一种偶氮芳香化合物及其应用和一种用于增强拉曼散射信号的试剂
1.本发明涉及分析化学领域,具体涉及一种偶氮芳香化合物及其应用和一种用于增强拉曼散射信号的试剂。
背景技术:
2.现代光谱学与显微技术的发展,推动人们更灵敏和特异地观察生命体系分子变化的动态过程。具体地,直接观察细胞内部大量不同分子种类的能力对于理解复杂系统和过程尤为重要,而在细胞和亚细胞水平上高灵敏度、高选择性地对许多物种进行成像仍然具有挑战性。与其他广泛应用的成像技术(尤其是荧光成像)相比,拉曼显微镜已成为一种有用的工具,用于进行多色活细胞成像。
3.由于与入射光耦合的分子振动的固有性质,拉曼信号提供了特征的化学键信息,其信号稳定、谱线狭窄,没有光漂白现象,允许在典型的拉曼光谱范围内进行多色活细胞成像,并通过多色成像实现活细胞动态过程的可视化研究。然而与荧光成像相比,拉曼成像较低的灵敏度限制了其广泛应用。拉曼散射的截面较小,成为活细胞中内源性成分或外源性探针灵敏成像的主要障碍。
4.目前,已经开发了多种策略和技术,例如表面增强拉曼散射(sers),相干反斯托克斯拉曼散射(cars),受激拉曼散射(srs)和尖端增强拉曼散射(ters)来增强被测化合物的拉曼信号。表面增强拉曼(sers)需要借助纳米颗粒的表面等离激元,其重现性有待提高。sers为细胞成像提供了显著的灵敏度和多样性,但是与小分子探针相比,sers颗粒可能导致对活细胞较低的渗透率和不利的生物学干扰。ters需要精确控制金属纳米尖端与表面的相对位置,以实现信号的增强。非线性光学的方法包括相干反斯托克斯拉曼(cars)以及受激拉曼(srs)需要搭建复杂昂贵的仪器。因此,如果在分子层面将拉曼染料自身固有的信号大幅增强,这将对拉曼领域的研究和应用产生重大影响。当使用典型的商业拉曼显微镜系统进行检测时,便捷的细胞拉曼成像对于具有固有强拉曼散射的小分子信号化合物的需求很大。
5.自从基于5-乙炔基-2'-脱氧尿苷(edu)的细胞拉曼成像的研究被报道以来,许多含炔的分子已被用作生物正交拉曼探针,用于自发或非线性拉曼显微成像。然而,当使用自发拉曼显微镜进行高质量的拉曼成像时,c≡c伸缩振动的拉曼强度仍然不能满足灵敏度的需求。目前,苯基封端的炔类信号分子作为srs成像的多色拉曼报告分子被合成出来,但是,当多炔链长增加时,分子的化学稳定性存在问题。同时,它们的溶解度也随之降低,限制了细胞靶向与染色的效率。
6.共振拉曼策略通过生色团耦合电子和振动能级的跃迁,提供了增强拉曼灵敏度的方法。在这种情况下,振动能级跃迁与电子能级跃迁存在一定程度的匹配,导致拉曼散射信号的显著放大。最近,一种基于聚二乙炔类型的拉曼染料在共振时相对于edu的相对拉曼强度达到4个数量级以上。该策略基于增加共轭长度使吸收波长红移。然而,该研究结果无法
提供拉曼频率的可调性,并且该信号分子在电子共振激发时呈现较强的荧光背景,干扰此时的拉曼信号检测。
7.也就是说,当激发波长与化合物的吸收波长相近时,电子跃迁可以提高拉曼散射的几率,使拉曼信号增强,但是由此导致的常见问题是,当激发波长位于化合物的吸收峰附近时,伴随而来的较强荧光背景,使得拉曼信号无法被准确检测。
技术实现要素:
8.本发明的目的是为了克服现有技术存在的上述缺陷,提供一类新的增强拉曼散射的分子。
9.为了实现上述目的,本发明的第一方面提供一种偶氮芳香化合物,该化合物具有式(i)或式(ii)所示的结构:
10.式(i):
11.式(ii):
12.式(i1):
13.式(i2):
14.式(i3):
15.式(i4):
16.式(i5):
17.式(i6):
18.其中,在式(i)和式(ii)中,
19.r
11
、r
12
、r
14
、r
15
各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基、羧基取代的c
1-6
的烷氧基中的至少一种;r
13
选自r
1-conh-、nh
2-、(r1)(r2)n-、c
1-6
的烷氧基、r
1-coo-、r
1-oco-、硝基、氰基;或者,r
11
和r
15
各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基、羧基取代的c
1-6
的烷氧基中的至少一种,r
12
、r
13
和r
14
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在选自c
1-6
的烷基中的至少一种取代基;
20.r
21
、r
22
、r
24
和r
25
各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基;r
23
选自h、式(i1)所示的基团、式(i2)所示的基团、硝基、r
1-coo-、r
1-oco-、nc-、(r1)(r2)n-、氰基;或者r
21
和r
25
各自独立地选自h、c
1-6
的烷基、r
1-coo-、r
1-oco-、c
1-6
的烷氧基;r
22
、r
23
和r
24
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在选自c
1-6
的烷基中的至少一种取代基;
21.l选自式(i3)所示的基团、式(i4)所示的基团、式(i5)所示的基团;
22.r1和r2各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基-c
1-3
的亚烷基-、羟基-c
1-3
的亚烷基-、式(i6)所示的基团;
23.r3和r4各自独立地选自h、苯基、由r
1-oco-取代的苯基;
24.n、m、x、y和z各自独立地为1-10的整数。
25.本发明第二方面提供前述第一方面中所述的化合物在拉曼散射信号中的应用。
26.本发明第三方面提供一种用于增强拉曼散射信号的试剂,该试剂中含有信号增强的功能化合物,所述功能化合物中的功能基团由前述第一方面中所述的化合物提供。
27.本发明的发明人发现,本发明的化合物中,通过引入共轭偶氮苯基团,能够促使化合物的紫外可见吸收波长发生红移,并且能够与拉曼激发波长相匹配,产生共振拉曼效应。另外,由于本发明中引入的偶氮苯结构在激发态时能够产生顺反异构化的现象,使得激发态的能量能够通过非辐射跃迁的方式释放出去,从而有效地淬灭共生的荧光,使增强的拉曼信号明显地显现出来。
28.与5-乙炔基2'-脱氧尿苷(edu)相比,本发明提供的偶氮芳香化合物在用于增强拉曼散射信号时,灵敏度提高了2-4个数量级以上,并且具有10个不同光谱带的频率可调性。本发明还开发了用于6色成像和7多重编码应用的活细胞细胞器靶向探针。
29.本发明提供的偶氮芳香化合物增强了电子和振动跃迁之间的耦合和/或改善了振动模式的对称性。这些效应导致了本发明的偶氮芳香化合物的超灵敏拉曼信号,为设计和合成小分子拉曼探针提供了平台,这些探针能够通过自发拉曼显微镜观察到多色靶向和成像活细胞中的特定细胞器/生物分子。
30.本发明提供的偶氮芳香化合物形成的探针在单个图像中,在拉曼位移的不同光谱带上可视化线粒体、溶酶体等不同类型的细胞器。
31.本发明提供的偶氮芳香化合物为将来在细胞内和/或细胞间转化过程中同时追踪功能性生物分子,细胞器和/或细胞类型之间的反应开辟了新的前景。
32.更具体地,本发明提供的偶氮芳香化合物子可通过自发拉曼显微镜实现活细胞成像的超灵敏性(rie约为104级)和多色功能(从10个频率选项中进行选择)。这些分子出现了频率可调性,并开发了用于6色成像的活细胞细胞器靶向探针。
附图说明
33.图1是100μm的c3.1在溶剂dmf中的拉曼光谱。
34.图2是化合物e3.1的光谱性质。其中,图2a为5.0μm的e3.1的吸收光谱和荧光光谱。图2b为100μm的e3.1分别在532nm和785nm激光下苯环碳-偶氮氮的伸缩振动ν(c
ph-n)模式的拉曼光谱。图2c为e3.1在1122cm-1
处拉曼信号相对于dmf溶剂峰(在867cm-1
处)的强度与其浓度之间的线性关系。
35.图3是用e3.1作为信号源进行免疫检测的结果。图3a为功能化聚苯乙烯微球结合免疫球蛋白后在532nm激光下的拉曼光谱。免疫球蛋白的浓度分别为10-10
、10-11
