1mmol中间体1-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,
用乙酸乙酯重结晶得中间体1-5的摩尔量为0.67mmol,反应收率为:67%,ms(asap)=425.7。
[0137]
化合物(1)的合成:
[0138]
向250ml的三口烧瓶中加入10mmol中间体1-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为48.4%,ms(asap)=355.6。
[0139]
实施例2
[0140]
化合物(2)的合成路线如下所示:
[0141][0142]
中间体2-3的合成:
[0143]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体2-1与10mmol中间体2-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体2-3摩尔量为8.31mmol,产率:83.1%。ms(asap)=277.1。
[0144]
中间体2-5的合成:
[0145]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体2-3与1mmol中间体2-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体2-5的摩尔量为0.63mmol,反应收率为:63%,ms(asap)=575.7。
[0146]
化合物(2)的合成:
[0147]
向250ml的三口烧瓶中加入10mmol中间体2-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2
小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为57.2%,ms(asap)=505.4。
[0148]
实施例3
[0149]
化合物(18)的合成路线如下所示:
[0150][0151]
中间体18-3的合成:
[0152]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体18-1与10mmol中间体18-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体18-3摩尔量为7.64mmol,产率:76.4%。ms(asap)=277.1。
[0153]
中间体18-5的合成:
[0154]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体18-3与1mmol中间体18-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体18-5的摩尔量为0.54mmol,反应收率为:54%,ms(asap)=625.7。
[0155]
化合物(18)的合成:
[0156]
向250ml的三口烧瓶中加入10mmol中间体18-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷
却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为46.8%,ms(asap)=555.4。
[0157]
实施例4
[0158]
化合物(31)的合成路线如下所示:
[0159][0160]
中间体31-3的合成:
[0161]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体31-1与10mmol中间体31-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体31-3摩尔量为6.93mmol,产率:69.3%。ms(asap)=327.1。
[0162]
中间体31-5的合成:
[0163]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体31-3与1mmol中间体31-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体31-5的摩尔量为0.84mmol,反应收率为:84%,ms(asap)=541.1。
[0164]
化合物(31)的合成:
[0165]
向250ml的三口烧瓶中加入10mmol中间体31-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷
却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为24.6%,ms(asap)=471.2。
[0166]
实施例5
[0167]
化合物(32)的合成路线如下所示:
[0168][0169]
中间体32-3的合成:
[0170]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体32-1与10mmol中间体32-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体32-3摩尔量为8.83mmol,产率:88.3%。ms(asap)=327.1。
[0171]
中间体32-5的合成:
[0172]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体32-3与1mmol中间体32-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体32-5的摩尔量为0.76mmol,反应收率为:76%,ms(asap)=633.7。
[0173]
化合物(32)的合成:
[0174]
向250ml的三口烧瓶中加入10mmol中间体32-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋
蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为38.6%,ms(asap)=563.4。
[0175]
实施例6
[0176]
化合物(35)的合成路线如下所示:
[0177][0178]
中间体35-3的合成:
[0179]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体35-1与10mmol中间体35-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体35-3摩尔量为8.16mmol,产率:81.6%。ms(asap)=327.1。
[0180]
中间体35-5的合成:
[0181]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体35-3与1mmol中间体35-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体35-5的摩尔量为0.50mmol,反应收率为:50%,ms(asap)=550.2。
[0182]
化合物(35)的合成:
[0183]
向250ml的三口烧瓶中加入10mmol中间体35-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为29.7%,ms(asap)=480.5。
[0184]
实施例7
[0185]
化合物(41)的合成路线如下所示:
[0186][0187]
中间体41-3的合成:
[0188]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体41-1与10mmol中间体41-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体41-3摩尔量为4.58mmol,产率:45.8%。ms(asap)=377.1。
[0189]
中间体41-5的合成:
[0190]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体41-3与1mmol中间体41-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体41-5的摩尔量为0.58mmol,反应收率为:58%,ms(asap)=525.3。
[0191]
化合物(41)的合成:
