一种帕瑞昔布衍生物及其制备方法和应用与流程
o-so
2-or3;所述r1独立的选自氢、卤素、和c1-c3烷基;r2独立的选自c1-c12烷基、c3-c8环烷基、c1-c5烷氧基;r3独立的选自氢、c1-c12烷基、c3-c8环烷基;且所述r1、r2、r3可以独立地任选被卤素、羟基、胺基、c1-c3烷代胺基、c1-c3二烷代胺基、c1-c3烷氧基、羧基、氰基取代;
[0008]
进一步地,所述r1选自h、甲基、乙基、丙基、异丙基;所述r2选自羟甲基、乙基、丙基、甲氧基、乙氧基、丙氧基、异丙基、环丙基、ch3och
2-、-ch2f、n(ch3)
2-ch
2-、nh(ch3)-ch
2-、nh2ch
2-;所述r3选自甲基、乙基、异丙基、-ch
2 ch2cl;
[0009]
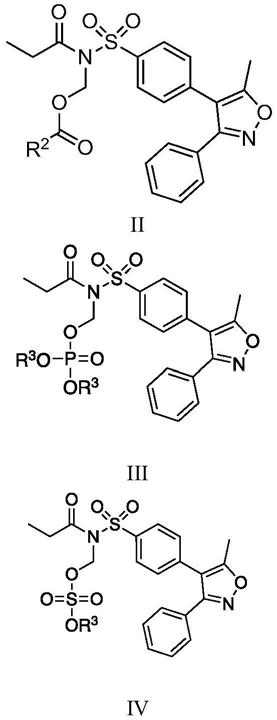
进一步地化合物选自式ii、式iii、式iv:
[0010][0011]
其中,r2独立的选自c1-c5烷基、c3-c6环烷基;r3独立的选自c1-c5烷基、c3-c6环烷基;且所述r2、r3可以独立地任选被卤素、羟基、胺基、c1-c3烷代胺基、c1-c3二烷代胺基、c1-c3烷氧基、羧基、氰基取代;
[0012]
进一步地化合物选自式v:
[0013][0014]
其中,r3选自c1-c3烷基,且r3可以任选被卤素、羟基、胺基、c1-c3二烷代胺基、c1-c3烷氧基取代;
[0015]
根据本发明的实施方案,所述r3选自甲基、乙基、丙基;
[0016]
根据本发明的实施方案,所述式i、ii、iii、iv和v所示的化合物及其消旋体、立体异构体、互变异构体、同位素标记物、溶剂化物、多晶型物、酯或其药学上可接受的盐中,非限制性的具体实例如下所示:
[0017]
[0018]
本发明还提供式i所示的化合物及其消旋体、立体异构体、互变异构体、同位素标记物、溶剂化物、多晶型物、酯或其药学上可接受的盐的制备方法,但不仅限于以下描述的方法。所有的原料都是根据符合通式规律的目标分子的基团特征,并通过这些路线中的方案、有机化学领域普通技术人员熟知的方法制备或者直接购买的。可将用下述方法和合成有机化学领域中已知的合成方法或本领域技术人员意识到的有关改变方法结合在一起,合成本发明化合物。本领域技术人员可知,根据特定的目标结构,可以任选采用下述一种或几种方案进行结合,或者一种或几种方案中的任意步骤进行组合得到合成方案。
[0019]
本发明式i化合物的可根据方案1制备,所述r如式i定义,这里所述x表示cl、br、i、-otf、-oms、-ots等基团,将帕瑞昔布在适当条件下与r-x反应,反应可选k2co3、csco3、csf、dipea、dbu、lihmds、py等碱进行:
[0020][0021]
式ii化合物可根据方案2制备,其中r2如式ii定义,所述x表示cl、br、i、-otf、-oms、-ots等基团,将帕瑞昔布在适当条件下与r
2-c(o)-o-ch
2-x反应,反应可选k2co3、csco3、csf、dipea、dbu、lihmds、py等碱进行:
[0022][0023]
式iii化合物可根据方案3制备,r3如式iii定义,所述x表示cl、br、i、-otf、-oms、-ots等基团,将帕瑞昔布在适当条件下与x-ch
2-o-p(o)-(or3)2反应,反应可选k2co3、csco3、csf、dipea、dbu、lihmds、py等碱进行:
[0024][0025]
式iv化合物可根据方案4制备,r3如式iv定义,所述x表示cl、br、i、-otf、-oms、-ots等基团,将帕瑞昔布在适当条件下与x-ch
2-o-so
2-or3反应,反应可选k2co3、csco3、csf、dipea、dbu、lihmds、py等碱进行:
[0026][0027]
式v化合物可根据方案5制备,其中r3如式iii定义,所述x表示cl、br、i、-otf、-oms、-ots等基团,将帕瑞昔布在适当条件下与r3o-c(o)-o-ch
2-x反应,反应可选k2co3、csco3、csf、dipea、dbu、lihmds、py等碱进行:
[0028][0029]
本发明进一步提供一种药物组合物,其包含本发明所述的式i化合物及其消旋体、立体异构体、互变异构体、同位素标记物、溶剂化物、多晶型物、酯或其药学上可接受的盐和药学上可接受的辅料和载体。
[0030]
所述药物组合物中的载体为“可接受的”,其可与组合物的活性成分相容(并且优选地,能够稳定活性成分)并且对被治疗的受试者不是有害的。可以使用一种或多种增溶剂作为药物赋形剂用于递送活性化合物。