、10-12
、10-13
mol/l。图3b为免疫球蛋白在10-13
至10-6
mol/l浓度范围的相对拉曼强度曲线。相对拉曼强度为1122cm-1
处拉曼信号与聚苯乙烯1001cm-1
处拉曼信号的比值。图3c为相对拉曼信号与浓度之间的线性曲线。
36.图4是偶氮增强拉曼探针与荧光探针共染海拉细胞的结果。其中,图4a为线粒体探针的共染结果。图4b为溶酶体探针的共染结果。
37.图5是偶氮增强拉曼探针染色海拉细胞的多色拉曼成像。其中,图5a为6种偶氮增强拉曼探针的化学结构。图5b为细胞的六色拉曼成像图。图5c为6种拉曼探针在细胞中的拉曼光谱。图5d为6种拉曼探针的细胞毒性测试结果。
具体实施方式
38.在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
39.本发明所述c
1-6
的烷基表示碳原子总数为1-6的烷基,包括直链烷基、支链烷基和环烷基,包括但不限于甲基、乙基、正丙基、异丙基、环丙基、正丁基、异丁基、叔丁基、环丁基、正戊基、异戊基、环戊基、正己基、环己基。
40.本发明所述c
1-6
的烷氧基表示碳原子总数为1-6的烷氧基,包括直链烷氧基、支链烷氧基和环烷氧基,包括但不限于甲氧基、乙氧基、正丙氧基、异丙氧基、环丙氧基、正丁氧基、异丁氧基、叔丁氧基、环丁氧基、正戊氧基、异戊氧基、环戊氧基、正己氧基、环己氧基。针对“羧基取代的c
1-6
的烷氧基”具有与此相似的解释,所不同的是,“羧基取代的c
1-6
的烷氧基”表示至少一个羧基取代了c
1-6
的烷氧基上的至少一个h。
41.本发明所述c
1-6
的烷氧基-c
1-3
的亚烷基-表示r
31-r
32-所示的基团,其中的r
31
表示c
1-6
的烷氧基,其中的r
32
表示c
1-3
的亚烷基。“羟基-c
1-3
的亚烷基
‑”
具有与此相似的解释。
42.本发明所述由r
1-oco-取代的苯基表示苯基中的至少一个h原子由r
1-oco-所示的取代基取代,并且对具体的取代位置没有特别的限定。
43.如前所述,本发明的第一方面提供了一种偶氮芳香化合物,该化合物具有式(i)或式(ii)所示的结构:
44.式(i):
45.式(ii):
46.式(i1):
47.式(i2):
48.式(i3):
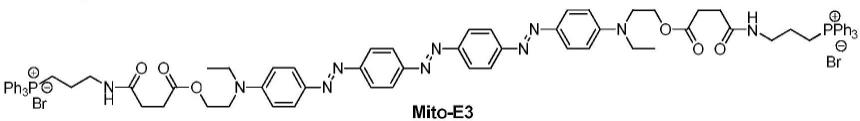
49.式(i4):
50.式(i5):
51.式(i6):
52.其中,在式(i)和式(ii)中,
53.r
11
、r
12
、r
14
、r
15
各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基、羧基取代的c
1-6
的烷氧基中的至少一种;r
13
选自r
1-conh-、nh
2-、(r1)(r2)n-、c
1-6
的烷氧基、r
1-coo-、r
1-oco-、硝基、氰基;或者,r
11
和r
15
各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基、羧基取代的c
1-6
的烷氧基中的至少一种,r
12
、r
13
和r
14
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在选自c
1-6
的烷基中的至少一种取代基;
54.r
21
、r
22
、r
24
和r
25
各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基;r
23
选自h、式(i1)所示的基团、式(i2)所示的基团、硝基、r
1-coo-、r
1-oco-、nc-、(r1)(r2)n-、氰基;或者r
21
和r
25
各自独立地选自h、c
1-6
的烷基、r
1-coo-、r
1-oco-、c
1-6
的烷氧基;r
22
、r
23
和r
24
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在选自c
1-6
的烷基中的至少一种取代基;l选自式(i3)所示的基团、式(i4)所示的基团、式(i5)所示的基团;
55.r1和r2各自独立地选自h、c
1-6
的烷基、c
1-6
的烷氧基-c
1-3
的亚烷基-、羟基-c
1-3
的亚烷基-、式(i6)所示的基团;
56.r3和r4各自独立地选自h、苯基、由r
1-oco-取代的苯基;
57.n、m、x、y和z各自独立地为1-10的整数。
58.优选情况下,在式(i)和式(ii)中,
59.r
11
、r
12
、r
14
、r
15
各自独立地选自h、c
1-4
的烷基、c
1-4
的烷氧基、羧基取代的c
1-6
的烷氧基中的至少一种;r
13
选自r
1-conh-、nh
2-、(r1)(r2)n-、c
1-6
的烷氧基、r
1-coo-、r
1-oco-、硝基、氰基;或者,r
11
和r
15
各自独立地选自h、c
1-4
的烷基、c
1-4
的烷氧基、羧基取代的c
1-6
的烷氧基中的至少一种,r
12
、r
13
和r
14
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在选自c
1-4
的烷基中的至少一种取代基;
60.r
21
、r
22
、r
24
和r
25
各自独立地选自h、c
1-4
的烷基、c
1-4
的烷氧基;r
23
选自h、式(i1)所示的基团、式(i2)所示的基团、硝基、r
1-coo-、r
1-oco-、nc-、(r1)(r2)n-、氰基;或者r
21
和r
25
各自独立地选自h、c
1-4
的烷基、r
1-coo-、c
1-4
的烷氧基;r
22
、r
23
和r
24
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在选自c
1-4
的烷基中的至少一种取代基;l选自式(i3)所示的基团、式(i4)所示的基团、式(i5)所示的基团;
61.r1和r2各自独立地选自h、c
1-4
的烷基、c
1-4
的烷氧基-c
1-3
的亚烷基-、羟基-c
1-3
的亚烷基-、式(i6)所示的基团;
62.r3和r4各自独立地选自h、苯基、由r
1-oco-取代的苯基;
63.n、m、x、y和z各自独立地为1-6的整数。
64.更优选情况下,在式(i)和式(ii)中,
65.r
11
、r
12
、r
14
、r
15
各自独立地选自h、甲基、甲氧基、-o(ch2)5cooh中的至少一种;r
13
选自(ch3ch2)(ch3och2ch2)n-、(ch3ch2)(hoch2ch2)n-、ch3conh-、nh
2-、ch3o-、ch3coo-、硝基、氰基;或者,r
11
和r
15
各自独立地选自h、甲基、甲氧基、-o(ch2)5cooh中的至少一种,r
12
、r
13
和r
14
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在甲基取代基,且所述含n稠合三环的非苯环的环结构均为六元环;
66.r
21
、r
22
、r
24
和r
25
各自独立地选自h、甲氧基、式(i6)所示的基团-coo-;r
23
选自h、式(i1)所示的基团、式(i2)所示的基团、硝基、ch3oco-、nc-、(ch3ch2)(ch3och2ch2)n-、(ch3ch2)(hoch2ch2)n-;或者r
21
和r
25
各自独立地选自h、甲氧基、式(i6)所示的基团-coo-;r
23
选自h、式(i1)所示的基团、式(i2)所示的基团、硝基、ch3oco-、nc-、(ch3ch2)(ch3och2ch2)n-、(ch3ch2)(hoch2ch2)n-、氰基;r
22
、r
23
和r
24
与三者所在的母核苯环一起形成含n稠合三环,所述含n稠合三环的非苯环的环结构上任选存在甲基取代基,且所述含n稠合三环的非苯环的环结构均为六元环;l选自式(i3)所示的基团、式(i4)所示的基团、式(i5)所示的基团;
67.r3和r4各自独立地选自h、苯基、由ch3oco-取代的苯基;
68.n、m、x、y和z各自独立地为1、2、3、4、5或6。
