一种利用微通道器制备依他佐辛中间体的方法与流程
1.本发明属于化学合成与医药技术领域,具体涉及一种利用微通道器制备依他佐辛中间体的方法。
背景技术:
2.氢溴酸依他佐辛是由日本科研株式会社开发,主要用于治疗手术后疼痛以及癌症疼痛等。氢溴酸依他佐辛为阿片受体的部分激动剂,作用于k受体,选择性拮抗的方式阻断突触后受体,阻断传递疼痛信息的信使。在镇痛效果方面,依他佐辛的镇痛疗效是喷他佐辛的1-2倍,临床效果良好。
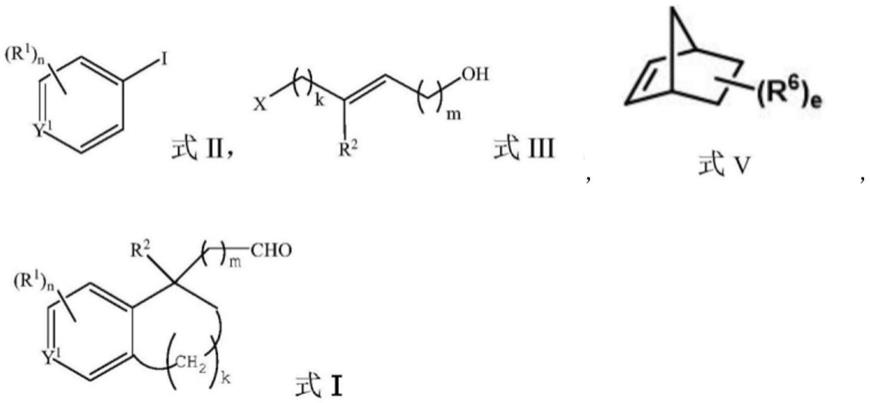
3.专利cn108530241a公开了一种具有苄位季碳中心的苯并环衍生物的合成方法:包括以下步骤:在惰性气体保护下,将式ii所示芳香碘代物、式iii所示烷基卤化物、钯催化剂、膦配体、碱、式v所示降冰片烯衍生物于30~120℃的有机溶剂中搅拌反应,反应结束后分离提纯,得到式i。
[0004][0005]
在该发明的合成具有苄位全碳季碳中心的1,2,3,4-四氢化萘化合物的方法的基础上,得到高效合成氢溴酸依他佐辛的方法,该方法只需要四步,大大减少了合成步骤,提高了合成效率。
[0006][0007]
但该发明所公开的合成1,2,3,4-四氢化萘化合物的方法,合成规模较小,仅仅是100mg规模,且使用了较昂贵的钯盐和相关的膦的配体,完全不适合工业生产。参考其说明书具体实施示例中以式iii所示烷基卤化物计,钯盐和膦的配体约0.05当量和0.1当量,在合成1,2,3,4-四氢化萘化合物中将占据较大的物料成本,所公开的方法中也并未涉及对钯盐和膦的配体进行回收与利用。
[0008]
该发明所公开的合成1,2,3,4-四氢化萘化合物的方法中,用于构建关键化合物(a)的片段,使用了式iii所示烷基卤化物如反式6-溴-3-甲基-2-己烯-1-醇、反式6-碘-3-甲基-2-己烯-1-醇等,此类结构的化合物并无商业化供应的原料;同理,用于构建关键化合物(a)的另一片段,使用了式ii所示芳香碘代物,如2-苄氧基碘苯和叔丁基(2-碘苄氧基)等也存在相同的问题。
技术实现要素:
[0009]
本发明要解决的技术问题是提供一种原料可及性高、无昂贵重原料和催化试剂、简单且适合产业化生产的依他佐辛中间体的制备方法。本发明所述依他佐辛中间体为1-乙醛基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘。
[0010]
为解决上述技术问题,本发明公开了一种依他佐辛中间体化合物4的制备方法,其包括通过dibal-h(二异丁基氰化铝)为氢源还原剂,在微通道反应器中将化合物3还原制得:
[0011][0012]
其中所用溶剂为甲苯、四氢呋喃或甲基叔丁基醚,所述微通道反应器中的温度为-50℃~0℃。
[0013]
该反应路线中酯基还原成醛基,由于醛基极为敏感,趋向于进一步还原生产相应的醇,通用的技术需要控制极低的反应温度如-70℃至-80℃,且反应的放热进一步导致醛基的转化,因而在反应条件和控制上要求较为苛刻。
[0014]
本发明利用微通道反应器,结合反应参数设计,可以实现反应物料的快速接触反
应,能够较好地避免反应放热和副反应,通过合理地控制投料比例,反应及时转移并淬灭。本技术可以在相对更温和温度条件下实现连续化反应,适合产业化生产放大。
[0015]
反应中优选化合物3与dibal-h的摩尔比为1:1~1.2;微通道反应器中的温度为-45~-15℃。
[0016]
其中化合物3可通过化合物2与硅烷试剂在路易斯酸催化条件下偶联制得:
[0017][0018]
其中所用溶剂为氯仿、二氯甲烷、1,2-二氯乙烷或甲苯,所用偶联硅试剂为1-乙氧基乙氧基(三甲基)硅烷,所用路易斯酸催化剂为三氟化硼、三氯化铝、三氯化铟、三氯化铁、四氯化钛、五氯化锑或四异丙氧基钛,反应温度为-10~40℃。
[0019]
