含有咔啉结构的铂(II)配合物及其制备方法和应用、有机光电器件与流程
含有咔啉结构的铂(ii)配合物及其制备方法和应用、有机光电器件
技术领域
1.本发明涉及磷光发光材料技术领域,具体涉及含有咔啉结构的铂(ii)配合物及其制备方法和应用、有机光电器件。
背景技术:
2.oled即有机发光二极管(organic light-emitting diode)或有机发光器件(organic light-emitting device),又被称为有机电致发光器件。有机电致发光是指在顺向偏压电场的作用下有机小分子、金属有机配合物分子或聚合物分子发光材料将电能直接转化为光能的一种发光现象。
3.早期研究的环金属铂(ii)配合物磷光材料多为含有双齿配体和三齿配体的金属有机分子,其刚性较低,两个双齿配体易扭曲、振动而使其磷光量子效率低下;含有三齿配体的环金属铂(ii)配合物由于分子需要第二个配体(如cl-、苯氧基负离子、炔负离子、卡宾等),会使配合物的化学稳定性降低,故双齿和三齿环金属铂(ii)配合物磷光材料均不利于制备稳定而高效的oled器件。而含有四齿配体的环金属铂(ii)配合物分子结构的刚性大大提高,使其磷光量子效率也大为提高,甚至高达100%,且其热稳定性和电化学稳定性亦非常好,因此基于此类磷光材料的oled器件性能也得到了巨大的提高,非常有希望达到了商业化应用的要求。
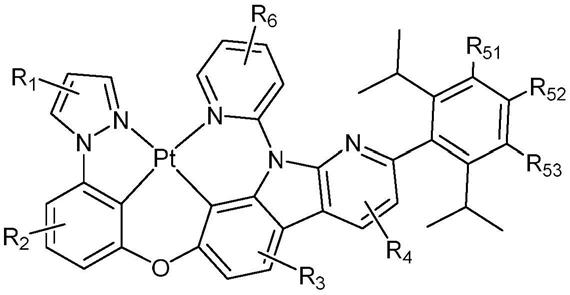
4.feiling yu等在“blue-phosphorescent pt(ii)complexes of tetradentate pyridyl-carbolinyl ligands:synthesis,structure,photophysics,and electroluminescence inorganic chemistry https://dx.doi.org/10.1021/acs.inorgchem.0c02244”中公开了三个含有咔啉结构的铂(ii)配合物,这三个配合物在用于溶液法制备有机发光二极管器件中时,具有蓝磷光电致发光的效果,但是,这三个配合物还具有器件发光效率低的限制,最高外量子效率仅为3.8%。
5.因此,虽然目前金属有机小分子磷光材料已取得了长足的发展,但是还没有能满足实际显示应用需求蓝磷光材料和器件,因此需要开发新型的蓝磷光材料,并不断提高其器件的发光色纯度、发光效率等。
技术实现要素:
6.本发明的目的是为了克服现有技术存在的oled器件缺少能够发射稳定高效蓝光的络合物发光材料的不足。
7.为了实现上述目的,本发明的第一方面提供一种含有咔啉结构的铂(ii)配合物,所述铂(ii)配合物为式(i)所示的配合物:
8.式(i):
9.其中,在式(i)中,
10.r1、r2、r3、r4、r
51
、r
52
、r
53
和r6各自独立地选自h、c
1-c
15
的烷基、苯基、由c
1-c
20
的烷基中的至少一种取代的苯基中的至少一种。
11.本发明的第二方面提供一种制备前述含有咔啉结构的铂(ii)配合物的方法,该方法包括:
12.(1)在保护气和第一铜试剂存在下,将式(ii)所示的1h-吡唑和式(iii)所示的1,3-二碘苯进行第一偶联反应,得到式(iv)所示的1-(3-碘苯基)-1h-吡唑;
13.(2)在保护气和第二铜试剂存在下,将式(iv)所示的1-(3-碘苯基)-1h-吡唑与式(v)所示的化合物进行第二偶联反应,得到式(vi)所示的化合物;
14.(3)在铂试剂存在下,将式(vi)所示的化合物进行环金属化反应,得到式(i)所示的含有咔啉结构的铂(ii)配合物;
15.式(i):
16.式(ii):式(iii):式(iv):
17.式(v):
18.式(vi):
19.其中,式(i)、式(ii)、式(iii)、式(iv)、式(v)和式(vi)中的基团的定义与本发明前文中的定义对应相同。
20.本发明的第三方面提供一种所述的含有咔啉结构的铂(ii)配合物在有机光电器件中的应用。
21.本发明的第四方面提供一种有机光电器件,该器件中含有基体、阳极层、空穴传输层、发光层、电子传输层和金属阴极层,所述发光层、所述电子传输层和所述空穴传输层中的至少一层中包含前述含有咔啉结构的铂(ii)配合物。