69.根据一种优选的具体实施方式,所述偶氮芳香化合物具有式(i)所示的结构。
70.更优选情况下,所述偶氮芳香化合物选自以下化合物中的任意一种:
71.化合物a2.1:
72.化合物a2.2:
73.化合物b2.1:
74.化合物c2.1:
75.化合物d2.1:
76.化合物d2.2:
77.化合物d2.3:
78.化合物d2.4:
79.化合物e1.1:
80.化合物e1.2:
81.化合物e1.3:
82.化合物e2.1:
83.化合物e2.2:
84.根据另一种优选的具体实施方式,所述偶氮芳香化合物具有式(ii)所示的结构。
85.更优选情况下,所述偶氮芳香化合物选自以下化合物中的任意一种:
86.化合物a3.1:
87.化合物a3.2:
88.化合物b3.1:
89.化合物b3.2:
90.化合物c3.1:
91.化合物c3.2:
92.化合物e3.1:
93.化合物f2.1:
94.本发明对制备前述偶氮芳香化合物的具体制备方法没有特别的限制,本领域技术人员可以根据本发明提供的具体结构式结合有机合成领域内的已知合成方法和本发明的
后文的实例中提供的部分具体的实施例中的制备方法获得本发明全部偶氮芳香化合物的具体制备方法。本文中不再对制备前述偶氮芳香化合物的通用制备方法进行详述,本领域技术人员不应理解为对本发明的限制。
95.如前所述,本发明的第二方面提供了前述偶氮芳香化合物在增强拉曼散射信号中的应用。
96.如前所述,本发明的第三方面提供了一种用于增强拉曼散射信号的试剂,该试剂中含有信号增强的功能化合物,所述功能化合物中的功能基团由前述第一方面所述的化合物提供。
97.优选情况下,在所述试剂中,所述功能化合物的浓度为0.1-1000微摩尔/升。
98.以下将通过实例对本发明进行详细描述。以下实例中,在没有特别说明的情况下,使用的原料均为普通市售品。
99.制备例1:a组化合物的合成(核心基团为二炔类)
[0100][0101]
向25ml单口瓶中加入3ml三氯甲烷和1ml的1,4-二氧六环,随后称取3mmol苯乙炔和1mmol的4-乙炔基苯甲酸甲酯溶解于其中,加入0.05mmol铜粉和0.2mmol四甲基乙二胺。将该混合体系置于50℃下搅拌12h。随后加入20ml二氯甲烷,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏掉溶剂,经柱层析后得到黄色固体产物a1.1.(收率65%)1h nmr(400mhz,cdcl3)δ7.99(d,j=8.3hz,2h),7.55(d,j=8.3hz,2h),7.39-7.30(d,j=8.3hz,2h),6.65-6.54(d,j=8.3hz,2h),3.92(s,3h).
13
c nmr(101mhz,cdcl3)δ166.5,147.9,134.2,132.2,129.9,129.5,127.0,114.6,110.2,84.5,79.8,77.5,71.8,52.4.hrms(esi):calcd for c
18h14
no
2+
[m+h]
+
276.1019,found 276.1017.
[0102]
称取0.15mmol产物a1.1溶于2ml无水乙醇,于冰浴条件下加入1.5mmol浓硫酸和0.6mmol亚硝酸钾(溶于1ml水中),冰浴30min后,用醋酸钠调节ph值至5.0后,加入n-乙基n-羟乙基苯胺(0.3mmol)的乙醇溶液,继续反应3h。反应完毕后,加入20ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到红色固体产物a2.2(收率47%)。1h nmr(400mhz,chloroform-d)δ8.01(d,j=8.1hz,2h),7.86(d,j=8.8hz,2h),7.81(d,j=8.3hz,2h),7.63(d,j=8.2hz,2h),7.59(d,j=8.1hz,2h),6.78(d,j=8.8hz,2h),4.32(t,j=6.3hz,2h),3.93(s,3h),3.67(t,j=6.3hz,2h),3.50(t,j=6.9hz,2h),1.25(d,j=7.0hz,3h).hrms(esi):calcd for c
28h26
n3o
3+
[m+h]
+
452.1969,found 452.1972.
[0103][0104]
重复a1.1相同的合成步骤得到中间产物a1.2,淡黄色固体.1h nmr(400mhz,cdcl3)
δ7.31(d,j=8.4hz,4h),6.58(d,j=8.4hz,4h),3.88(s,4h).
13
c nmr(101mhz,cdcl3)δ147.3,133.9,114.7,111.2,81.8,72.4.hrms(esi):calcd for c
16h13n2+
[m+h]
+
233.1073,found 233.1083.
[0105]
称取产物a1.2(0.15mmol,1eq)溶于2ml无水乙醇,于冰浴条件下加入浓硫酸(1.5mmol,10eq)和亚硝酸钾(0.6mmol,4eq)(溶于1ml水中),冰浴30min后,用醋酸钠调节ph至5.0后,加入n-乙基n-羟乙基苯胺(0.3mmol)的乙醇溶液,继续反应3h。反应完毕后,加入20ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到红色固体产物a3.1(收率47%)。1h nmr(400mhz,chloroform-d)δ7.88(d,j=8.5hz,4h),7.82(t,j=6.9hz,4h),7.63(t,j=9.1hz,4h),6.83(d,j=8.6hz,4h),4.35(t,j=6.0hz,4h),3.71(t,j=6.1hz,4h),3.54(q,j=7.1hz,4h),1.24(d,j=7.2hz,6h).hrms(esi):calcd for c
36h37
n6o
2-[m+h]
+
585.2973,found 585.2980.
[0106]
制备例2:b组化合物的合成(核心基团为三炔类)
[0107][0108]
向50ml单口瓶中加入9ml三氯甲烷和3ml的1,4-二氧六环,随后称取三甲基硅乙炔(15mmol,3eq)和4-乙炔基苯甲酸甲酯(5mmol,1eq)溶解于其中,加入铜粉(0.25mmol,0.05eq)和四甲基乙二胺(1mmol,0.2eq)。将该混合体系置于50℃下搅拌12h。随后加入40ml二氯甲烷,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏掉溶剂,经柱层析后得到浅黄色固体产物1(收率64%)。1h nmr(400mhz,chloroform-d)δ8.03
–
7.91(m,2h),7.53(d,j=8.4hz,2h),3.91(s,3h),0.23(s,9h).
13
c nmr(101mhz,cdcl3)δ166.3,132.6,130.4,129.5,126.1,92.5,87.4,76.8,75.6,52.4,0.4.hrms(esi):calcd for c
15h16
o2sina
+
[m+na]
+
279.0812,found 279.0825.
[0109]
将819mg的产物1用25ml乙腈溶解,称量水(6.4mmol)和氟化银(3.2mmol),该混合体系于避光条件下搅拌20min后,加入n-溴代丁二酰亚胺(3.2mmol),继续搅拌3h。反应完毕后,减压蒸馏掉溶剂,用30ml乙酸乙酯稀释,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到浅黄色固体产物2(收率85%)。1h nmr(400mhz,chloroform-d)δ7.98(d,j=8.1hz,2h),7.54(d,j=8.2hz,2h),3.91(s,3h).
13
c nmr(101mhz,cdcl3)δ166.3,132.8,130.5,129.5,125.7,76.9,73.0,65.1,52.4,46.4.hrms(esi):calcd for c
12
h8bro
2+
[m+h]
+
262.9702,found 262.9712.