[0031]
本发明进一步提供所述式i化合物及其消旋体、立体异构体、互变异构体、同位素标记物、溶剂化物、多晶型物、酯或其药学上可接受的盐或所述药物组合物在制备用于治疗炎症或疼痛的药物中的用途。
[0032]
根据本发明的实施方案,所述疼痛选自炎症性疼痛、牙痛、肩周炎疼痛、骨关节炎疼痛、妇科疼痛、肌痛、创伤疼痛、癌症引起的疼痛、手术疼痛和其它急性中重度疼痛。
[0033]
根据本发明的实施方案,所述联合给药包括将提供所述药物联合的有益效果的、以顺序方式给药的方案,还包括以基本上同时的方式共同给药,例如以含有固定比例的这些活性物质的同一制剂或以多个独立含有各个药物的制剂形式给药。本发明药物组合物可与其他治疗剂连用或协同药用。
[0034]
所述其他治疗剂可选自可待因、双氢可待因、氢吗啡酮、羟考酮、美沙酮、吗啡、芬太尼、哌替啶(杜冷丁)或其它阿片类镇痛药,通过联合给药可以提高药效和降低阿片类镇痛药的使用剂量,使得治疗有更少的副作用或潜在的风险。
[0035]
所述其他治疗剂可选自麻醉止疼药物或麻醉剂。
[0036]
本发明的单个剂型可适用于经口服或局部外用给药。
[0037]
药用组合物和剂型一般包括一种或多种赋形剂。药剂学领域技术人员熟知合适的赋形剂,本文所提供的合适的赋形剂的实例并不限于此。一种特殊的赋形剂是否适合加至药用组合物或剂型中,将根据本领域熟知的多种因素,包括但不限于将所述剂型给予患者的途径等确定。例如,口服剂型如片剂可包含不适用于胃肠外给药剂型的赋形剂。由于给药
方便,片剂和胶囊剂为最常用的其中使用固体赋形剂的口服剂型。如果需要,可通过标准的技术对片剂包衣,可通过任何药剂学方法制备这些剂型。
[0038]
按照常规药学配混技术,将活性组分与至少一种赋形剂充分混匀,制备本发明的代表性口服剂型。根据给药需要的制剂形式,赋形剂可为各种各样的形式。例如,适用于液体口服剂型或气雾剂的赋形剂包括但不限于水、二元醇、油、醇、矫味剂、防腐剂和着色剂。适用于固体口服剂型(例如散剂、片剂、胶囊剂和胶囊形片剂)的赋形剂实例包括但不限于淀粉、糖、微晶纤维素、稀释剂、造粒剂、增溶剂、稳定剂、润滑剂、粘合剂和崩解剂。
[0039]
适用于本文公开的药用组合物和剂型的填充剂的实例包括但不限于:滑石粉,碳酸钙(例如颗粒或粉末),微晶纤维素,粉状纤维素,葡萄糖结合剂,高岭土,甘露醇,硅酸,山梨醇,淀粉,预胶化淀粉,及其混合物。本发明的药用组合物中的粘合剂或填充剂一般占所述药用组合物或剂型的约50%-99%重量。
[0040]
适合形式的微晶纤维素包括但不限于:以avicel-ph-101、avicel-ph-103avicel rc-581、avicel-ph-105(来自fmc corporation,american viscose division,avicel sales,marcus hook,pa)出售的商品,及其混合物。具体的粘合剂为以avicel rc-581出售的微晶纤维素和羧甲基纤维素钠的混合物。合适的无水或低含水量的赋形剂或添加剂包括avicel-ph-103tm和starch 1500lm。
[0041]
当暴露在含水环境中时,用于本发明的组合物的崩解剂可使片剂崩解,应该使用足量的崩解剂制备本发明的固体口服剂型。
[0042]
本发明的不含乳糖的组合物可包含本领域熟知的各种赋形剂,以及列在例如美国药典(usp)sp(xxi)/nf(xvi)中的赋形剂。不含乳糖的组合物一般包括活性组分、药学相容并且药学可接受量的粘合剂/填充剂和润滑剂,优选的不含乳糖的剂型包含活性组分、微晶纤维素、预胶化淀粉和硬脂酸镁。
[0043]
本发明进一步包括包含活性组分的无水药用组合物和剂型,参见jens t.carstensen,drug stability:principles&practice,2版,marcel dekker,纽约,1995年,379-80页。使用无水或含水量低的组分并在低水分或低湿度的环境中,可制备本发明的无水药用组合物和剂型。无水药用组合物的制备和储存应该保持其无水性质。相应地,优选无水组合物使用已知可防水的材料包装,以便它们可用合适药剂盒包装。合适的包装实例包括但不限于密封箔、塑料、单剂量容器(例如管瓶)、泡罩包装和对开包装。
[0044]
本发明进一步包括含有一种或多种可降低活性组分分解速率的化合物的药用组合物和剂型。本文中称为“稳定剂”的这些化合物包括但不限于抗氧化剂,如抗坏血酸、ph缓冲剂或盐缓冲剂。
[0045]