22.通过上述技术方案,本发明至少具有以下技术效果:
23.本发明提供的含有咔啉结构的铂(ii)配合物能够作为有机蓝光发光体发射稳定高效且长波长的蓝光,且其发出的蓝光具有能量低、光谱稳定的特点。
附图说明
24.图1是根据本发明的一种实施方式的配合物1分别在dcm溶液和pmma薄膜中的光致发光光谱图;
25.图2是根据本发明的一种实施方式的对比配合物1分别在dcm溶液和pmma薄膜中的光致发光光谱图;
26.图3是根据本发明的一种实施方式的对比配合物2分别在dcm溶液和pmma薄膜中的光致发光光谱图;
27.图4是根据本发明的一种实施方式的配合物1的紫外可见吸收光谱图;
28.图5是根据本发明的一种实施方式的对比配合物1的前沿轨道分布图;
29.图6是根据本发明的一种实施方式的对比配合物2的前沿轨道分布图;
30.图7是根据本发明的一种实施方式的配合物1的前沿轨道分布图;
31.图8是根据本发明的一种实施方式的有机光电器件的结构示意图;
32.图9是根据本发明的一种实施方式的有机光电器件的断面图;
33.图10是根据本发明的一种实施方式的器件1的电致发光光谱图;
34.图11是根据本发明的一种实施方式的器件1的光电转换电流效率图;
35.图12是根据本发明的一种实施方式的器件1的功率效率图;
36.图13是根据本发明的一种实施方式的器件1的外量子效率图。
37.附图标记说明
38.1000
ꢀꢀ
有机光电器件
ꢀꢀꢀꢀꢀꢀꢀꢀ
1002
ꢀꢀ
基体
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1004
ꢀꢀ
阳极层
39.1006
ꢀꢀ
空穴传输层
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1008
ꢀꢀ
发光层
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1010
ꢀꢀ
电子传输层
40.1012
ꢀꢀ
金属阴极层
具体实施方式
41.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
42.如前所述,本发明的第一方面提供了一种含有咔啉结构的铂(ii)配合物,所述铂(ii)配合物为式(i)所示的配合物:
43.式(i):
44.其中,在式(i)中,
45.r1、r2、r3、r4、r
51
、r
52
、r
53
和r6各自独立地选自h、c
1-c
15
的烷基、苯基、由c
1-c
20
的烷基中的至少一种取代的苯基中的至少一种。
46.本发明中,c
1-c
20
的烷基表示碳原子总数为1-20的烷基基团,可以为直链的、支链的或者环状的烷基基团,例如可以为碳原子数分别为1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20的烷基,示例性地,c
1-c
20
的烷基可以为甲基、乙基、正丙基、异丙基、异丁基、叔丁基和环己基中的一种。本发明中,对于“c
1-c
15
的烷基”、“c
1-c
10
的烷基”的定义与前述的“c
1-c
20
的烷基”的定义相似,仅仅是碳原子数不同。
47.根据一种优选的具体实施方式:在式(i)中,
48.r1、r2、r3、r4、r
51
、r
52
、r
53
和r6各自独立地选自h、c
1-c
10
的烷基、苯基、由c
1-c
15
的烷基中的至少一种取代的苯基中的至少一种。
49.根据另一种优选的具体实施方式,在式(i)中,
50.r1、r2、r3、r4、r
51
、r
52
、r
53
和r6各自独立地选自h、c
1-c6的烷基、苯基、由c
1-c
10
的烷基中的至少一种取代的苯基中的至少一种。
51.根据还有一种优选的具体实施方式,在式(i)中,
52.r1、r2、r3、r4、r
51
、r
52
、r
53
和r6各自独立地选自h、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、正己基、苯基、由取代基x取代的苯基中的至少一种;所述x选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、正己基中的至少一种。