[0110]
称取产物2(1mmol)、4-乙炔基苯胺(1.5mmol)、双(三苯基膦)氯化钯(ii)(0.1mmol)和碘化亚铜(0.1mmol)于25ml两口瓶中,反复抽充氮气3次后,加入5ml四氢呋喃和170μl三乙胺,该混合体系于50℃下搅拌12h。反应完毕后,减压蒸馏除去溶剂,用20ml乙酸乙酯稀释,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到黄色固体产物b1.1(收率65%)。1h nmr(400mhz,chloroform-d)δ
7.99(dd,j=8.3,1.8hz,2h),7.60
–
7.50(m,2h),7.39
–
7.30(m,2h),6.65
–
6.54(m,2h),5.38(s,2h),3.92(d,j=1.7hz,3h).
13
c nmr(101mhz,dmso-d6)δ150.8,134.8,132.8,130.6,129.6,125.7,114.2,105.7,82.6,78.7,77.5,72.3,69.2,68.5,65.5,52.4.hrms(esi):calcd for c
20h13
no
2+
[m]
+
299.0941,found 299.0988.
[0111]
将b1.1进行与a1.1的相同的重氮化步骤即得到b2.1,橙色固体。1h nmr(400mhz,chloroform-d)δ8.01(d,j=8.1hz,2h),7.86(d,j=8.8hz,2h),7.81(d,j=8.3hz,2h),7.63(d,j=8.2hz,2h),7.59(d,j=8.1hz,2h),6.78(d,j=8.8hz,2h),4.32(t,j=6.3hz,2h),3.93(s,3h),3.67(t,j=6.3hz,2h),3.50(t,j=6.9hz,2h),2.67(d,j=6.3hz,2h),2.63(d,j=6.2hz,2h),1.25(d,j=7.0hz,3h).hrms(esi):calcd for c
30h26
n3o
3+
[m+h]
+
476.1969,found 476.1975.
[0112][0113]
向25ml单口瓶中加入5ml四氢呋喃,称取4-乙炔基苯胺(6mmol)和二碳酸二叔丁酯(18mmol)溶解于其中,回流搅拌12h,反应完毕后减压蒸馏除去溶剂。向该体系加入6ml三氯甲烷、2ml的1,4-二氧六环和3ml四甲基乙二胺,称量三甲基硅乙炔(14mmol)和铜粉(0.6mmol),于50℃下搅拌12h。随后加入20ml二氯甲烷,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到淡黄色固体产物3(收率70%)。1h nmr(400mhz,chloroform-d)δ7.41(dd,j=8.7,2.3hz,2h),7.34(dd,j=8.6,2.3hz,2h),1.51(d,j=2.3hz,9h),0.23(d,j=2.4hz,9h).
13
c nmr(101mhz,cdcl3)δ152.3,139.5,133.6,118.0,115.2,90.2,88.0,81.0,76.9,73.4,28.3,0.4.hrms(esi):calcd forc
18h24
no2si
+
[m+h]
+
314.1571,found 314.1582.
[0114]
将1.34g产物3用30ml乙腈溶解,称量水(8.4mmol)和氟化银(4.2mmol),该混合体系于避光条件下搅拌20min后,加入n-溴代丁二酰亚胺(4.2mmol),继续搅拌3h。反应完毕后,减压蒸馏掉溶剂,用40ml乙酸乙酯稀释,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到浅黄色固体产物4(收率81%)。1h nmr(400mhz,chloroform-d)δ7.41(dt,j=9.9,6.2hz,2h),7.33(t,j=6.9hz,2h),1.49(d,j=6.6hz,9h).
13
c nmr(101mhz,cdcl3)δ146.8,139.6,133.8,117.3,114.9,85.2,81.1,65.5,60.4,44.0,28.3.hrms(esi):calcd for c
15h13
brno
2-[m-h]-318.0135,found 318.0148.
[0115]
称取产物4(1mmol)、4-乙炔基苯胺(1.5mmol)、双(三苯基膦)氯化钯(ii)(0.1mmol)和碘化亚铜(0.1mmol)于25ml两口瓶中,反复抽充氮气3次后,加入5ml四氢呋喃
和170μl三乙胺,该混合体系于50℃下搅拌12h。反应完毕后,减压蒸馏除去溶剂,用20ml乙酸乙酯稀释,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到黄色固体产物5(收率60%)。1h nmr(400mhz,chloroform-d)δ7.48
–
7.40(m,2h),7.37
–
7.29(m,4h),6.58(d,j=8.0hz,2h),1.51(d,j=1.6hz,9h).
13
c nmr(101mhz,cdcl3)δ152.3,148.0,139.7,134.7,133.9,133.3,118.0,114.6,109.6,81.1,79.9,78.4,74.1,72.9,67.0,66.1,28.23.hrms(esi):calcd for c
23h21
n2o
2+
[m+h]
+
357.1598,found 357.1580.
[0116]
将产物5溶于3ml二氯甲烷,加入1ml三氟乙酸,于室温下搅拌3h后,减压蒸馏除去溶剂,加入足量三乙胺后,溶于10ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到黄色固体产物b1.2(收率83%)。1h nmr(400mhz,chloroform-d)δ7.37
–
7.28(m,4h),6.62
–
6.53(m,4h),3.93(s,4h).
13
c nmr(101mhz,cdcl3)δ147.8,134.6,114.7,109.9,79.6,73.1,66.5.hrms(esi):calcd for c
18h13n2+
[m+h]
+
257.1073,found 257.1070.
[0117]
将b1.2进行与a2.1的相同的重氮化步骤即得到b3.1。1h nmr(400mhz,chloroform-d)δ7.88(d,j=8.5hz,4h),7.82(t,j=6.9hz,4h),7.63(t,j=9.1hz,4h),6.83(d,j=8.6hz,4h),4.35(t,j=6.0hz,4h),3.71(t,j=6.1hz,4h),3.54(q,j=7.1hz,4h),1.24(d,j=7.2hz,6h).hrms(esi):calcd for c
38h37
n6o2[m+h]
+
609.2973,found 609.2978.
[0118]
制备例3:c组化合物的合成(核心基团为四炔类)
[0119][0120]
向50ml单口瓶中加入9ml三氯甲烷和3ml的1,4-二氧六环,随后称取三甲基硅乙炔(15mmol)和4-乙炔基苯胺(5mmol)溶解于其中,加入铜粉(0.25mmol)和四甲基乙二胺(1mmol)。将该混合体系置于50℃下搅拌12h。随后加入40ml二氯甲烷,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏掉溶剂,经柱层析后得到棕黄色固体产物6(收率60%)。1h nmr(400mhz,chloroform-d)δ7.28(d,j=8.4hz,2h),6.55(d,j=8.4hz,2h),3.90(s,2h),0.21(s,9h).
13
c nmr(101mhz,cdcl3)δ147.7,134.3,114.6,110.1,89.5,88.5,78.1,72.4,0.3.hrms(esi):calcd for c
13h16
nsi
+
[m+h]
+
214.10465,found 214.10468.
[0121]
称取产物6(1mmol)溶于2ml二氯甲烷和2ml甲醇中,加入无水碳酸钾(10mmol),于室温下搅拌3h。反应完毕后,加入20ml二氯甲烷稀释,先后用蒸馏水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸馏掉溶剂至1ml备用。另称取产物2(1mmol)、双(三苯基膦)氯化钯(ⅱ)(0.1mmol)和碘化亚铜(0.1mmol)于25ml两口瓶中,反复抽充氮气3次后,加入产物6的溶液、5ml四氢呋喃和三乙胺(1.2mmol),该混合体系于50℃下搅拌12h。反应完毕后,减压蒸馏除去溶剂,用20ml乙酸乙酯稀释,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫
酸钠干燥,减压蒸馏掉溶剂,经柱层析后得到黄色固体产物c1.1(收率52%)。1hnmr(400mhz,chloroform-d)δ7.99(dd,j=8.3,1.8hz,2h),7.60
–
7.50(m,2h),7.39
–
7.30(m,2h),6.65
–
6.54(m,2h),5.38(s,2h),3.92(s,3h).
13
cnmr(101mhz,dmso)δ165.7,151.6,135.3,133.4,131.1,129.7,124.9,114.1,104.3,82.2,77.1,72.6,69.1,66.8,66.2,63.5,60.5,52.6.hrms(esi):calcd forc
22h13
no
2+
[m]
+
323.0941,found 323.0962.