[0192]
向250ml的三口烧瓶中加入10mmol中间体41-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为54.7%,ms(asap)=455.1。
[0193]
实施例8
[0194]
化合物(53)的合成路线如下所示:
[0195][0196]
中间体53-3的合成:
[0197]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体53-1与10mmol中间体53-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体53-3摩尔量为5.86mmol,产率:58.6%。ms(asap)=377.1。
[0198]
中间体53-5的合成:
[0199]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体53-3与1mmol中间体53-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体53-5的摩尔量为0.87mmol,反应收率为:87%,ms(asap)=709.3。
[0200]
化合物(53)的合成:
[0201]
向250ml的三口烧瓶中加入10mmol中间体53-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为57.6%,ms(asap)=639.2。
[0202]
实施例9
[0203]
化合物(63)的合成路线如下所示:
[0204][0205]
中间体63-3的合成:
[0206]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体63-1与10mmol中间体63-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体104-3摩尔量为6.73mmol,产率:67.3%。ms(asap)=427.6。
[0207]
中间体63-5的合成:
[0208]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体63-3与1mmol中间体63-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体63-5的摩尔量为0.59mmol,反应收率为:59%,ms(asap)=697.4。
[0209]
化合物(63)的合成:
[0210]
向250ml的三口烧瓶中加入10mmol中间体63-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为47.6%,ms(asap)=627.8。
[0211]
实施例10
[0212]
化合物(78)的合成路线如下所示:
[0213][0214]
中间体78-3的合成:
[0215]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体78-1与10mmol中间体78-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体78-3摩尔量为7.63mmol,产率:76.3%。ms(asap)=427.6。
[0216]
中间体78-5的合成:
[0217]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体78-3与1mmol中间体78-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体78-5的摩尔量为0.49mmol,反应收率为:49%,ms(asap)=716.5。
[0218]
化合物(78)的合成:
[0219]
向250ml的三口烧瓶中加入10mmol中间体78-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为25.9%,ms(asap)=646.6。
[0220]
实施例11
[0221]
化合物(92)的合成路线如下所示:
[0222][0223]
中间体92-3的合成:
[0224]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体92-1与10mmol中间体92-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体92-3摩尔量为4.96mmol,产率:49.6%。ms(asap)=551.1。
[0225]
中间体92-5的合成:
[0226]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体92-3与1mmol中间体92-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体92-5的摩尔量为0.62mmol,反应收率为:62%,ms(asap)=731.5。
[0227]
化合物(92)的合成:
[0228]
向250ml的三口烧瓶中加入10mmol中间体92-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为35.3%,ms(asap)=661.4。
[0229]
实施例12
[0230]
化合物(110)的合成路线如下所示:
[0231][0232]
中间体110-3的合成:
[0233]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体110-1与10mmol中间体110-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体110-3摩尔量为8.84mmol,产率:88.4%。ms(asap)=433.2。
[0234]
中间体110-5的合成:
[0235]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体110-3与1mmol中间体110-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体110-5的摩尔量为0.47mmol,反应收率为:47%,ms(asap)=672.4。
[0236]
化合物(110)的合成:
[0237]
向250ml的三口烧瓶中加入10mmol中间体110-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为87.3%,ms(asap)=602.1
[0238]
实施例13
[0239]
化合物(120)的合成路线如下所示:
[0240][0241]
中间体120-3的合成:
[0242]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体120-1与10mmol中间体120-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体120-3摩尔量为6.63mmol,产率:66.3%。ms(asap)=513.3。
[0243]
中间体120-5的合成:
[0244]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体120-3与1mmol中间体120-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体120-5的摩尔量为0.57mmol,反应收率为:57%,ms(asap)=839.1。
[0245]
化合物(120)的合成:
[0246]
向250ml的三口烧瓶中加入10mmol中间体120-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为77.4%,ms(asap)=769.2。
[0247]
实施例14
[0248]
化合物(121)的合成路线如下所示:
[0249][0250]
中间体121-3的合成:
[0251]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体121-1与10mmol中间体121-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体120-3摩尔量为4.59mmol,产率:45.9%。ms(asap)=579.3。