53.根据还有一种优选的具体实施方式,在式(i)中,
54.r1、r2、r3、r4、r
51
、r
52
、r
53
和r6各自独立地选自h、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、苯基、甲基取代的苯基中的至少一种。
55.根据还有一种优选的具体实施方式,在式(i)中,
56.r1、r2、r3、r4和r6各自独立地选自h、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、苯基、甲基取代的苯基中的至少一种;
57.r
51
、r
52
和r
53
各自独立地选自h和异丙基。
58.根据还有一种优选的具体实施方式,所述铂(ii)配合物选自以下中的至少一种:
59.配合物1:
60.配合物2:
61.配合物3:
62.配合物4:
63.配合物5:
64.配合物6:
65.配合物7:
66.本发明的发明人发现:咔啉片段2位含有大位阻2,6-二异丙基取代基时器件性能能够得到较大幅度的提高,更适合作为磷光掺杂材料制备蓝磷光器件。示例性地,同样溶液法制备的含有本发明的配合物1的器件的最高电流效率和外量子效率分别达到22.1cd/a和10.9%,是同类型材料论文报道中pt(ppzoclpy-ipr)所制备器件的两倍以上。
67.本发明对制备所述含有咔啉结构的铂(ii)配合物的方法没有特别的限制,本领域技术人员能够根据本发明提供的结构式,结合有机合成领域内的已知合成方法获得合适的步骤以合成本发明所述的配合物,本发明的后文中示例性地提供了几种具体化合物的制备方法,本领域技术人员不应理解为对本发明的限制。并且,本领域技术人员也可以参照feiling yu等在“blue-phosphorescent pt(ii)complexes of tetradentate pyridyl-carbolinyl ligands:synthesis,structure,photophysics,and electroluminescence,inorganic chemistry https://dx.doi.org/10.1021/acs.inorgchem.0c02244”中公开的具体制备方法,通过替换原料而获得具体的制备配合物的方法。
68.但是,为了使得本发明的铂(ii)配合物的收率和纯度更高,如前所述,本发明的第二方面还提供了一种制备所述的含有咔啉结构的铂(ii)配合物的方法,该方法包括:
69.(1)在保护气和第一铜试剂存在下,将式(ii)所示的1h-吡唑和式(iii)所示的1,3-二碘苯进行第一偶联反应,得到式(iv)所示的1-(3-碘苯基)-1h-吡唑;
70.(2)在保护气和第二铜试剂存在下,将式(iv)所示的1-(3-碘苯基)-1h-吡唑与式(v)所示的化合物进行第二偶联反应,得到式(vi)所示的化合物;
71.(3)在铂试剂存在下,将式(vi)所示的化合物进行环金属化反应,得到式(i)所示的含有咔啉结构的铂(ii)配合物;
72.式(i):
73.式(ii):式(iii):式(iv):
74.式(v):
75.式(vi):
76.其中,式(i)、式(ii)、式(iii)、式(iv)、式(v)和式(vi)中的基团的定义与本发明的前述定义对应相同。
77.优选地,在步骤(1)中,所述第一偶联反应的反应条件包括:温度为110-140℃,时间为4-6h。
78.更优选地,在步骤(1)中,所述第一铜试剂为卤化亚铜,更优选为碘化亚铜。
79.优选地,在步骤(1)中,式(ii)所示的1h-吡唑、式(iii)所示的1,3-二碘苯和第一铜试剂的投料摩尔比为1:(2-5):(0.1-0.5)。
80.在本发明的一个优选的实施方式中,所述第一偶联反应的步骤包括:将式(ii)所示的1h-吡唑、式(iii)所示的1,3-二碘苯、碘化亚铜、l-脯氨酸(作为催化剂配体)、碳酸钾和dmso混合均匀后,在保护气体存在下,例如在氮气环境中加热到110-140℃下搅拌反应4-6h。
81.优选地,在步骤(2)中,所述第二偶联反应的反应条件包括:温度为110-140℃,时间为4-6h。