[0122]
将c1.1进行与a2.1相同的重氮化步骤即得到c2.1。红色固体,1h nmr(400mhz,chloroform-d)δ8.01(d,j=8.1hz,2h),7.86(d,j=8.8hz,2h),7.81(d,j=8.3hz,2h),7.63(d,j=8.2hz,2h),7.59(d,j=8.1hz,2h),6.78(d,j=8.8hz,2h),4.32(t,j=6.3hz,2h),3.93(s,3h),3.67(t,j=6.3hz,2h),3.50(t,j=6.9hz,2h),1.25(d,j=7.0hz,3h).hrms(esi):calcd for c
32h26
n3o
3+
[m+h]
+
500.1969,found 500.1975.
[0123][0124]
称取产物6(1.2mmol)溶于7ml二氯甲烷和7ml哌啶中,加入一水合醋酸铜(2.4mmol),于室温下搅拌3h。反应完毕后,减压蒸馏除去溶剂,用10ml乙酸乙酯稀释,先后用饱和氯化铵、蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到黄色固体产物c1.2(收率42%)。1h nmr(400mhz,chloroform-d)δ7.37
–
7.28(m,4h),6.62
–
6.53(m,4h),3.93(s,4h).
13
c nmr(101mhz,cdcl3)δ147.4,133.9,114.8,108.9,79.6,74.1,68.3,62.5.hrms(esi):calcd for c
20h13n2+
[m+h]
+
281.1073,found 281.1087.
[0125]
将c1.2进行与a2.1的重氮化步骤即得到c3.1。深红色固体1h nmr(400mhz,dmso-d6)δ7.79(d,j=5.1hz,12h),6.88(d,j=8.5hz,4h),4.24(d,j=5.8hz,4h),3.68(s,4h),3.51(s,4h),1.23(s,6h).hrms(esi):calcd for c
40h37
n6o
2+
[m+h]
+
633.2973,found 633.2979.
[0126]
制备例4:d组化合物的合成(核心基团为硝基类)
[0127][0128]
将4-硝基苯胺进行与a2.1相同的重氮化步骤即得到d2.3.
[0129]
d2.3(收率73%)红色固体。1h nmr(400mhz,cdcl3)δ8.30(d,j=8.8hz,2h),7.90(d,j=8.8hz,2h),7.40(s,1h),6.35(s,1h),4.65(s,2h),3.98(s,3h),3.90(s,3h).
13
c nmr(101mhz,cdcl3)δ157.3,156.3,147.1,144.5,142.0,133.9,124.7,122.6,97.8,97.3,56.7,55.9.hrms(esi):calcd for c
14h15
n4o
4+
[m+h]
+
303.1088,found 303.1090.
[0130]
将4-硝基苯胺进行与1,2,4-三甲氧基苯进行与d2.3相同的重氮化步骤即得到d2.1.
[0131][0132]
橙色固体1h nmr(400mhz,cdcl3)δ8.34(d,j=8.9hz,2h),7.96(d,j=8.9hz,2h),7.45(s,1h),6.64(s,1h),4.08(s,3h),4.02(s,3h),3.93(s,3h).
13
c nmr(101mhz,cdcl3)δ156.68,155.19,147.85,144.23,135.39,124.72,123.07,111.88,98.76,97.38,57.54,56.30,56.21.hrms(esi):calcd for c
15h16
n3o
5+
[m+h]
+
318.1085,found 318.1086.
[0133]
将4-硝基苯胺进行与6-(8-羟基久洛尼定基)-己酸进行与d2.3相同的重氮化步骤即得到d2.4。
[0134][0135]
紫色固体1h nmr(400mhz,cdcl3)δ8.30(d,j=8.5hz,2h),7.83(d,j=8.5hz,2h),7.46(s,1h),4.21(t,j=6.5hz,2h),3.31(t,j=5.6hz,4h),2.90-2.70(m,4h),2.41(t,j=7.4hz,2h),2.00-1.90(m,4h),1.90-1.80(m,2h),1.79-1.68(m,2h),1.65-1.55(m,2h).
13
c nmr(101mhz,cdcl3)δ179.3,157.5,157.1,148.5,146.6,136.0,124.8,122.2,117.7,114.9,112.8,76.08,50.3,49.9,33.9,30.3,27.5,25.8,24.6,21.6,21.2,20.9.hrms(esi):calcd for c
24h27
n4o
5-[m-h]-451.1987,found 451.1952.
[0136]
制备例5:e组化合物的合成(核心基团为偶氮类)
[0137][0138]
称取4-乙炔基苯胺(3mmol,1eq)溶于10ml无水乙醇中,于冰浴条件下加入浓硫酸(18mmol)和亚硝酸钾(18mmol)(溶于3ml水中),冰浴30min后,加入氨基磺酸(18mmol),5min后,用醋酸钠调节ph至5.0后,加入2,5-二甲氧基苯胺(4.5mmol)的乙醇溶液,继续反应3h。反应完毕后,加入60ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到红色固体产物e1.1。1h nmr(400mhz,chloroform-d)δ8.16
–
8.06(m,2h),7.85(dd,j=8.5,1.9hz,2h),7.40(d,j=1.7hz,1h),6.35(d,j=1.7hz,1h),4.53(s,2h),3.96(d,j=1.8hz,3h),3.93(d,j=1.7hz,3h),3.88(d,j=1.7hz,3h).
13
c nmr(101mhz,cdcl3)δ166.9,156.4,155.4,143.4,141.9,133.7,130.5,122.1,97.9,56.8,55.8,52.2,21.1,14.2.hrms(esi):calcd for c
16h18
n3o
4+
[m+h]
+
316.1292,found 316.1298.
[0139]
经上述相同操作得到以下产物:
[0140]
[0141]
红色固体。1h nmr(400mhz,cdcl3)δ7.86(d,j=8.4hz,2h),7.71(d,j=8.4hz,2h),7.39(s,1h),6.35(s,1h),4.60(s,2h),3.96(s,3h),3.89(s,3h).
13
c nmr(101mhz,cdcl3)δ156.0,155.9,144.1,141.9,133.8,133.1,122.8,119.2,111.4,97.8,97.5,56.7,55.9.hrms(esi):calcd for c
15h15
n4o
2+
[m+h]
+
283.1190,found 283.1209.
[0142][0143]
紫色固体。1h nmr(400mhz,cdcl3)δ7.81(d,j=8.5hz,2h),7.71(d,j=8.5hz,2h),7.43(s,1h),4.19(t,j=6.4hz,2h),3.29(t,j=5.8hz,4h),2.81(t,j=6.4hz,2h),2.73(t,j=6.3hz,2h),2.40(t,j=7.2hz,2h),2.00-1.90(m,4h),1.85(p,j=6.6hz,2h),1.68-1.77(m,2h),1.65-1.54(m,2h).
13
c nmr(101mhz,cdcl3)δ179.4,156.8,156.1,148.2 135.7,133.1,122.4,119.3 117.5,114.8,112.8,110.7,75.9,50.2,49.9,34.0,27.5,25.8,24.6,21.6,21.3,21.1,21.0.hrms(esi):calcd for c
25h28
n4o
3-[m]-432.2167,found 432.2181.
[0144][0145]
称取产物e1.1(0.15mmol)溶于2ml无水乙醇中,于冰浴条件下加入10wt%的亚硝酰硫酸(0.3mmol),冰浴30min后,用醋酸钠调节ph至5.0后,加入n-乙基n-羟乙基苯胺(0.3mmol)乙醇溶液,继续反应3h。反应完毕后,加入20ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到紫色固体产物e2.1(收率60%)。h nmr(400mhz,chloroform-d)δ7.89(d,j=8.1hz,2h),7.62(d,j=8.0hz,2h),7.54
–
7.43(m,3h),4.43(s,1h),4.23(t,j=6.4hz,2h),4.04(d,j=4.1hz,6h),3.28(t,j=5.7hz,4h),2.38(t,j=7.3hz,2h),1.96(s,4h),1.89(t,j=7.3hz,2h),1.72(t,j=7.6hz,2h),1.59(d,j=8.4hz,2h).hrms(esi):calcd for c
26h30
n5o
5+
[m+h]
+
492.2242,found 492.2248.