使用本发明的化合物或组合物治疗疾病的治疗活性化合物的给药量和剂量方案取决于多种因素,包括患者的年龄、体重、性别和健康状况,炎症或疾病相关的炎症的严重程度,给药途径和频率以及使用的特定化合物等。前药组合物应包含与母体化合物相类似的剂量。药物组合物可含有的活性成分为约0.1-1000mg,优选为约1-300mg,最优选为约20-200mg。每日剂量为约0.01-100mg/kg体重,优选为约0.05-20mg/kg体重,最优选为约0.1-10mg/kg体重是适宜的。该每日剂量每天可分一至多次给药。
[0046]
在某些病例中,可能需要使用超出本文公开的剂量范围的活性组分。在结合个体患者的反应后,临床医师或主治医师将知道如何和何时中止、调节或终止治疗。
[0047]
本发明的化合物还可应用于包括哺乳动物、啮齿类动物等的配对(companion)动物、野生动物和农场动物的兽医治疗。较优选的动物对象包括马、猪、养、兔、狗和猫。
[0048]
通常优选本发明的活性组分不同时或不通过相同的途径给予患者。因此,本发明进一步提供一种药剂盒,当临床医师使用时,所述药剂盒可使适量的活性组分对患者的给药简单化。
[0049]
所述药剂盒所述式i化合物及其消旋体、立体异构体、互变异构体、同位素标记物、溶剂化物、多晶型物、酯或其药学上可接受的单位剂型,以及所述其他治疗剂的单位剂型。
[0050]
术语解释:
[0051]
除非另有说明,本申请说明书和权利要求书中记载的基团和术语定义,包括其作为实例的定义、示例性的定义、优选的定义、表格中记载的定义、实施例中具体化合物的定义等,可以彼此之间任意组合和结合。这样的组合和结合后的基团定义及化合物结构,应当属于本申请说明书记载的范围。
[0052]
本文所述任选的被取代基所取代的情形涵盖了无取代以及被一个或多个取代基所取代的情形。
[0053]
术语“卤素”指f、cl、br和i。
[0054]
术语“烷基”的碳原子数优选为1~12,还可以为1~6,或1~5,进一步的优选范围为1~3,具体可包括但不限于如下基团:甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基;
[0055]
术语“环烷基”的碳原子数优选为3-8,进一步的优选范围为3~6,例如可选自环丙基、环丁基、环戊基或环己基。
[0056]
术语“有效量”或者“治疗有效量”是指足以实现预期应用(包括但不限于如下定义的疾病治疗)的本发明所述化合物的量。治疗有效量可以取决于以下因素而改变:预期应用(体外或者体内),或者所治疗的受试者和疾病病症如受试者的重量和年龄、疾病病症的严重性和给药方式等,其可以由本领域普通技术人员容易地确定。具体剂量将取决于以下因素而改变:所选择的特定化合物、所依据的给药方案、是否与其它化合物组合给药、给药的时间安排、所给药的组织和所承载的物理递送系统。
[0057]
术语“可药用盐”可以是常规任意酸加成盐或碱加成盐,只要它是可药用的即可。适宜的无机酸可选自氢氯酸、氢溴酸、氢碘酸、硝酸、碳酸、硫酸和磷酸。适宜的有机酸可选自脂族、环脂族、芳族、芳脂族、杂环基羧酸和磺酸类的有机酸。本发明化合物的适宜的可药用碱加成盐包括金属盐或有机盐。更优选的金属盐包括,但不限于合适的碱金属(ⅰa族)盐、碱土金属(ⅱa族)盐和其他生理上可耐受的金属盐。这类盐可由铝、钙、锂、镁、钾、钠和锌制备。优选的有机盐可由叔胺和季胺盐制备,部分包括trometamine、二乙胺、n,n
’-
二苄基乙二胺、氯普鲁卡因、胆碱、二乙醇胺、乙二胺、甲葡胺(n-甲基葡萄糖胺)和普鲁卡因。所有这些盐均可以采用常规方法由本发明化合物与合适的酸或碱反应来制备。
[0058]
术语“溶剂化物”是本发明的化合物的那些形式,其以固体或液体的状态通过与溶剂分子的配位作用形成配合物。水合物是溶剂化物的特定形式,其中配位作用是与水进行。在本发明中,优选的溶剂化物是水合物。进一步的,本发明通式i化合物的药学上可接受的溶剂化物(水合物)是指化合物i与化学计量学的一个或多个分子的水或其他溶剂形成的共晶和包合物。可用于溶剂化物的溶剂包括但不限于:水、甲醇、乙醇、乙二醇和醋酸。
[0059]
有益效果
[0060]
本发明提供了一种方便和高顺应性的帕瑞昔布衍生物可选择一种口服或外用给药的镇痛药(止疼药)。在急性疼痛治疗,特别是急性中重度镇痛、创伤引起的疼痛、手术止疼等需要的药物快速起效,相对于帕瑞昔布的注射给药,有更加稳定的血药浓度和更长药物半衰期的有益效果。相对于注射给药的帕瑞昔布,本发明的帕瑞昔布衍生物提供了一种可以选择口服给药的形式,有基本等同的药物效果,并且有更好的病患依从性和操作安全性(附图i)。