82.更优选地,在步骤(2)中,所述第二铜试剂为卤化亚铜,更优选为碘化亚铜。
83.本发明中,优选地,在步骤(2)中,式(v)所示的化合物与式(iv)所示的1-(3-碘苯
(0.03mmol),磷酸钾(0.5mmol),dmso(8ml)和h2o(2ml),接着在氮气环境中加热到110℃下搅拌18h,冷却至室温,加入水和乙酸乙酯萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-2,橙色固体,产率为67%。
114.(3)制备中间体1-3(2-(2,6-二异丙基苯基)吡啶1-氧化物)
[0115][0116]
向25ml的史莱克管中加入中间体1-2(0.42mmol),3-氯苯并过氧酸(0.63mmol)和dcm(5ml),接着在氮气环境中室温下搅拌12h,加入碳酸氢钠饱和溶液中和至中性,接着用二氯甲烷萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-3,白色液体,产率为93.4%。
[0117]
(4)制备中间体1-4(2-氯-6-(2,6-二异丙基)吡啶)
[0118][0119]
向25ml的史莱克管中加入中间体1-3(0.4mmol),草酰氯(0.56mmol),三乙胺(0.56mmol)和ch2cl2(0.2ml),接着在氮气环境中室温下搅拌12h,冷却至室温,加入碳酸氢钠饱和溶液中和,用二氯甲烷萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-4,浅黄色液体,产率为56%。
[0120]
(5)制备中间体1-5(n-(2-溴-5-甲氧基苯基)-6-(2,6-二异丙基苯基)吡啶-2-胺)
[0121][0122]
向150ml的史莱克管中加入中间体1-4(13mmol),2-溴-5-甲氧基苯胺2-溴-5-甲氧基苯胺(6.5mmol),pd2(dba)3(0.325mmol),binap(0.325mmol)、叔丁醇钠(19.5mmol)和甲苯(60ml)溶液,接着在氮气环境中加热到110℃下搅拌24h,冷却至室温,加入水和乙酸乙酯萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-5,浅黄色液体,产率为43%。
[0123]
(6)制备中间体1-6(2-(2,6-二异丙基苯基)-7-甲氧基-9h-吡啶并[2,3-b]吲哚)
[0124][0125]
向75ml的封管中加入中间体1-5(2.5mmol),醋酸钯(0.5mmol),三苯基膦(0.5mmol),dbu(10mmol)和dmac(20ml),接着在氮气环境中加热到130℃下搅拌5h,冷却至室温,加入水和乙酸乙酯萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-6,橙色固体,产率为47%。
[0126]
(7)制备中间体1-7(2-(2,6-二异丙基苯基)-7-甲氧基-9-(吡啶-2-基)-9h-吡啶并[2,3-b]吲哚)
[0127][0128]
上式中,l为(1r,2r)-环己烷-1,2-二胺。
[0129]
向25ml的史莱克管中加入中间体1-6(1mmol),2-溴吡啶(1.2mmol),碘化亚铜(0.2mmol),(1r,2r)-环己烷-1,2-二胺(0.2mmol),磷酸钾(4mmol)和dmso(8ml),接着在氮气环境中加热到120℃下搅拌24h,冷却至室温,加入水和乙酸乙酯萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-7,白色固体,产率为50%。
[0130]
(8)制备中间体1-8(2-(2,6-二异丙基苯基)-9-(吡啶-2-基)-9h-吡啶并[2,3-b]吲哚-7-醇)
[0131][0132]
向10ml的史莱克管中加入中间体1-7(0.25mmol)和hbr(2ml),接着在氮气环境中加热到120℃搅拌12h,冷却至室温,加入碳酸氢钠饱和溶液中和至中性,用乙酸乙酯萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-8,黄色固体,产率为83%。
