一种帕布昔利布中间体的制备方法与流程
1.本发明涉及一种帕布昔利布中间体的制备方法,属于药物合成领域。
背景技术:
2.帕布昔利布(palbociclib)自2015年获得美国fda批准后,相继与2016年和2017年获得欧盟ema和日本pdma认可,帕布昔利布胶囊获批当年销售额为7.32亿美元,2016年销售额为21.35亿美元,2017年销售额为31.26亿美元,预计2020年销售额可达50亿美元以上标志着该药已经得到患者的认可,成为重磅炸弹级新药。帕布昔利布(palbociclib),化学名为2
‑
[(4
‑
哌啶基)苄基]
‑6‑
乙酰基
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,由辉瑞公司研制开发,2015年2月在美国率先批准上市,是一种周期蛋白
‑
依赖激酶(cdk)4和6的抑制剂,主要通过调节细胞周期、抑制(cdk)4和6活性来阻止细胞有g1期到s期进而抑制dna的合成;临床主要用于治疗晚期乳腺癌患者。
[0003]
式(1)化合物6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮是合成帕布昔利布的关键中间体,英文名称为6
‑
bromo
‑2‑
chloro
‑8‑
cyclopentyl
‑5‑
methylpyrido[2,3
‑
d]py
‑
rimidin
‑
7(8h)
‑
one,cas号为1016636
‑
76
‑
2,结构式为:
[0004][0005]
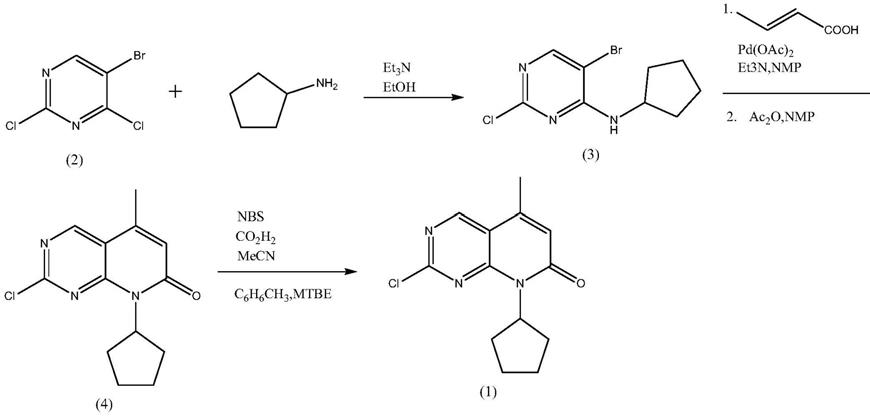
文献报道的6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的合成过程中,b
·
p
·
柴卡尔等人在原研(辉瑞)专利cn105008357a中报道了首先以5
‑
溴
‑
2,4
‑
二氯嘧啶为起始原料,在缚酸剂三乙胺的作用下,以乙醇做溶剂,与环戊胺反应得到5
‑
溴
‑2‑
氯
‑
n
‑
环戊胺嘧啶
‑
4胺(式(3)化合物,式(3)化合物在n
‑
甲基吡咯烷酮中,以0.03当量的醋酸钯为催化剂,三乙胺为缚酸剂,与巴豆酸进行heck反应,再在乙酸酐的脱水作用下环合得到2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮(式(4)化合物),式(4)化合物用nbs溴化,通过甲苯、mtbe混合溶剂精制得到式(1)化合物。反应式如下:
[0006][0007]
该合成工艺采用5
‑
溴
‑
2,4
‑
二氯嘧啶为起始原料,廉价易得,但制备式(3)化合物采用乙醇做溶剂,三乙胺做缚酸剂,经加水分离产品后所得的废水因含大量的三乙胺盐酸盐及乙醇,污水量大,成分复杂,不利于污水的处理,给产品的工业化带来一定的困难;制备式(4)化合物,采用0.03当量的醋酸钯,由于醋酸钯价格较高,且不利于回收利用,因此,占式(1)化合物成本比例偏高,约占总成本的60~70%,不利于终产物帕布昔利布的产品普及。
[0008]
wo2016030439a1报道了以二异丙基乙胺为缚酸剂,以双(氰基苯)二氯化钯为催化剂,以三(邻甲基苯基)磷为配体,由式(3)制备式(4)的heck反应路线,但钯催化剂的量仍达到0.02当量。
[0009]
随着中国政府和医药化工企业对环境保护意识的逐年增强,开发一种低成本、高收率、环境友好的6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的合成方法就成为一种重要的课题。
技术实现要素:
[0010]
本发明克服了上述现有技术的不足,提供一种帕布昔利布中间体的制备方法,利用2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮为原料,通过草酸和醋酸酐催化通过nbs溴化,降温析晶离心可以获得6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,同时对制备过程中产生的废液进行充分再利用减少三废产生,提供了一条高质量、低成本、对环境友好、适合工业化生产的制备方法。
[0011]
一种帕布昔利布中间体的制备方法,包括如下步骤:
[0012]
1)将100
‑