通过路易斯酸参与反应,将较难离去的羟基转化为易离去的氢氧根并结合到路易斯酸分子中,形成的碳正离子能够较好地接受具备亲核性的硅试剂的进攻,本技术可以在较温和条件下实现较高的反应转化率,所使用的催化剂路易斯酸和硅试剂相对价格低廉,原料具备可及性,适合产业化放大生产。
[0020]
反应中优选化合物2与路易斯酸催化剂的摩尔比为1:0.3~1.0。
[0021]
其中化合物2可通过甲基溴化镁溶液为甲基化亲核试剂,将化合物1加成制得:
[0022][0023]
其中所用甲基溴化镁溶液为1.0m的甲基溴化镁四氢呋喃溶液、3.0m的甲基溴化镁乙醚溶液、3.0m的甲基溴化镁2-甲基四氢呋喃溶液或1.4m的甲基溴化镁甲苯溶液,反应温度为-10~0℃。反应中优选甲基溴化镁与化合物1的摩尔比1~4:1。
[0024]
优选地,微通道反应器中反应原料的流速为40~60ml/min,反应停留时间为15~30s。
[0025]
优选地,微通道反应器内反应停留15~30s后,搅拌淬灭且控制温度在-10℃至0℃直至反应结束。
[0026]
本发明所公开的技术方案使用了价格相对低廉易得的可商业化物料如化合物1作为合成化合物4的起始物料,在合成工艺过程中,所使用的物料和试剂等均具备商业化供应的特点,不涉及较昂贵试剂的使用诸如重金属催化剂和膦配体等。同时反应时间短,几十秒钟内可以完成反应,反应体系的温度为-50~0℃,相比较报道的反应温度需-70℃至-80℃,降低了能耗,反应体系温度条件相对温和,转化率高,适合产业化放大的需求。
具体实施方式
[0027]
以下通过具体实施例再对本发明的上述内容作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅局限于以下的实施例。在不脱离本发明上述技术思想的情况
下,根据本领域普通技术知识和惯用手段做出的各种替换或变更,均应包括在本发明的范围内。
[0028]
实施例1 1-羟基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物2)的制备
[0029]
量取2.0l的3.0m甲基溴化镁2-甲基四氢呋喃溶液,加入到5.0l的四口烧瓶中,降温至-10℃下搅拌,随后控制反应体系内温0到-10℃下缓慢滴加350g化合物1:7-甲氧基-1-酮-1,2,3,4-四氢化萘(cas:6836-19-7)和1000g四氢呋喃的混合澄清液,滴加完毕后,料液保温反应0.5h,随后缓慢滴加1500g的饱和氯化铵溶液淬灭反应,分液,分出有机相,有机相使用1500g饱和氯化钠溶液洗涤,分液,得有机相,有机相减压浓缩至无液体流出后加入500g正庚烷,60℃下搅拌浆洗1h,随后降温至0℃下搅拌3h,过滤,滤饼减压干燥后得化合物2:367.3g,收率96.3%,纯度98.2%。
[0030]
实施例2 1-羟基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物2)的制备
[0031]
量取5.7l的1.4m甲基溴化镁甲苯溶液,加入到10.0l的四口烧瓶中,降温至-10℃下搅拌,随后控制反应体系内温0到-10℃下缓慢滴加350g化合物1和1000g四氢呋喃的混合澄清液,滴加完毕后,料液保温反应1.0h,随后缓慢滴加1500g的饱和氯化铵溶液淬灭反应,分液,分出有机相,有机相使用1500g饱和氯化钠溶液洗涤,分液,得有机相,有机相减压浓缩至无液体流出后加入500g正庚烷,60℃下搅拌浆洗1h,随后降温至0℃下搅拌3h,过滤,滤饼减压干燥后得化合物2:348.2g,收率91.2%,纯度94.6%。
[0032]
实施例3 1-羟基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物2)的制备
[0033]