[0061]
附图说明
[0062]
图1示意为帕瑞昔布(parecoxib)(1mg/kg,i.v.)在雄性icr小鼠体内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0063]
图2示意为帕瑞昔布(parecoxib)(1mg/kg,i.v.)在雄性icr小鼠体内监测原型帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0064]
图3示意为化合物1(1mg/kg,i.v.)在雄性icr小鼠体内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0065]
图4示意为化合物1(1mg/kg,i.v.)在雄性icr小鼠体内监测帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0066]
图5示意为化合物1(1mg/kg,i.v.)在雄性icr小鼠体内监测化合物1的血浆浓度(ng/ml)(未检出);
[0067]
图6示意为化合物1(10mg/kg,p.o.)在雄性icr小鼠体内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0068]
图7示意为化合物1(10mg/kg,p.o.)在雄性icr小鼠体内监测帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0069]
图8示意化合物1(10mg/kg,p.o.)在雄性icr小鼠体内监测化合物1的血浆浓度(ng/ml)(未检出);
[0070]
图9示意为化合物2(1mg/kg,i.v.)在雄性icr小鼠体内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0071]
图10示意为化合物2(1mg/kg,i.v.)在雄性icr小鼠体内监测帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0072]
图11示意为化合物2(1mg/kg,i.v.)在雄性icr小鼠体内监测化合物2的血浆浓度
(ng/ml)(未检出);
[0073]
图12示意为化合物2(10mg/kg,p.o.)在雄性icr小鼠体内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0074]
图13示意为化合物2(10mg/kg,p.o.)在雄性icr小鼠体内监测帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0075]
图14示意化合物2(10mg/kg,p.o.)在雄性icr小鼠体内监测化合物2的血浆浓度(ng/ml)(未检出)。
[0076]
图15示意为parecoxib(2mg/kg,i.v.)在雄性食蟹猴体内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0077]
图16示意为parecoxib(2mg/kg,i.v.)在雄性食蟹猴体内监测帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0078]
图17示意为化合物2(15mg/kg,p.o.)在雄性食蟹猴内监测伐地昔布(valdecoxib)的血浆浓度(ng/ml);
[0079]
图18示意为化合物2(15mg/kg,p.o.)在雄性食蟹猴体内监测帕瑞昔布(parecoxib)的血浆浓度(ng/ml);
[0080]
图19示意为化合物2(15mg/kg,p.o.)在雄性食蟹猴体内监测化合物2的血浆微量浓度(ng/ml)。
[0081]
注:i.v.表示“静脉注射”给药,p.o.表示“口服”给药
具体实施方式
[0082]
下文将结合具体实施例对本发明的技术方案做更进一步的详细说明。应当理解,下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明上述内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0083]
除非另有说明,以下实施例中使用的原料和试剂均为市售商品,或者可以通过已知方法制备。
[0084]
合成实例1.
