一种在大肠杆菌中表达的对苯二酚降解酶基因组及其应用的制作方法
[0001]
本发明属于基因工程领域,具体涉及一种在大肠杆菌中表达的对苯二酚降解酶基因组及其应用。
背景技术:
[0002]
对苯二酚又名氢醌,是一种重要的化工原料,其应用广泛,是医药、农药、染料和橡胶等的重要原料、中间体和助剂,主要用于显影剂、蒽醌染料、偶氮颜料、橡胶防老剂和单体组聚剂、食品稳定剂和涂料抗氧化剂、石油抗凝剂、合成氨催化剂等方面。目前,对苯二酚的应用领域正在逐渐拓展,消费需求也在逐年扩大。
[0003]
随着对苯二酚的大量生产和消费,该物质不可避免的会通过各种途径污染地下水、地表水和土壤,使得对苯二酚不断地在废水、河流、湖泊、土壤、空气和地下水中检出。由于对苯二酚具有毒性,对皮肤、粘膜有强烈的腐蚀作用,可抑制中枢神经系统或损害肝、皮肤功能,甚至可致死,因此已被美国环保局(epa)列为优先控制污染物之一,我国已于2011年将该物质划为第6.1类毒害品。
[0004]
对苯二酚具有一定的水溶性,用传统的处理污染物的物理和化学方法进行处理工艺复杂且不能完全清除污染物。而微生物由于具有极强的变异性和适应性等特点,在环境的选择压力下可以降解硝基苯酚,因而成为自然环境中降解对苯二酚的主要成员。
[0005]
目前,对苯二酚的降解研究主要源于对硝基苯酚的降解,因前者是后者的降解中间产物。降解对硝基苯酚的途径主要有两条:对苯二酚途径和偏苯三酚途径。前者在革兰氏阴性菌中较多,而后者普遍存在于革兰氏阳性菌中。对苯二酚途径中,中间产物对苯二酚在对苯二酚1,2-双加氧酶的作用下开环生成γ-羟基粘康酸半醛,随后转化为马来酰乙酸,并继续还原成β-酮己二酸。
技术实现要素:
[0006]
本发明的目的在于提供一种能在大肠杆菌中表达的对苯二酚降解酶基因组及其应用,利用合成生物学技术将源于假单胞菌(pseudomonas putida)的不同基因片段进行设计、改造,将完整的异源分解代谢途径引入到大肠杆菌中,从而使大肠杆菌具备降解和耐受对苯二酚的能力,提高细菌对对苯二酚的降解效率和耐受能力,丰富工业应用降解菌资源。
[0007]
为了达到上述目的,本发明提供如下技术方案:
[0008]
一种在大肠杆菌中表达的对苯二酚降解酶基因组,包括源于假单胞菌并按大肠杆菌的密码子偏好性优化后获得的pnpcs、pnpds和pnpes基因。
[0009]
进一步,按大肠杆菌的密码子偏好性优化后,pnpcs基因的核苷酸序列如seq id no.1所示;pnpds基因的核苷酸序列如seq id no.2所示;pnpes基因的核苷酸序列如seq id no.3所示。
[0010]
本发明提供所述对苯二酚降解酶基因组在大肠杆菌中的应用。
[0011]
一种多基因大肠杆菌转化载体,包括大肠杆菌表达载体,以及源于假单胞菌并按
大肠杆菌的密码子偏好性优化后获得的pnpcs、pnpds和pnpes基因分别与t7启动子和终止子融合的基因表达盒。
[0012]
优选地,所述大肠杆菌表达载体为pet-28a。
[0013]
一种可完全降解对苯二酚的转基因大肠杆菌的获得方法,包括以下步骤:
[0014]
1)根据大肠杆菌的密码子偏好性,对源于假单胞菌的pnpc基因、pnpd基因和pnpe基因按大肠杆菌表达模式优化,优化后分别获得pnpcs基因、pnpds基因和pnpes基因,利用重叠延伸pcr技术,将优化后的五段基因分别与t7启动子和终止子融合,分别构建基因表达盒;
[0015]
2)将步骤1)中构建的三个基因表达盒依次按pnpcs、pnpds、pnpes的顺序连接入大肠杆菌表达载体,获得三基因表达盒的多基因大肠杆菌转化载体;
[0016]
3)将步骤2)所述多基因大肠杆菌转化载体转入大肠杆菌,获得可完全降解对苯二酚的大肠杆菌。
[0017]
优选地,步骤1)中,所述t7启动子的核苷酸序列如seq id no.4所示;所述t7终止子序列如seq id no.5所示。
[0018]
优选地,步骤2)中,所述大肠杆菌表达载体为pet-28a。
[0019]
又,步骤3)中,所述大肠杆菌为bl21-ai。
[0020]
本发明中,根据大肠杆菌密码子的偏好性对假单胞菌(pseudomonas putida)的pnpc、pnpd和pnpe基因进行优化,优化按照以下原则进行:(一)优化基因密码子,根据大肠杆菌密码子偏爱,提高基因翻译效率;(二)消除基因内部的限制性内切酶ecori和hindiii的识别位点,便于表达盒构建;(三)消除与t7启动子或终止子相邻100bp以内的逆向重复序列和茎环结构;(四)消除两基因间相邻200bp以内的逆向重复序列和茎环结构;(五)消除转录终止信号,使基因内部的gc/at均衡,提高rna的稳定性;(六)使基因编码蛋白符合n端原则,以提高翻译蛋白的稳定性;(七)优化mrna二级结构自由能,以提高基因表达效率。