[0146]
经上述相同操作得到以下产物:
[0147][0148]1h nmr(400mhz,chloroform-d)δ8.37(d,j=8.5hz,2h),8.04(d,j=8.3hz,2h),7.93(d,j=8.7hz,2h),7.48(d,j=16.1hz,2h),6.79(d,j=8.8hz,2h),4.33(s,2h),4.07(d,j=18.5hz,6h),3.69(d,j=8.8hz,4h),1.25(s,3h).hrms(esi):calcd forc
24h27
n6o
5+
[m+h]
+
479.2037,found 479.2042.
[0149][0150]
称取4,4'-偶氮二苯胺(1mmol)溶于3ml无水乙醇中,于冰浴条件下加入浓硫酸(12mmol)和亚硝酸钾(12mmol)(溶于3ml水中),冰浴30min后,用醋酸钠调节ph至5.0后,加入n-乙基n-羟乙基苯胺(4mmol)的乙醇溶液,继续反应3h。反应完毕后,加入30ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到紫色固体产物e3.1(收率56%)。1h nmr(400mhz,chloroform-d)δ8.06(d,j=8.3hz,4h),7.98(d,j=7.8hz,4h),7.93
–
7.85(m,4h),6.79(d,j=9.0hz,4h),4.33(t,j=6.3hz,4h),3.68(s,4h),3.52(d,j=7.5hz,4h),1.25
–
1.22(m,6h).
13
c nmr(101mhz,dmso)δ172.7,154.5,152.5,151.3,143.4,126.0,124.4,123.4,112.0,62.0,48.7,45.4,12.5.hrms(esi):calcd for c
32h37
n8o
2+
[m+h]
+
565.3034,found 565.3029.
[0151]
制备例6:f组化合物的合成(核心基团为多烯类)
[0152][0153]
化合物8(26.3mmol)溶于50ml无水乙醚,冰浴中逐滴加入三溴化磷(18mmol)。滴加完毕后,反应物搅拌过夜。反应物倒入冰水中,并用碳酸钠中和。产物用乙醚萃取,有机相用水与饱和食盐水洗涤,硫酸钠干燥。过滤后减压蒸馏得到化合物9(收率84%)。1h nmr(400mhz,cdcl3)δ6.35-6.25(m,2h),6.0-5.85(m,2h),4.12(d,j=7.0hz,4h).
[0154]
化合物9(9.0mmol)溶于30ml甲苯,加入亚磷酸三乙酯(20mmol),室温下搅拌12h。减压蒸馏除去溶剂和过量的原料,得到淡黄色产物10(收率80%)。1h nmr(400mhz,cdcl3)6.25-6.15(m,2h),5.76-5.66(m,2h),4.20-4.05(m,8h),2.68(dd,j=9.6hz,4.0hz,4h),1.32(t,j=7.0hz,12h).
[0155][0156]
称取4-氨基苯甲醛(1mmol)溶于3ml无水乙醇中,于冰浴条件下加入浓硫酸(12mmol)和亚硝酸钾(12mmol)(溶于3ml水中),冰浴30min后,用醋酸钠调节ph至5.0后,加入n-乙基n-羟乙基苯胺(3mmol)乙醇溶液,继续反应3h。反应完毕后,加入30ml乙酸乙酯,先后用蒸馏水和饱和食盐水洗涤,无水硫酸镁干燥,减压蒸馏掉溶剂,经柱层析后得到紫色固
体产物11,化合物10(0.22mmol)与11(0.1mmol)混合溶解于5ml的乙二醇二甲醚中,加入叔丁醇钾(0.3mmol)的乙二醇二甲醚溶液。加热反应,温度控制在回流温度以下,12小时以后,加入水,产物从水中析出,经柱层析分离得到红色固体产物f2.1(收率53%)。1h nmr(400mhz,dmso-d6)δ7.76(t,j=8.0hz,8h),7.47(d,j=8.1hz,4h),6.83(d,j=8.9hz,4h),5.32(t,j=5.7hz,2h),4.84(t,j=5.3hz,2h),4.58(d,j=4.5hz,4h),3.63
–
3.58(m,4h),3.54
–
3.45(m,8h),1.15(t,j=6.9hz,6h).
13
c nmr(101mhz,dmso-d6)151.9,150.9,144.6,142.6,127.5,125.4,122.1,111.6,63.0,58.8,52.6,45.5,12.5.
[0157]
测试例1:化合物拉曼光谱性质的表征
[0158]
采用共聚焦拉曼光谱仪测试表1中所示化合物的拉曼位移和相对拉曼强度。各化合物均在dmf溶液中进行测试,以dmf的867cm-1
的峰为内标,调整化合物的浓度使得被测峰的峰强与dmf 867cm-1
的峰的比例为0.1-10之间。激发光波长为532nm,除测试罗丹明b和6g的激光强度为0.025mw,积分时间为10次、每次1秒之外,测试其余化合物的激光强度为5mw,积分时间为10次、每次1秒。所得结果列于表1中。
[0159]
表1代表化合物的拉曼位移和拉曼强度
[0160][0161][0162]
从表1的结果可知,本发明提供的化合物的拉曼信号强度与edu相比,有不同程度的增加,强度最高的信号分子达到了edu信号的104倍左右,例如b3.2、c3.1、c3.2、e3.1。
[0163]
进一步地,图1中示出了化合物c3.1在dmf中的拉曼光谱图(浓度10-4
m,532nm激发)。图2示出了有代表特征的化合物e3.1的紫外可见吸收、荧光以及拉曼光谱的性质。可以看到化合物e3.1的最大吸收波长在532nm,较好的契合了商用拉曼光谱仪的常用激发光波
长。在532nm的光源激发下,该化合物荧光信号背景极低。其10-4
m的溶液在532nm和785nm激发光下激发的拉曼信号证明了1120cm-1
处的拉曼信号在电子共振的情况下信号大幅增强。该化合物的拉曼信号在1.0
–
500μm浓度下,以dmf溶剂峰为内标,具有良好的线性关系,显示了其可用于定量分析的能力。其中,图2a为5μm的化合物e3.1的紫外可见吸收光谱;图2b为100μm的化合物e3.1使用532nm和785nm激光激发并与溶剂dmf相比较的拉曼光谱;图2c为化合物e3.1对于dmf溶剂峰的相对拉曼强度与浓度之间的线性关系图。
[0164]
测试例2:将该类拉曼分子作为信号标记用于免疫检测
[0165]
免疫分析技术是定量检测生化指标如蛋白质最为有效的工具之一。免疫分析的精确性和可操作性促进了对疾病诊断、食品安全和环境保护等问题的深入研究。通常免疫检测需要非常灵敏的信号源,可以将得到的共振拉曼化合物作为信号发射基团,连接到抗体上,采用三明治夹心免疫检测的方法,检验其在免疫检测方面的效果。
[0166]
首先将带化合物e3.1标记到羊抗兔igg上。称取1mg(0.0013mmol)合成的化合物e3.1、1mg(0.0087mmol)nhs和1mg(0.0052mmol)edc溶于200μl dmf,加入20μl mes缓冲溶液,于室温下活化30min。取1mg羊抗兔igg溶于200μl pbs中,震荡摇匀后取20μl于0.5ml离心管中,再加入20μl nahco3溶液,将a中活化完毕后的溶液用dmf稀释10倍,并移取4μl,室温下反应5h。反应完毕后,将所得溶液转移至透析袋中,加入500μl nahco3溶液,透析3次,透析总时长24h以上(透析液为50mmol/l nahco3溶液)。透析完毕后,将袋中溶液转移至离心管中定容至1ml,即得到浓度为100μg/ml的带拉曼标记的羊抗兔igg。
[0167]
以聚苯乙烯微球为载体,其直径为13μm。微球分散在溶液中,浓度为0.025g/ml,微球上氨基浓度为0.1mmol/g。具体操作步骤如下:(i)用力震荡含聚苯乙烯微球的溶液,使其均匀分散在该溶液中,用移液枪移取100μl(氨基含量为2.5