[0133]
(8)制备中间体1-9(7-(3-(1h-吡唑-1-基)苯氧基)-2-(2,6-二异丙基苯基)-9-(吡啶-2-基)-9h-吡啶并[2,3-b]吲哚)
[0134][0135]
上式中,l为n,n-二甲基甘氨酸;
[0136]
向25ml的史莱克管中加入中间体1-8(0.8mmol),(3-碘苯基)-1h-吡唑(0.96mmol),碘化亚铜(0.08mmol),n,n-二甲基甘氨酸(0.08mmol),碳酸铯(2.4mmol)和dmso(5ml),接着在氮气环境中加热到120℃下搅拌24h,冷却至室温,加入水和乙酸乙酯萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到中间体1-9,白色固体,产率为58%。
[0137]
(10)配合物1的合成:
[0138][0139]
向48ml的封管中加入中间体1-9(0.58mmol),k2ptcl4(0.64mmol)和dmf(60ml),接着在氮气环境中室温下搅拌1天,然后将混合物加热至120℃搅拌48h,冷却至室温后,加入水和二氯甲烷萃取,有机相合并,无水na2so4干燥,旋干得粗产物。通过柱色谱法纯化粗产物,得到浅黄色固体,产率为48%。
[0140]
配合物1:1h nmr(400mhz,dmso-d6)δ9.51(d,j=8.9hz),9.27(d,j=5.8hz),9.05,8.91(d,j=2.7hz),8.64(d,j=7.8hz),8.21(ddd,j=8.9,7.2,1.8hz),8.10(d,j=2.2hz),8.00(d,j=8.3hz),7.53(d,j=7.8hz),7.30(d,j=8.3hz),7.24(t,j=7.9hz),7.13(d,j=2.4hz),6.99(ddd,j=8.3,3.6,1.7hz),6.88(dd,j=5.3,3.0hz),6.67(d,j=8.3hz),2.64-2.52(m),1.10(d,j=6.8hz).
13
c nmr(75mhz,cdcl3)δ153.01,150.30,148.17,146.88,145.92,138.82,137.96,137.52,128.21,126.54,126.46,124.47,124.41,123.78,123.23,122.38(2c),119.46,118.55,117.85,115.73,115.68,113.84,113.60,106.80,104.97,31.36,30.22,29.43(3c),24.11(2c),23.70(2c).ms(esi):757.59[m+h]
+
.
[0141]
配合物2:1h nmr(400mhz,dmso-d6)δ9.49(d,j=8.9hz),9.26(d,j=5.8hz),9.03(s),8.91(d,j=2.7hz),8.64(d,j=7.8hz),8.21(ddd,j=8.9,7.2,1.8hz),8.10(d,j=2.2hz),7.67(s),7.53(d,j=7.8hz),7.30(d,j=8.3hz),7.13(d,j=2.4hz),6.99(ddd,j=8.3,3.6,1.7hz),6.88(dd,j=5.3,3.0hz),6.67(d,j=8.3hz),2.63-2.52(m),1.21(d,j=6.8hz).ms(esi):799.35[m+h]
+
.
[0142]
配合物3:1h nmr(400mhz,dmso-d6)δ9.51(d,j=8.9hz),9.05(s),8.64(d,j=7.8hz),8.21(ddd,j=8.9,7.2,1.8hz),8.10(d,j=2.2hz),8.00(d,j=8.3hz),7.53(d,j=7.8hz),7.28(d,j=8.3hz),7.20(t,j=7.9hz),7.13(d,j=2.4hz),6.59(ddd,j=8.3,3.6,1.7hz),6.78(dd,j=5.3,3.0hz),6.48(d,j=8.3hz),2.64-2.52(m),1.10(d,j=6.8hz),2.31(s),2.70(s).ms(esi):785.23[m+h]
+
.