[0021]
与现有技术相比,本发明具有如下有益效果:
[0022]
本发明中,将pnpc基因、pnpd基因和pnpe基因结合,优化后在大肠杆菌中成功表达,获得的阳性菌株可在8小时之内完全降解1mm的对苯二酚,24小时内完全降解5mm的对苯二酚,24小时内可降解5%的10mm的对苯二酚。
[0023]
本发明的对苯二酚降解酶基因组基因组合,可用于制备降解对苯二酚的微生物,在废水处理和环境修复等领域具有应用潜力。
附图说明
[0024]
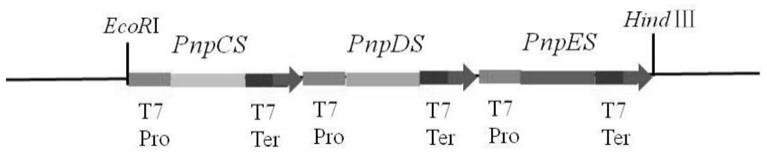
图1为本发明实施例1中三基因pnpcs、pnpds和pnpes的大肠杆菌转化载体结构示意图。
[0025]
图2为本发明实施例3中阳性克隆质粒dna的外源基因的pcr检测结果。
[0026]
图3为本发明实施例3中阳性菌株的外源基因rt-pcr检测结果。
[0027]
图4为本发明实施例4中阳性菌株对对苯二酚的去除效果。
[0028]
图5为本发明实施例4中阳性菌株中生成的最终产物β-酮己二酸的质谱图。
具体实施方式
[0029]
下面结合说明书附图和具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。
[0030]
实施例中所用大肠杆菌由上海市农业科学院生物技术研究所植物基因工程研究室保存,所使用的试验方法如无特殊说明,均为常规分子生物学方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料;实施例中的hplc分析采用安捷伦1100高效液相色谱系统;gc-ms检测采用agilent 7890b-7000c气质联用仪。
[0031]
实施例1
[0032]
1、三基因的优化合成
[0033]
以假单胞菌(pseudomonasputida)的pnpc、pnpd和pnpe基因(genbank no.fj376608.2)为模板,按照以下原则对上述三段基因进行优化:(一)优化基因密码子,根据大肠杆菌密码子偏爱,提高基因翻译效率;(二)消除基因内部的限制性内切酶ecori和hindiii的识别位点,便于表达盒构建;(三)消除与t7启动子或终止子相邻100bp以内的逆向重复序列和茎环结构;(四)消除两基因间相邻200bp以内的逆向重复序列和茎环结构;(五)消除转录终止信号,使基因内部的gc/at均衡,提高rna的稳定性;(六)使基因编码蛋白符合n端原则,以提高翻译蛋白的稳定性;(七)优化mrna二级结构自由能,以提高基因表达效率。
[0034]
优化后,以假单胞菌(pseudomonasputida)的pnpc、pnpd和pnpe基因(genbank no.fj376608.2)为模板,分别合成获得seq id no.1、seq id no.2、seq id no.3所示dna序列pnpcs、pnppds和pnpes,分别克隆到质粒载体,测序确定其序列。
[0035]
序列的合成方法参照nucleic acids research,2004,32(12)e98。
[0036]
实施例2构建多基因大肠杆菌转化载体
[0037]
1.构建三个基因表达盒元件
[0038]
1.1以t7启动子为模板,合成获得seq id no.4所示dna序列t7启动子,克隆到质粒载体,测序确定其序列;以t7终止子为模板,合成获得seq id no.5所示dna序列t7终止子,克隆到质粒载体,测序确定其序列,序列的合成方法参照nucleic acids research,2004,32(12)e98。
[0039]
按照改良的“重叠延伸pcr”技术进行元件的拼接(appl microbiol biotechnol.2006,73(1):234-40)。
[0040]
1.2pnpcs基因表达盒的构建
[0041]
根据上述化学合成的t7启动子、pnpcs基因和t7终止子序列设计一对引物p1f和p1r进行串联,引物长度为60bp,引物p1f上有ecori酶切位点和启动子,p1r上有终止子,具体序列如下:
[0042]
p1f:5
’-
gaattctaatacgactcactataggatgactgatcattacaaggctgtggaggcactgat-3’;
[0043]
p1r:5
’-
caaaaaacccctcaagacccgtttagaggccccaaggggttatgctactctgcctccatc-3’。