×
10-4
mmol,1eq)该液体置于1ml离心管中,加入0.5ml二氯甲烷,17μl三乙胺和1.2mg dmap,置于摇床上室温震荡反应3h。反应完毕后离心3min(5000r/min),除去上层清液。(ii)称取11.2mg(0.1mmol)nhs和38.4mg(0.2mmol)edc,分别溶于1ml mes中,用移液枪移取100μl nhs溶液和200μl edc于(i)中所得聚苯乙烯微球,于摇床上室温震荡活化30min,活化完毕后离心3min(5000r/min),除去上层清液,再用mes洗涤三次。随后加入300μl pbs,移取30μl羊抗兔igg,于4℃下震荡反应3h。反应完毕后离心3min(5000r/min),除去上层清液。再用pbst清洗3次。(iii)称取50mg脱脂奶粉溶于1ml pbs中,移取500μl该溶液与(ii)中聚苯乙烯微球混合,于37℃下震荡封闭2h。封闭完毕后离心3min(5000r/min),除去上层清液。再用pbst清洗3次。(iv)配制一系列浓度梯度的兔igg,使其浓度分别为0、10-7
、10-8
、10-9
、10-10
、10-11
、10-12
、10-13
mol/l,将(iii)中所得聚苯乙烯微球定容至100μl,分别取5μl聚苯乙烯微球溶液于不同浓度的兔igg溶液中,并置于摇床上室温震荡反应3h。反应完毕后离心3min(5000r/min),除去上层清液,再用pbst清洗3次。(v)加入300μl合成的带拉曼信号的羊抗兔igg,浓度为10μg/ml。置于摇床上于室温下震荡反应3h。反应完毕后离心3min(5000r/min),除去上层清液,再用pbst清洗3次,最后用清水清洗一次。(vi)将(v)中所得的聚苯乙烯微球置于激光共聚焦拉曼显微镜下测试其拉曼光谱,激发波长为532nm,激光强度5mw,积分时间2s,每个样品测10个微球。
[0168]
用合成的化合物e3.1作为检出信号,来进行兔igg的定量检测,经免疫检测完毕后,测得的聚苯乙烯微球的光谱图如图3所示,其中,图3a为免疫检测后聚苯乙烯微球在激发波长为532nm,从上到下兔igg浓度分别为10-10
、10-11
、10-12
和10-13
m的拉曼光谱,所有谱图
以聚苯乙烯微球在1000cm-1
下的强度进行归一化。
[0169]
由于每个聚苯乙烯微球表面氨基的浓度具有差异性,为了确保结果的准确度,以聚苯乙烯微球在1000cm-1
下的特征峰作为参照,用化合物e3.1在1122cm-1
下的信号强度除以聚苯乙烯微球在1001cm-1
下的信号强度,作为相对拉曼信号强度),再以该值为纵坐标,以兔igg的浓度为横坐标,得到的结果如下图3所示。其中,图3b为兔igg浓度为10-13
~10-6
m的相对拉曼强度。3c为兔igg浓度为10-13
~10-10
m时相对强度与浓度之间的线性拟合曲线。
[0170]
由图3可知,本发明的化合物e3.1,其共振拉曼信号用于免疫检测时,利用线性曲线可以实现对浓度范围为10-10
~10-13
m的兔igg的定量检测。此外,当兔igg的浓度为10-13
m时,换算成质量浓度约为1.5
×
10-8
mg/ml,达到了痕量分析(待测组分含量低于百万分之一)的水平。由此可见,本发明合成的化合物e3.1信号非常灵敏,可以作为免疫检测的信号源。
[0171]
测试例3:将该类拉曼分子作为信号标记用于细胞成像
[0172]
测试例3.1:靶向细胞器的拉曼探针的合成
[0173]
线粒体(mitochondria)是具有双膜结构的细胞器,它是细胞呼吸并产生能量的主要场所,除此之外,它还参与钙循环、蛋白质的合成、细胞凋亡等生命代谢过程。但是这些历程会产生大量的活性氧(reactive oxygen species,ros),这类物质会造成线粒体功能的损伤。因此,合成一些mito的探针对监测线粒体功能、研究线粒体相关疾病起着重要作用。在线粒体呼吸过程中,线粒体膜内的质子泵将质子输送到线粒体膜间隙,导致跨膜负电位的形成,因此,一些带正电荷的阳离子化合物能够特异性的定位到线粒体上。这类定位基团包括三苯基膦(triphenylphosphonium,tpp)、带正电荷的吡啶和喹啉衍生物、罗丹明等。这里,我们以三苯基膦阳离子作为线粒体定位基团进行细胞成像分子的合成,具体合成步骤如下:
[0174][0175]
称取三苯基膦(3.82mmol)和3-溴丙胺盐酸盐(3.82mmol)溶于其中,加入5ml乙腈,加热回流反应16h。反应完毕后,冷却至室温,加入大量正己烷析出产物,过滤得到粗产物,再将其溶于异丙醇中,用石油醚重结晶获得产物12(收率92%)。1h nmr(400mhz,dmso-d6)δ7.96-7.75(m,15h),3.99-3.85(m,2h),3.12-3.05(m,2h),1.99-1.84(m,2h).
13
c nmr(101mhz,dmso-d6)δ135.6,134.1,130.9,118.5,39.3,20.6,18.9.hrms(esi):calcd for c
21h23
np
+
[m-br]
+
320.3872,found 320.3862.
[0176]
称取a2.2(0.05mmol)溶于5mldcm中,加入丁二酸酐(0.10mmol),dmap(0.01mmol),三乙胺(0.1mmol),回流反应3h。反应完毕减压蒸馏除去溶剂,产物用乙酸乙酯溶解,水与饱和食盐水洗涤,硫酸钠干燥,抽滤后旋干溶剂得到橙色固体中间产物。将此中间产物溶解于2ml的dmf中,加入hatu(0.2mmol)和diea(0.5mmol),于室温下活化10min,然后加入0.2mmol
产物12,继续反应12h。反应完毕加入10ml二氯甲烷,先后用蒸馏水和饱和食盐水洗涤三次,除去dmf,然后经氯仿/乙醚混合溶剂重结晶得到红色固体产物mito-a(收率40%)。1h nmr(400mhz,cdcl3)δ8.01(d,j=8.4hz,2h),7.81-7.74(m,7h),7.68-7.58(m,17h),6.71(d,j=9.2hz,2h),4.14(t,j=6.8hz,2h),3.93(s,3h),3.62(t,j=6.8hz,2h),3.51-3.45(m,2h),3.28-3.21(m,2h),2.66-2.64(m,2h),2.59-2.56(m,2h),2.03-1.90(m,4h),1.14(t,j=6.8hz,3h).
13
c nmr(101mhz,cdcl3)δ172.9,172.7,166.3,153.2,150.4,143.6,135.2,135.2,133.4,133.3,133.2,1324,130.6,130.5,130.3,129.5,125.6,122.3,118.4,117.5,111.3,83.2,77.2,76.8,75.1,61.2,52.3,48.6,45.7,30.4,29.7,29.3,22.4,19.4,12.3.hrms(esi):calcd for c
53h50
n4o5p
+
[m-br-]
+
853.3519,found 853.3512.
[0177]
重复上述步骤可得到如下所示的多个特征拉曼位移的线粒体拉曼染料。
[0178][0179]
红色固体。1h nmr(400mhz,cdcl3)δ8.84(s,2h),7.79(d,j=5.1hz,8h),7.75-7.55(m,34h),6.70(d,j=8.8hz,4h),4.14(t,j=6.0hz,4h),3.85-3.75(m,4h),3.60-3.50(m,4h),3.45-3.55(m,8h),2.40-2.68(m,8h),1.86(m,4h),1.21(t,j=6.0hz,6h).