[0085][0086]
将溶有parecoxib帕瑞昔布(200mg,0.54mmol,1.0eq)的dmf(2ml)混合物中分别添加k2co3(375mg,2.7mmol,5.0eq),氯乙酸甲酯(1.16g,10.8mmol,20eq),在室温下搅拌反应18小时。用h2o(20ml)稀释反应混合物,用etoac(20ml x2)萃取。合并有机相用饱和食盐水(15ml)洗涤,经na2so4干燥后过滤,真空浓缩。得到粗品残渣经tlc(pe/ea=1.7:1)纯化,得到50mg粗品,粗品用prep-hplc(acn中0.1%hcooh/h2o)进一步纯化,得到白色固体化合物1(16.65mg,产率6%)。
[0087]
lcms:(m+h)+:443.1
[0088]1h-nmr(400mhz,cdcl3)δ=8.00(m,2h),7.38(m,7h),5.91(s,2h),2.61(q,j=7.2hz,2h),2.52(s,3h),2.08(s,3h),1.10(t,j=7.2hz,3h).
[0089]
合成实例2.
[0090][0091]
将溶有parecoxib帕瑞昔布(200mg,0.541mmol,1.0eq)的dmf(5ml)混合物中分别添加k2co3(746mg,5.41mmol,1.54eq),氯甲基异丙基碳酸酯(4ml),在室温下搅拌反应18小时。用h2o(30ml)稀释反应混合物,用etoac(40ml
×
3)萃取。合并有机相用饱和盐水(30ml
×
3)洗涤组,用na2so4干燥过滤,浓缩得到粗品残渣,经tlc(etoac:pe=1:2)纯化,得到白色固体实例2(104.4mg,产率33.73%)。lcms:(m+h)+:487.2
[0092]1h-nmr(400mhz,dmso-d6)δ=8.01-7.94(m,2h),7.50-7.39(m,5h),7.35-7.30(m,2h),5.91(s,2h),4.87-4.71(m,1h),2.65(dd,j=7.1hz,2h),2.49(s,3h),1.23(d,j=6.2hz,6h),0.92(t,j=7.2hz,3h).
[0093]
合成实施例3.
[0094][0095]
将溶有parecoxib帕瑞昔布(200mg,0.540mmol,1.0eq)的乙腈(2ml)混合物中分别添加dipea(174mg,1.35mmol,2.5eq),氯甲基碳酸甲酯(134.4mg,1.08mmol,2.0eq),在室温下搅拌反应18小时。旋干后用h2o(3ml)稀释反应混合物,用etoac(3ml
×
3)萃取。合并有机相用饱和盐水(3ml
×
3)洗涤组,用na2so4干燥过滤,浓缩得到粗品残渣,经tlc(etoac:pe=1:2)纯化,得到白色固体实例2(178.2mg,产率72.1%)。
[0096]
lcms:(m+h)+:459.1
[0097]1h nmr(400mhz,meod)δ8.09
–
7.92(m,2h),7.51
–
7.26(m,7h),5.96(s,2h),3.77(s,3h),2.65(q,j=7.2hz,2h),2.51(s,3h),1.02(t,j=7.2hz,3h).
[0098]
合成实施例4.
[0099]
[0100]
将溶有parecoxib帕瑞昔布(200mg,0.540mmol,1.0eq)的乙腈(2ml)混合物中分别添加dipea(174.4mg,1.35mmol,2.5eq),氯甲基碳酸乙酯(149.6mg,1.08mmol,2.0eq),在室温下搅拌反应18小时。旋干后用h2o(3ml)稀释反应混合物,用etoac(3ml
×
3)萃取。合并有机相用饱和盐水(3ml
×
3)洗涤组,用na2so4干燥过滤,浓缩得到粗品残渣,经tlc(etoac:pe=1:2)纯化,得到白色固体实例2(194.1mg,产率76%)。
[0101]
lcms:(m+h)+:473.1
[0102]
合成实施例5.