[0044]
1.3 pnpds基因表达盒的构建:根据上述化学合成的t7启动子、pnpds基因和t7终止子序列设计一对引物p4f和p4r进行串联,引物长度为60bp,引物p2f上有启动子,p2r上有
终止子。具体序列如下:
[0045]
p2f:5
’-
taatacgactcactataggatgcaaaaccttcttttcatcgatggtcgttttgttgaggc-3’;
[0046]
p2r:5
’-
caaaaaacccctcaagacccgtttagaggccccaaggggttatgctaacgcttgaagtgt-3’。
[0047]
1.4 pnpes基因表达盒的构建:根据上述化学合成的t7启动子、pnpes基因和t7终止子序列设计一对引物p3f和p3r进行串联,引物长度为60bp,引物p3f上有启动子,p3r上有终止子和hindiii酶切位点,具体序列如下:
[0048]
p5f:5
’-
taatacgactcactataggatgaatccattcgtgtaccaatcactgccaactcgtgttgt-3’;
[0049]
p5r:5
’-
caaaaaacccctcaagacccgtttagaggccccaaggggttatgctatgcaggagaccaa-3’。
[0050]
2.构建多基因大肠杆菌转化载体
[0051]
将上述合成的三个基因表达盒按pnpcs、pnpds、pnpes的顺序依次进行连接,获得pnpcs-pnpds-pnpe重组基因表达盒。
[0052]
将上述获得的pnpcs-pnpds-pnpes基因表达盒重组质粒用ecori和hindiii酶切并连接到经同样酶切的pet-28a载体上,即可获得含有三个基因的多基因大肠杆菌表达载体pet-hq,其结构可表示为pet-pnpcs-pnpds-pnpes(如图1所示)。
[0053]
实施例3大肠杆菌的转化
[0054]
1.转基因大肠杆菌的获得与鉴定
[0055]
1.1大肠杆菌的准备及转化
[0056]
1)挑取大肠杆菌单菌落接种于20ml lb培养基中,37℃振荡培养过夜。
[0057]
2)按1%接种量接种20ml lb培养基中,37℃230转/min振荡培养3h。
[0058]
3)取1ml培养物,冰浴30min,4℃4000r/min离心3min,去上清。
[0059]
4)加500μl体积的冰冷的0.1mol/l cacl2溶液,重新悬浮细菌沉淀,4℃4000r/min离心3min,去上清。
[0060]
5)加100μl体积的冰冷的0.1mol/l cacl2溶液,重新悬浮细菌沉淀,4℃4000r/min离心3min,去上清。
[0061]
6)50μl体积的冰冷的0.1mol/l cacl2溶液,重新悬浮细菌沉淀,该菌液可立即使用或冻于-70℃存放。
[0062]
7)在1.5ml的eppendorf中,加入80μl菌液和1-2μl的质粒dna(0.4pg-0.3μg),冰上放置20分钟后,放入42℃水浴锅中热击90s,后迅速置于冰上。
[0063]
8)热击转化后,即在菌体中加入1.0ml表达培养液,在29℃下培养1小时,涂于加入抗菌素的2yt平板(卡那霉素50μg/ml,x-gel 30μg/ml),37℃过夜培养后得到阳性克隆。利用以上转化程序,将大肠杆菌表达载体pet-hq热击转入大肠杆菌bl21-ai,获得大肠杆菌阳性株系bl-hq。
[0064]
2.转基因大肠杆菌的鉴定
[0065]
2.1阳性克隆中质粒的dna序列测定
[0066]
对转入多基因大肠杆菌表达载体pet-hq获得的阳性克隆,用碱裂解法抽提质粒,
以此为模板,用pcr扩增的方法,分别检测外源基因pnpcs、pnpds、pnpes。
[0067]
所用引物如下:
[0068]
pnpcs:f:5
’-
atgactgatcattacaaggctg-3’,
[0069]
r:5
’-
ctccatcacgaactcgtagt-3’。
[0070]
pnpds:f:5
’-
atgcaaaaccttcttttcatcg-3’;
[0071]
r:5
’-
acgcttgaagtgtgcaggaatag-3’。
[0072]
pnpes:f:5
’-
atgaatccattcgtgtaccaatc-3’;
[0073]
r:5
’-
tgcaggagaccaaccgttccatg-3’。
[0074]
所用扩增程序:94℃30s,54℃30s,72℃120s,共45个循环,最后72℃再延伸10min,以只转化空载体的菌株(bl-control)为对照菌株,结果参见图2。