量取2.0l的1.0m甲基溴化镁四氢呋喃溶液,加入到5.0l的四口烧瓶中,降温至-5℃下搅拌,随后控制反应体系内温0到-5℃下缓慢滴加350g化合物1和1000g四氢呋喃的混合澄清液,滴加完毕后,料液保温反应1.5h,随后缓慢滴加1500g的饱和氯化铵溶液淬灭反应,分液,分出有机相,有机相使用1500g饱和氯化钠溶液洗涤,分液,得有机相,有机相减压浓缩至无液体流出后加入500g正庚烷,60℃下搅拌浆洗1h,随后降温至0℃下搅拌3h,过滤,滤饼减压干燥后得化合物2:360.8g,收率94.5%,纯度92.9%。
[0034]
实施例4 7-甲氧基-1-甲基-1,2,3,4-四氢化萘-1-乙酰乙酯(化合物3)的制备
[0035]
称量192.3g化合物2到3l的三口烧瓶中,加入1923g二氯甲烷,室温条件下滴加94.8g的四氯化钛,随后滴加192.4g的1-乙氧基乙氧基(三甲基)硅烷,滴加完毕后保温室温条件下反应6h,随后加入800g的5%盐酸溶液淬灭反应,所得有机相依次通过800g的水溶液、800g的10%碳酸氢钠溶液、500g的饱和氯化钠溶液洗涤,所得有机相减压浓缩得化合物3,呈淡黄色透明油状物,重量241.4g,收率92.0%,纯度94.3%。
[0036]
实施例5 7-甲氧基-1-甲基-1,2,3,4-四氢化萘-1-乙酰乙酯(化合物3)的制备
[0037]
称量192.3g化合物2到3l的三口烧瓶中,加入1923g二氯甲烷,降温至-10℃,保温-10℃~10℃下滴加190.0g的四氯化钛,随后-10℃~10℃条件下滴加192.4g的1-乙氧基乙氧基(三甲基)硅烷,滴加完毕后保温-10℃~10℃条件下反应12h,随后加入800g的5%盐酸溶液淬灭反应,所得有机相依次通过800g的水溶液、800g的10%碳酸氢钠溶液、500g的饱和氯化钠溶液洗涤,所得有机相减压浓缩得化合物3,呈淡黄色透明油状物,重量245.3g,收率93.5%,纯度95.6%。
[0038]
实施例6 7-甲氧基-1-甲基-1,2,3,4-四氢化萘-1-乙酰乙酯(化合物3)的制备
[0039]
称量192.3g化合物2到3l的三口烧瓶中,加入1923g二氯甲烷,保温30℃~40℃下
滴加85.4g的四异丙氧基钛,随后30℃~40℃条件下滴加192.4g的1-乙氧基乙氧基(三甲基)硅烷,滴加完毕后保温40℃条件下反应3h,随后加入800g的5%盐酸溶液淬灭反应,所得有机相依次通过800g的水溶液、800g的10%碳酸氢钠溶液、500g的饱和氯化钠溶液洗涤,所得有机相减压浓缩得化合物3,呈淡黄色透明油状物,重量235.6g,收率89.8%,纯度92.6%。
[0040]
实施例7 1-乙醛基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物4)的制备
[0041]
1、称量262.35g化合物3,加入甲苯配制成1000ml的甲苯溶液,使用低温冷凝循环泵降温至内温-45℃,随后连接管路i。
[0042]
2、量取800ml的1.4m二异丁基氰化铝的甲苯溶液,使用低温冷凝循环泵降温至内温-45℃,随后连接管路ii。
[0043]
3、通过两台恒流泵分别将管路i和管路ii所连的料液以50ml/min和40ml/min的体积流量,流入到微反应器中进行料液混合反应,反应停留时间为30s,反应温度-45℃,所得流出液流入到保温在-10℃的1100g的20%甲醇水溶液中,搅拌淬灭且控制温度在-10℃至0℃直至反应结束,分液,分出有机相,有机相使用1000g饱和氯化钠溶液洗涤,分液,所得有机相减压浓缩得粗品,粗品经过减压蒸馏得化合物4,重量175g,收率80.1%,纯度97.5%。
[0044]
实施例8 1-乙醛基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物4)的制备
[0045]
1、称量262.35g化合物3,加入甲苯配制成1000ml的甲苯溶液,使用低温冷凝循环泵降温至内温-15℃,随后连接管路i。
[0046]
2、量取938ml的1.4m二异丁基氰化铝的甲苯溶液,使用低温冷凝循环泵降温至内温-15℃,随后连接管路ii。