13
c nmr(101mhz,cdcl3)δ173.0,172.7,143.7,135.0,135.0,133.8,133.4,133.3,131.6,130.5,130.4,125.6,125.0,122.3,119.7,118.6,117.7,111.3,110.1,61.1,48.7,45.7,42.2,30.5,29.4,23.4,22.6,12.3.hrms(esi):calcd for c
88h86
n8o6p
2+
[m-2br-]
2+
706.3073,found 706.3052.
[0180][0181]
紫色固体。1h nmr(400mhz,cdcl3)δ8.28(d,j=8.8hz,2h),7.83(d,j=8.8hz,2h),7.80-7.63(m,15h),4.20(t,j=6.5hz,2h),3.65-3.61(m,2h),3.48-3.45(m,2h),3.30(t,j=5.6hz,4h),2.82(t,j=6.4hz,2h),2.73(t,j=6.4hz,2h),2.41(t,j=7.4hz,2h),2.00-1.90(m,4h),1.90-1.68(m,6h),1.65-1.55(m,2h).
13
c nmr(101mhz,cdcl3)δ174.6,157.5,146.4,136.1,135.1,135.1,133.5,133.4,130.6,130.5,124.8,122.2,118.6,117.7,117.5,114.8,113.0,76.3,50.2,50.0,36.2,30.4,29.8,29.3,27.5,27.2,25.9,25.7,25.5,21.6,21.2,20.9.hrms(esi):calcd for c
45h49
n5o4p
+
[m-br-]
+
754.3522,found 754.3513.
[0182][0183]
紫色固体。1h nmr(400mhz,cdcl3)δ7.87-7.74(m,5h),7.69-7.39(m,15h),4.15(t,j=6.4hz,2h),3.42-3.38(m,2h),3.27(t,j=5.8hz,4h),2.78(t,j=6.4hz,2h),2.70(t,j=6.3hz,2h),2.34(t,j=7.2hz,2h),2.15-1.98(m,4h),2.00-1.90(m,4h),1.83(p,j=
6.6hz,2h),1.77-1.68(m,2h),1.65-1.54(m,2h).
13
c nmr(101mhz,cdcl3)δ174.6,156.9,156.1,148.2,135.7,135.3,133.3,133.2,133.0,130.7,130.5,122.4,119.3,118.2,117.4,117.4,114.6,113.0,110.5,70.8,50.2,49.9,39.1,39.0,36.1,30.3,27.5,25.9,25.6,22.5,22.4,21.6,21.2,20.9,20.3,19.8.hrms(esi):calcd for c
46h49
n5o2p
+
[m-br-]
+
734.3624,found 734.3618.
[0184][0185]
紫色固体。1h nmr(400mhz,cdcl3)δ8.17(d,j=8.5hz,2h),7.96(d,j=8.5hz,2h),7.84(d,j=9.2hz,2h),7.68-7.60(m,15h),7.48(s,1h),7.44(s,1h),6.71(d,j=9.2hz,2h),4.14-4.12(m,2h),4.11(s,3h),4.01(s,3h),3.95(s,3h),3.64-3.59(m,2h),3.53-3.39(m,4h),3.30-3.18(m,2h),2.67-2.60(m,2h),2.59-2.53(m,2h),1.90-1.80(m,2h),1.20(t,j=6.8hz,3h).
13
c nmr(101mhz,cdcl3)δ172.9,172.7,166.6,155.7,153.0,151.7,150.9,150.6,146.0,144.4,142.3,135.3,135.2,133.31,133.30,133.22,133.20,131.5,130.62,130.58,130.5,126.3,126.1,122.8,118.4,117.5,111.3,110.2,101.0,100.3,61.2,56.8,56.7,52.3,48.6,45.8,30.4,29.3,22.4,22.4,19.4,12.3.hrms(esi)calcd for c
51h54
n6o7p
+
[m-br-]
+
893.3792,found 893.3789.
[0186][0187]
深紫色固体。1h nmr(400mhz,cdcl3)δ8.99(s,2h),8.03(d,j=8.7hz,4h),7.94(d,j=8.7hz,4h),7.83(d,j=9.0hz,4h),7.79-7.52(m,30h),6.74(d,j=9.0hz,4h),4.18(t,j=6.4hz,4h),3.80-3.70(m,4h),3.62(t,j=6.3hz,4h),3.51-3.47(m,8h),2.75(t,j=6.3hz,4h),2.65(t,j=6.3hz,4h),2.02-1.97(m,4h),1.20(t,j=6.4hz,6h).
13
c nmr(101mhz,cdcl3)δ173.1,172.7,154.6,152.8,150.4,143.8,135.0,135.0,133.5,133.4,130.5,130.4,125.7,123.9,123.0,118.7,117.9,111.3,61.1,48.8,45.8,30.6,29.6,23.5,22.5,20.2,12.3.hrms(esi):calcd for c
82h86n10
o6p
22+
[m-2br-]
2+
684.3104,found 684.3077.
[0188]
另外,我们改变细胞器的识别基团,合成了具有溶酶体靶向的小分子拉曼探针lyso-d。
[0189][0190]
深红色固体。1h nmr(400mhz,cdcl3)δ8.28(d,j=8.5hz,2h),7.83(d,j=8.5hz,2h),7.44(s,1h),6.32(s,1h),4.19(t,j=6.5hz,2h),3.34(dd,j=11.4,5.3hz,2h),3.30-3.25(m,4h),2.80(t,j=6.0hz,2h),2.72(t,j=6.0hz,2h),2.46(t,j=6.0hz,2h),2.26(s,6h),2.21(t,j=7.2hz,2h),1.99-1.90(m,4h),1.88-1.79(m,2h),1.76-1.67(m,2h),
1.59-1.51(m,2h).
13
c nmr(101mhz,cdcl3)δ173.1,157.5,157.1,148.5,146.5,136.0,124.7,122.2,117.6,114.9,112.8,76.1,57.9,50.2,49.9,44.9,36.5,36.4,30.3,27.4,25.9,25.6,21.6,21.2,20.9;hrms(esi)calcd for c
28h39
n6o
4+
[m+h]
+
523.3027,found 523.3031.
[0191]
测试例3.2:细胞共定位成像:
[0192]
经过细胞毒性测试发现我们合成的各类不同拉曼位移的细胞成像分子对细胞的毒性很小,因此我们将它们应用于接下来的细胞器成像实验当中。我们选取mito-e3.1和lyso-d为例分别对hela细胞的线粒体和溶酶体进行成像,结合目前成熟的商业化荧光染料mitotracker red和lysotracker red得到了如下图4所示的细胞成像图:从左到右分别是拉曼成像效果图(左)、荧光效果成像效果图(中)和二者叠加效果图(右)。其中图4a拉曼成像使用1120cm-1
处信号成像,激发波长532nm,5μmazo-3-mito染色1h;荧光成像使用610nm波长信号,200nm mitotracker染色1h。图4b拉曼成像使用1340cm-1
处信号成像,激发波长532nm,5μm lyso-d染色1h;荧光成像600nm波长信号,100nmlysotracker red染色1h。
[0193]
测试例3.3:细胞多色成像
[0194]
将我们合成的具有线粒体靶向的拉曼染料mito-a,mito-b,mito-d,mito-e1,mito-e2,mito e3对hela细胞进行染色,得到以下的成像图,说明此类偶氮增强的多色拉曼信号分子强度强,谱线狭窄且特征,可以很好的用于细胞成像。
[0195]
图5为使用超强拉曼分子的多色拉曼成像图,其中,图5a为分子结构mito-e2,mito-e3,mito-e1,mito-d,mito-b and mito-a;图5b为使用以上拉曼染料对海拉细胞线粒体进行染色后得到的拉曼成像图,孵育时间2h,激光强度5mw,每个像素的成像时间为30ms。图5c为细胞的累计拉曼成像光谱图,图5d为拉曼染料的毒性实验,染色浓度5μm孵育时间2h.
[0196]
通过本发明的结果可以看出,采用本发明的中的共轭连接的偶氮基团对于多种官能团的内源性拉曼信号具有明显的增强效果,将此类信号分子用于拉曼成像时具有信号强,成像时间短,光漂白与淬灭效应弱等优异效果。
[0197]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1