[0143]
配合物4:1h nmr(400mhz,dmso-d6)δ9.51(d,j=8.9hz),9.27(d,j=5.8hz),9.05,8.91(d,j=2.7hz),8.55(s),8.22(ddd,j=8.9,7.2,1.8hz)8.10(d,j=2.2hz),8.00(d,j=8.3hz),7.52(s),7.30(d,j=8.3hz),7.17(s),7.13(d,j=2.4hz),,6.80(dd,j=5.3,3.0hz),6.64(d,j=8.3hz),2.64-2.52(m),1.32(s),1.10(d,j=6.8hz).ms(esi):813.61[m+h]
+
.
[0144]
配合物5:1h nmr(400mhz,dmso-d6)δ9.43(d,j=8.9hz),9.00(d,j=5.8hz),9.05,8.91(d,j=2.7hz),8.64(d,j=7.8hz),8.10(d,j=2.2hz),8.00(d,j=8.3hz),7.53(d,j=7.8hz),7.30(d,j=8.3hz),7.24(t,j=7.9hz),7.13(d,j=2.4hz),6.99(ddd,j=8.3,3.6,1.7hz),6.80(dd,j=5.4,3.0hz),6.67(d,j=8.3hz),2.60-2.51(m),1.33(s),1.12(d,j=6.8hz).ms(esi):813.50[m+h]
+
.
[0145]
配合物6:1h nmr(400mhz,dmso-d6)δ9.43(d,j=8.9hz),9.20(d,j=5.8hz),9.05,8.91(d,j=2.7hz),8.64(d,j=7.8hz),8.10(d,j=2.2hz),8.00(d,j=8.3hz),7.53(d,j=7.8hz),7.30(d,j=8.3hz),7.24(t,j=7.9hz),7.13(d,j=2.4hz),7.00(s),6.92(ddd,j=8.3,3.6,1.7hz),6.88(dd,j=5.3,3.0hz),6.67(d,j=8.3hz),2.64-2.52(m),2.46(s),1.10(d,j=6.8hz).ms(esi):874.36[m+h]
+
.
[0146]
配合物7:1h nmr(400mhz,dmso-d6)δ9.49(d,j=8.9hz),9.26(d,j=5.8hz),9.05,8.88(d,j=2.7hz),8.51(d,j=7.8hz),8.21(ddd,j=8.9,7.2,1.8hz),8.10(d,j=2.2hz),8.00(d,j=8.3hz),7.47(d,j=7.8hz),7.30(d,j=8.3hz),7.18(t,j=7.9hz),7.10(d,j=2.4hz),6.99(ddd,j=8.3,3.6,1.7hz),6.88(dd,j=5.3,3.0hz),2.76(s,3h)2.64-2.52(m),1.10(d,j=6.8hz).ms(esi):770.44[m+h]
+
.