[0047]
3、通过两台恒流泵分别将管路i和管路ii所连的料液以50ml/min和47ml/min的体积流量,流入到微反应器中进行料液混合反应,反应停留时间为18s,反应温度-15℃,所得流出液流入到保温在-10℃的1100g的20%甲醇水溶液中,搅拌淬灭且控制温度在-10℃至0℃直至反应结束,分液,分出有机相,有机相使用1000g饱和氯化钠溶液洗涤,分液,所得有机相减压浓缩得粗品,粗品经过减压蒸馏得化合物4,重量168g,收率76.9%,纯度94.8%。
[0048]
实施例9 1-乙醛基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物4)的制备
[0049]
1、称量262.35g化合物3,加入四氢呋喃配制成1000ml的四氢呋喃溶液,使用低温冷凝循环泵降温至内温-45℃,随后连接管路i。
[0050]
2、量取1120ml的1.0m二异丁基氰化铝的正己烷溶液,使用低温冷凝循环泵降温至内温-45℃,随后连接管路ii。
[0051]
3、通过两台恒流泵分别将管路i和管路ii所连的料液以50ml/min和56ml/min的体积流量,流入到微反应器中进行料液混合反应,反应停留时间为30s,反应温度-45℃,所得流出液流入到保温在-10℃的1100g的20%甲醇水溶液中,搅拌淬灭且控制温度在-10℃至0℃直至反应结束,料液中补加50g固体氯化钠至溶解澄清,分液,分出的有机相使用1000g饱和氯化钠溶液洗涤,分液,所得有机相减压浓缩得粗品,粗品经过减压蒸馏得化合物4,重量159g,收率72.8%,纯度96.2%。
[0052]
实施例10 1-乙醛基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物4)的制备
[0053]
1、称量262.35g化合物3,加入四氢呋喃配制成1000ml的四氢呋喃溶液,使用低温冷凝循环泵降温至内温-45℃,随后连接管路i。
[0054]
2、量取800ml的1.4m二异丁基氰化铝的甲苯溶液,使用低温冷凝循环泵降温至内温-45℃,随后连接管路ii。
[0055]
3、通过两台恒流泵分别将管路i和管路ii所连的料液以50ml/min和40ml/min的体积流量,流入到微反应器中进行料液混合反应,反应停留时间为30s,反应温度-45℃,所得流出液流入到保温在-10℃的1100g的20%甲醇水溶液中,搅拌淬灭且控制温度在-10℃至0℃直至反应结束,料液中补加50g固体氯化钠至溶解澄清,分液,分出的有机相使用1000g饱和氯化钠溶液洗涤,分液,所得有机相减压浓缩得粗品,粗品经过减压蒸馏得化合物4,重量172g,收率78.8%,纯度95.3%。
[0056]
实施例11 1-乙醛基-7-甲氧基-1-甲基-1,2,3,4-四氢化萘(化合物4)的制备
[0057]
1、称量262.35g化合物3,加入甲基叔丁基醚配制成1000ml的甲基叔丁基醚溶液,使用低温冷凝循环泵降温至内温-20℃,随后连接管路i。
[0058]
2、量取940ml的1.0m二异丁基氰化铝的正己烷溶液,使用低温冷凝循环泵降温至内温-20℃,随后连接管路ii。
[0059]
3、通过两台恒流泵分别将管路i和管路ii所连的料液以50ml/min和47ml/min的体积流量,流入到微反应器中进行料液混合反应,反应停留时间为20s,反应温度-20℃,所得流出液流入到保温在-10℃的1100g的20%甲醇水溶液中,搅拌淬灭且控制温度在-10℃至0℃直至反应结束,料液中补加50g固体氯化钠至溶解澄清,分液,分出的有机相使用1000g饱和氯化钠溶液洗涤,分液,所得有机相减压浓缩得粗品,粗品经过减压蒸馏得化合物4,重量163g,收率74.7%,纯度95.1%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1