[0252]
中间体121-5的合成:
[0253]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体121-3与1mmol中间体121-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体121-5的摩尔量为0.43mmol,反应收率为:43%,ms(asap)=741.3。
[0254]
化合物(121)的合成:
[0255]
向250ml的三口烧瓶中加入10mmol中间体121-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为75.3%,ms(asap)=671.4。
[0256]
实施例15
[0257]
化合物(125)的合成路线如下所示:
[0258][0259]
中间体125-3的合成:
[0260]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol中间体125-1与10mmol中间体125-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到中间体125-3摩尔量为7.42mmol,产率:74.2%。ms(asap)=447.6。
[0261]
中间体125-5的合成:
[0262]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol中间体125-3与1mmol中间体125-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得中间体125-5的摩尔量为0.63mmol,反应收率为:63%,ms(asap)=849.1。
[0263]
化合物(125)的合成:
[0264]
向250ml的三口烧瓶中加入10mmol中间体125-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为67.3%,ms(asap)=779.3。
[0265]
对比例2(bd-ref2)
[0266][0267]
化合物对比例2的合成路线如下所示:
[0268][0269]
对比例中间体2-3的合成:
[0270]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入10mmol对比例中间体2-1与20mmol对比例中间体2-2、0.2mmol醋酸钯、0.2mmol三叔丁基膦、20mmol碳酸钾,加入150ml的甲苯使其溶解,加热至80℃至反应液回流,反应12小时,待反应完全,加水淬灭反应,同时用二氯甲烷萃取有机相,合并多次洗涤有机相,用无水硫酸镁干燥,过滤,旋蒸干溶剂得粗产品,用快速柱层析色谱法纯化得到对比例中间体2-3摩尔量为6.11mmol,产率:61.1%。ms(asap)=338.1
[0271]
对比例中间体2-5的合成:
[0272]
氮气保护氛围下,在一干燥的三口烧瓶中,分别加入1mmol对比例中间体2-3与1mmol对比例中间体2-4,倒入100ml的dmso作为溶剂,加入干燥k2co3作碱,120℃条件下反应8小时,tlc监测反应,待反应完全后,将反应液冷却至室温,依次加入水与二氯甲烷,用水洗涤反应液多次,同时用二氯甲烷萃取水相多次,合并有机相,用无水na2co3干燥,过滤,旋干反应液,得粗产品,用乙酸乙酯重结晶得对比例中间体2-5的摩尔量为0.57mmol,反应收率为:57%,ms(asap)=579.3
[0273]
对比例(2)的合成:
[0274]
向250ml的三口烧瓶中加入10mmol对比例中间体2-5以及100ml干燥叔丁基苯,在n2气氛中,冷却至-30摄氏度,逐滴加入30.6mmol t-buli正已烷溶液。升高温度至60摄氏度反应2小时,减压蒸除其中的正已烷溶剂。将反应液再次冷却至-30摄氏度,加入10.5mmol三溴化硼溶液,升至室温下搅拌0.5小时,然后将反应液冷却至0摄氏度,加入21mmol n,n-二异丙基乙基胺,待滴加完毕,升温至室温搅拌,再继续升温至120摄氏度搅拌3小时,将反应液冷却至室温。加入碳酸钠水溶液与乙酸乙酯淬灭反应。水相用乙酸乙酯萃取并合并有机
相,旋蒸除掉其中的溶剂,得到粗品,用快速硅胶柱纯化得到纯品。用甲苯与乙酸乙酯重结晶,得产品淡黄色固体粉末。收率为41.7%,ms(asap)=509.2
[0275]
2、化合物的能级计算
[0276]
有机化合物材料的能级可通过量子计算得到,比如利用td-dft(含时密度泛函理论)通过gaussian09w(gaussian inc.),具体的模拟方法可参见wo2011141110。首先用半经验方法“ground state/semi-empirical/default spin/am1”(charge 0/spin singlet)来优化分子几何结构,然后有机分子的能量结构由td-dft(含时密度泛函理论)方法算得“td-scf/dft/default spin/b3pw91”与基组“6-31g(d)”(charge 0/spin singlet)。homo和lumo能级按照下面的校准公式计算,s1,t1和谐振因子f(s1)直接使用。
[0277]
homo(ev)=((homo(g)
×
27.212)-0.9899)/1.1206
[0278]
lumo(ev)=((lumo(g)
×
27.212)-2.0041)/1.385
[0279]
其中homo(g)和lumo(g)是gaussian 09w的直接计算结果,单位为hartree。结果如表1所示:
[0280]
表1
[0281][0282][0283]
3、oled器件的制备与表征
[0284]
器件结构为:
[0285][0286]
上述材料bh、et、liq、bd-ref1均是商业够得,其合成方法为现有技术,详见现有技术中的参考文献,在此不再赘述。其中bh作为主体材料,et作为电子传输材料,liq作为电子注入材料。
[0287]
下面通过具体实施例来详细说明采用上述化合物的oled器件的制备过程,该oled器件的结构为:ito/hil/htl/eml/etl/阴极,制备步骤如下:
[0288]
a、ito(铟锡氧化物)导电玻璃基片的清洗:使用各种溶剂(例如氯仿、丙酮或异丙醇中的一种或几种)清洗,然后进行紫外臭氧处理;
[0289]
b、hil(空穴注入层,40nm):60nm的pedot(聚乙撑二氧噻吩,cleviostmai4083)作为hil在超净室旋转涂布而成,并在180℃的热板上处理10分钟;
[0290]
c、htl(空穴传输层,20nm):20nm的tfb或pvk(sigma aldrich,平均mn 25,000-50,000)是在氮气手套箱中通过旋转涂布而成,所用的溶液是加入至甲苯溶剂的tfb或pvk,溶液溶度5mg/ml,随后在180℃的热板上处理60分钟;
[0291]
d、eml(有机发光层,40nm):eml是在氮气手套箱中通过旋转涂布而成,所用的溶液不同主客体的苯甲酸甲酯溶液(主客体的重量比例为95:5),溶液溶度15mg/ml,随后在140℃的热板上处理10分钟,主体结构为bh,客体采用如表2所述的有机化合物,其他实施方案均相同。
[0292]
e、电子传输层和阴极:将热处理后的基板转移至真空腔体,接着将et和liq置于不同的蒸发单元,在高真空(1
×
10-6毫巴)中使其分别以50重量%的比例进行共沉积,在发光层上形成20nm的电子传输层,随后再沉积厚度为100nm的al阴极。
[0293]
f、封装:器件在氮气手套箱中用紫外线固化树脂封装。
[0294]
各oled器件的电流电压(j-v)特性通过表征设备来表征,同时记录重要的参数如效率,寿命及外部量子效率,如表2所示:
[0295]
表2
[0296][0297]
由表2可知,将本技术的化合物作为eml层的bd,制得的oled器件的效率及寿命方面均好于现有常用的bd材料bd-ref。特别是当材料选自化合物92、120、121和125时,其寿命提高约20%,同时器件效率也提高了约8%。这可能是本发明化合物有较大的增溶基团(在结构中引入了甲基、t-am或t-bu),分子尺寸变大,成膜后稳定性增大。
[0298]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0299]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1