[0075]
由图2可见,对照菌株不能扩出外源基因,而阳性克隆中都能扩增出上述三个基因,表明外源基因均完整地整合到了大肠杆菌基因组中。
[0076]
2.2阳性菌株的rt-pcr检测
[0077]
用生工生物公司的rna抽提试剂盒提取阳性菌株中的rna,并用全式金生物公司的逆转录试剂盒将提取的rna反转录为cdna,用如下引物和扩增条件进行外源pnpcs、pnpds、pnpes基因的rt-pcr检测,细菌16srrna作为内参,所用引物如下:
[0078]
pnpcs:f:5
’-
tggtcgttaccgttctgac-3’;
[0079]
r:5
’-
gtcttcgtacttcacttgc-3’。
[0080]
pnpds:f:5
’-
tgggtccactgacttctg-3’;
[0081]
r:5
’-
gtccacagaccagaaccca-3’。
[0082]
pnpes:f:5
’-
cgtggatgaccctgaaca-3’;
[0083]
r:5
’-
gggataccaagtttctcg-3’。
[0084]
16s rrna:f:5
’-
agagtttgatcctggctcag-3’;
[0085]
r:5
’-
taccttgttacgactt-3’。
[0086]
所用扩增程序:94℃30s,54℃30s,72℃30s,共45个循环,最后72℃再延伸10min,结果参见图3。
[0087]
结果显示,阳性菌株都能扩增出上述三个基因(参见图3),表明外源基因在转基因大肠杆菌中均进行了正确的转录表达,相对表达量在1.6倍以上。
[0088]
实施例4阳性菌株对对苯二酚的降解作用
[0089]
1.样品的准备
[0090]
将实施例3中的阳性菌株(bl-hq)和对照菌株(只转化空载体的菌株)分别接种于100毫升m9液体培养中,该培养基中含1%甘油和50μg/ml卡那霉素,37℃摇菌24小时(150rpm),离心去上清,菌体用灭菌的蒸馏水洗一遍,之后用10毫升m9液体培养基重悬,该培养基中含1%甘油、0.2%阿拉伯糖、50μg/ml卡那霉素和1mm iptg,向阳性菌株中添加浓度为1mm、5mm和10mm的对苯二酚,向对照菌株培养基中添加浓度为1mm的对苯二酚,37℃摇菌处理,不同时间取菌液,通过hplc检测其残余的对苯二酚含量;通过气相质谱检测最后生成的β-酮己二酸含量。
[0091]
2.对苯二酚的残留检测及产物分析
[0092]
2.1阳性菌株中对苯二酚的hplc分析及含量测定
[0093]
取1ml菌液,离心后收集上清,用0.22μm的有机滤膜过滤后备用。
[0094]
测对苯二酚的hplc条件为:c18柱(4.6
×
150mm,5μm);流动相为甲醇:水=30:70,流速为0.5ml/min;柱温为30℃;检测波长为270nm;进样量为20μl。
[0095]
检测结果参见图4,阳性菌株可在8小时内快速降解培养基中的对苯二酚,而对照菌株无法降解或利用对苯二酚。浓度更高的对苯二酚,如5mm,阳性菌株也可在24小时内完全降解;10mm的对苯二酚,阳性菌株也有较强的耐受性,并可在24小时内实现5%的降解。
[0096]
2.2阳性菌株中最终产物β-酮己二酸的gc-ms检测:
[0097]
取100μl样品于气相进样瓶,预冻后放置冻干机中进行冷冻干燥,在冻干后的样品中加入100μl的吡啶和100μl的bstfa,放置于60℃烘箱反应2小时,反应后的样品经0.22μm滤膜过滤后进样分析。
[0098]
气相色谱条件为:采用hp-5毛细管色谱柱(30m
×
0.25mm
×
0.25μm);载气he(99.999%),流速1ml/min;进样口温度280℃;柱温程序:初始温度80℃保持,20℃/min升至150℃,10℃/min升至280℃,进样量1μl,采用分流进样模式,分流比10:1。
[0099]
gc-ms质谱分析条件为:电子轰击离子源(ei),电离能70ev;离子源温度230℃,四级杆温度150℃。碰撞气n2,采用sim监测模式,监测离子169。
[0100]
质谱图参见图5,从质谱图中可以看出,在阳性菌株中明显检测出了β-酮己二酸的存在,表明对苯二酚已被完全降解,最终产物β-酮己二酸可进入微生物的三羧酸循环,参与微生物体内物质的合成。
[0101]
因此,转入了本发明对苯二酚降解酶基因组的大肠杆菌,其对对苯二酚的降解速率及浓度均大大提升。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1