[0147]
对比配合物1:
[0148][0149]1h nmr(300mhz,dmso)δ9.67(d,j=8.7hz,1h),9.27(d,j=4.8hz,1h),8.89(s,1h),8.45(d,j=7.8hz,1h),8.32(t,j=8.1hz,1h),8.08(s,1h),7.89(d,j=8.4hz,1h),7.51(d,j=7.5hz,1h),7.44(t,j=6.6hz,1h),7.37(d,j=7.8hz,1h),7.23(t,j=9.0hz,1h),6.97(d,j=8.1hz,1h),6.88(s,1h),3.25(dd,j=13.8,6.9hz,1h),1.40(d,j=6.9hz,1h).ms(esi):639.14[m+h+]。
[0150]
对比配合物2:
[0151][0152]1h nmr(300mhz,dmso)δ9.50(d,j=9.0hz,1h),9.28(d,j=6.2hz,1h),8.90(s,1h),8.63(d,j=7.8hz,1h),8.27(t,j=7.9hz,1h),8.09(s,1h),7.99(d,j=8.3hz,1h),7.52(d,j=7.7hz,1h),7.42(t,j=7.0hz,1h),7.35(d,j=7.8hz,1h),7.29(d,j=8.3hz,1h),7.24(t,j=7.9hz,1h),7.01(s,4h),6.88(s,1h),2.33(s,4h),2.07(s,8h).ms(esi):715.35[m+h
+
]
[0153]
测试例
[0154]
为了更详细地叙述本发明所述的配合物的优良性能,以下将本发明的部分配合物为例,进行示例性的说明。
[0155]
1、光致发光光谱
[0156]
测试条件:激发波长为340nm,溶剂为二氯甲烷(dcm),浓度2
×
10-5
m;仪器型号美国horiba公司fluorolog-3光谱测试平台。
[0157]
测试结果如表1和图1至图3所示。
[0158]
表1
[0159][0160]
其中,λ为峰值波长,fwhm为半峰宽。
[0161]
图1至图3示例性的给出了配合物的光致发光光谱,具体地,图1是配合物1分别在dcm溶液和pmma薄膜中的光致发光光谱图;图2是对比配合物1分别在dcm溶液和pmma薄膜中的光致发光光谱图;图3是对比配合物2分别在dcm溶液和pmma薄膜中的光致发光光谱图;通过表1和图1可以看出:实例1至实例7制备得到的配合物在dcm中的发光波长在469-492nm之间,半峰宽在52-70nm之间;在聚甲基丙烯酸甲酯(5wt%的pmma)中的发光波长在468-480nm之间。上述7种配合物的波长均在蓝光区,并且发光光谱的半峰宽较窄,说明该系列配合物是很好的蓝色发光材料。
[0162]
2、紫外可见吸收光谱
[0163]
测试仪器:紫外可见吸收光谱由日本岛津公司生产的uv-1750紫外可见分光光度计
[0164]
测试结果如图4至图7所示。
[0165]
图4给出了实例1的配合物1的紫外可见吸收光谱。
[0166]
通过图4可以看出:实例1制备得到的配合物1在长波区250-300nm区间的吸收很强,250-300nm可以归属为配合物中以咔啉为中心的π-π*跃迁。其中300nm以后的吸收峰可以归属为配合物中心金属离子与配体之间的价态转移跃迁(mlct)跃迁。
[0167]
3、材料的带隙值(eg)和光学性质测定
[0168]
测试方法:材料的带隙值(eg)、lumo和homo值采用循环伏安法(cv)测得,具体的测试方法为:整个测试过程在手套箱(lab2000,etelux)中的chi600d电化学工作站(上海辰华仪器公司)上进行,以pt柱为工作电极、以ag/agcl为参比电极,pt丝为辅助电极构成三电极系统,测试过程采用的介质是0.1m六氟磷酸四丁基胺(bu4npf6)的二甲基甲酰胺(dmf)溶液,所测电势均以加入的二茂铁(fc)作为内标。
[0169]
测试结果如表2所示。
[0170]
表2
[0171]
配合物e
homo
/eve
lumo
/eveg/eve
t1
/ev配合物1-5.21-2.213.002.70配合物2-5.21-2.213.002.70配合物3-5.21-2.222.992.68配合物4-5.17-2.212.962.70配合物5-5.21-2.123.092.70配合物6-5.22-2.252.972.58配合物7-5.13-2.222.912.60对比配合物1-5.20-2.193.052.73对比配合物2-5.23-2.213.022.69
[0172]
其中,三线态光子能量(e
t1
)为配合物在77k条件下的第一振动峰能量,由公式1240/λ