[0103]
参照实施例1合成以下化合物5-28
[0104]
具体实施例列表
[0105]
[0106]
[0107][0108]
本发明的范围中还包括一类包含所述活性化合物与一种或多种无毒的可药用载体和/或稀释剂和/或辅剂(下文统称为“载体”材料)混合的药物组合物,其中根据需要还可含有其他药物活性成分。本发明的活性化合物可通过口服的途径给药或者外用给药形式,优选以与给药途径相适应的药物组合形式和以治疗所需的有效剂量的口服给药。
[0109]
实施例6.药代动力学(pk)实验
[0110]
6.1icr小鼠的药代动力学(pk)研究
[0111]
实验材料和方法:
[0112]
静脉注射组:给药剂量:1mg/kg;溶媒:5%dmso+10%solutol+85%saline;动物数:icr雄性小鼠3只,禁食12小时后给药,给药后继续禁食4小时。样品采集时间点:5min,15min,30min,1h,2h,4h,8h,24h;
[0113]
制备标准曲线和qc样品后用lc-ms/ms测定样品中化合物浓度。
[0114]
口服给药组:给药剂量10mg/kg;溶媒:5%dmso+10%solutol+85%saline;动物数:icr雄性小鼠3只,禁食12小时后给药,给药后继续禁食4小时。样品采集时间点:15min,30min,1h,2h,4h,8h,24h;
[0115]
制备标准曲线和qc样品后用lc-ms/ms测定样品中化合物浓度。
[0116]
测试结果:
[0117]
经实验证实,本发明化合物,尤其是实施例化合物经口服给药后,在体内迅速转化成帕瑞昔布(parecoxib)及其药物活性成分伐地昔布(valdecoxib),且血药浓度基本与帕瑞昔布针剂给药后在体内转化成药物活性成分伐地昔布(valdecoxib)的血药浓度相匹配的,且不仅起效快,还表现出相较于帕瑞昔布针剂给药更平稳的血药浓度和更长的药物作用时间(t
1/2
延长),也即有更好的药物效果和作用时间。同时,在动物试验中发现,血液中原型化合物1或原型化合物2检测不到,这说明化合物1或化合物2能够在口服后快速转变为帕瑞昔布或其活性成分伐地昔布,与帕瑞昔布针剂给药有类似的效果,且更加平稳和更好患者依从性。具体可参见图3-图14所示化合物1和化合物2的测试结果。
[0118]
6.2食蟹猴药代动力学(pk)研究:
[0119]
实验材料和方法:
[0120]
口服给药组:
[0121]
给药剂量:15mg/kg,溶媒:5%dma+10%solutol+85%磺丁基环糊精;动物数:3只雄性食蟹猴;禁食12小时后给药,给药后继续禁食4小时。样品采集时间点:15min,30min,1h,2h,3h,5hr,8h,12h,24h。
[0122]
制备标准曲线和qc样品后用lc-ms/ms测定样品中化合物浓度。
[0123]
帕瑞昔布(parecoxib)的注射给药:
[0124]
给药剂量:2mg/kg,溶媒:5%dmso+10%solutol+85%saline;动物数:3只雄性食蟹猴;禁食12小时后给药,给药后继续禁食4小时。样品采集时间点:5min,15min,30min,1h,2h,3h,5h,8h,12h,24h。
[0125]
制备标准曲线和qc样品后用lc-ms/ms测定样品中化合物浓度。
[0126]
测试结果:
[0127]
经实验证实,尤其是实施例化合物(例如化合物2)的经口服给药后,在体内迅速转化成帕瑞昔布(parecoxib)及其药物活性成分伐地昔布(valdecoxib),且血药浓度是基本与帕瑞昔布针剂给药后在体内转化成之药物活性成分伐地昔布(valdecoxib)的血药浓度相匹配的,且不仅起效快,还表现出相较于帕瑞昔布针剂给药更平稳的血药浓度和更长的药物作用时间(t
1/2
延长),也即有更好的药物效果和作用时间。同时发现在动物血药浓度测试仅仅发现微量的原型化合物2的浓度。这说明化合物2能够在口服后及时转变为帕瑞昔布或其活性成分伐地昔布。口服给药化合物2与帕瑞昔布针剂给药有类似的效果,且更加平稳和更好的患者依从性,快速转变成帕瑞昔布和伐地昔布。具体可参见图15-图19所示化合物2的测试结果。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1