0-1
计算而来,其中,λ
0-1
是配合物在77k下第一个振动峰的波长。
[0173]
从表2可以看出,配合物1和对比配合物1的homo轨道的能级比配合物1的相当,说明在咔啉单元氮位置上增加位阻基团的共轭性对配合物的homo能级影响不大。同时也说明,可以在含有咔啉结构的铂(ii)配合物的结构中引入取代基对其能级及其发射光谱可以进行小范围的调控,从而获得具有最优的发光光谱区间的配合物。
[0174]
图5至图7示例性的给出了对比配合物1、2和实例配合物1的前沿轨道分布,具体地,图5是对比配合物1的前沿轨道分布图;图6是对比配合物2的前沿轨道分布图;图7是配合物1的前沿轨道分布图。
[0175]
图5至图7具体示出了配合物的最高占据轨道(homo)和最低空轨道(lumo)分布。从图5至图7中可以看出:对比配合物1、对比配合物2和配合物1的homo和lumo均显示为分离的模式,其中,homo轨道均主要分布在二价铂和咔啉以及氧桥相连的苯上,lumo均主要分布在吡啶和吡唑上。其中,明显的二异丙基苯基不提供前线轨道的作用,起到大位阻基团的作用。
[0176]
有机光电器件实例
[0177]
图8是根据本发明的一种实施方式的有机光电器件的结构示意图;图9是根据本发明的一种实施方式的有机光电器件的断面图;如图8至图9所示,所述有机光电器件由下至上依次包括:基体1002、阳极层1004、空穴传输层1006、发光层1008、电子传输层1010以及金属阴极层1012。
[0178]
器件1至器件4的制备例和测试例:本发明采用溶液法制备得到结构为ito/pedot:pss(40nm)/bcpo:配合物x(90:10,50nm)/dpepo(10nm)/tmpypb(50nm)/liq(1nm)/al(100nm)的有机光电器件,其中,各层的材料间用“/”隔开,括号内的数据为各层的厚度以及各层中物质的重量百分含量。
[0179]
表3示出了器件的发光性能数据。
[0180]
表3
[0181] 化合物λ/nmce/cd2a-1
pe/lmw-1
eqe/%cie(x,y)器件1配合物147322.110.510.90.199,0.364器件2配合物247322.010.212.30.194,0.359器件3配合物348524.015.118.70.239,0.406器件4配合物447923.512.515.90.200,0.385器件5配合物546921.310.011.80.185,0.268器件6配合物648524.014.616.30.282,0.473器件7配合物747720.710.011.30.205,0.363对比器件1对比配合物14675.52.33.00.181,0.283对比器件2对比配合物24736.23.22.70.211,0.401
[0182]
其中λ是发光波长,ce、pe和eqe分别是电流效率、能量效率和外量子效率。cie(x,y)是根据国际照明委员会标准的色度坐标参数。
[0183]
图10至图13示出了参杂配合物1用作发光材料的器件1的发光性能,具体地,图10是器件1的电致发光光谱图;图11是器件1的光电转换电流效率图;图12是器件1的功率效率图;图13是器件1的外量子效率图。
[0184]
从图10至图13和表3中可以看出:器件1的发光峰相对配合物1在pmma介质中的光致发光几乎没有位移,说明配合物1具有非常优异的发光稳定性,受到介质材料或主体材料影响小。器件1的最大电流效率(ce)为22.1cd/a、最大功率效率(pe)为10.5lm/w、最大外量子效率(eqe)为10.9%,且效率滚降较低,说明配合物1作为蓝光发光掺杂材料具有高效的、稳定发光的光转化性能。且利用对比例配合物制备的对比器件1-2的发光性能明显低于器件1-7,说明当咔啉二位有大位阻二异丙基苯基取代基时,配合物才具有良好的发光性能。
[0185]
通过对比器件1至器件2的电致发光光谱和其对应的配合物在薄膜中光致发光器件比较可知,相比配合物在薄膜中的光致发光光谱,发光器件的电致发光光谱稍有红移或蓝移,但波峰波长仍位于蓝光区域(469-485nm),光谱大部分也位于蓝光范围内,计算的色度坐标说明该发光器件属于蓝光发光器件。
[0186]
综上:器件1至器件7的电致发光波长主要由配合物本身的光致发光的波长决定,且与配合物本身的光致发光光谱的纯度和电致发光的光谱纯度直接相关。在同一条件下,器件的效率高低也与配合物本身的发光量子效率趋势一致,发光器件的发光的色纯度与掺
杂材料本身光激发下发射光的光谱色纯度直接关联。
[0187]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1