110g的2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮添加到在1000
‑
1100ml的乙腈中,再加入少量草酸和醋酸酐;
[0013]
2)在步骤1)的反应体系加入nbs,nbs的添加量80
‑
90g,升温到50
‑
60℃,反应10
‑
12h;
[0014]
3)反应完成后,降温到0
‑
10℃,保温3
‑
4h,过滤获得反应母液和滤渣a;
[0015]
4)在步骤3)获得的滤渣a中加入乙腈进行洗涤,烘干后即可获得6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮。
[0016]
进一步的,上述制备方法中步骤1)所述的草酸的添加量和2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮添加量的质量比为1:0.01~1:0.05。
[0017]
进一步的,所述醋酸酐添加量为为反应物2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮质量比为0.2~0.5倍。
[0018]
进一步的,上述制备方法中步骤4)中所述的乙腈的使用量为90
‑
100ml。
[0019]
进一步的,上述制备方法中的步骤3)中所述的反应母液的处理方法,包括如下步骤:
[0020]
s1将反应母液常压蒸馏至近干,获得浓缩液体;
[0021]
s2在步骤s1获得的浓缩液体中加入无机碱,常温搅拌1.8
‑
2h,过滤,获得无色滤液和滤渣b;
[0022]
s3将s2获得的无色滤液常压蒸馏得即可获得乙腈。
[0023]
进一步的,上述步骤s2中所述的无机碱为氢氧化钠、氢氧化钾、氧化钙、碳酸钠、碳酸钾等无机碱中的一种,其添加量与步骤s1获得的浓缩溶液的质量比为(2
‑
6):100。
[0024]
进一步的,上述步骤s2中所述的滤渣b的的处理方法,包括如下步骤:在滤渣b中加入水溶解,用氨水调ph值至8,减压蒸馏除水,获得得粘稠状固体,所述粘稠状固体为丁二酰亚胺。
[0025]
进一步的,上述溶解滤渣b的水的使用量为180
‑
200ml。
[0026]
有益效果:
[0027]
(1)本专利通过加入醋酸酐,有效避免了对产物收率影响较高的如下的式(2)杂质的产生,提高6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮制备步骤的反应收率,收率97%左右。
[0028][0029]
(2)对反应废液进行蒸馏,馏出液经碱处理,二次蒸馏得到乙腈,该乙腈满足制备6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的标准,可以再利用进行6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的制备,实现了回收利用,减少了三废的排放。
[0030]
(3)将残渣进行处理,同时回收副产物丁二酰亚胺,进一步减少了固废的排放,使该步骤三废的排放大大降低,对环境更加友好。
具体实施方式
[0031]
为了使本技术领域人员更好地理解本申请中的技术方案,下面结合实施例对本发明作进一步说明,所描述的实施例仅是本申请一部分实施例,而不是全部,本发明不受下述实施例的限制。
[0032]
实施例1 6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的制备
[0033]
向2000毫升反应瓶中依次加入110克(0.42mol)2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,1100毫升乙腈,2.7克草酸,55g醋酸酐及89.7克(0.50mol)nbs,升温至60℃反应12小时,反应毕,降温至0~10℃,保温4h,过滤,100ml乙腈洗涤,烘干得白色固体140克,收率98.1%,hplc纯度98.5%。
[0034]
将反应母液常压蒸馏至近干,的紫色液体1150ml,加入50g氢氧化钠固体,常温搅拌2h,过滤,得无色滤液,常压蒸馏得1105ml乙腈,该乙腈的gc检测99.9%,水分(卡氏法)0.2%。
[0035]
剩余物加200ml水溶解,用氨水调ph值至8,减压蒸馏除水,得粘稠状固体。高真空蒸馏(5mmhg),收集170~20℃馏分,得36g固体,收率72.1%,为丁二酰亚胺,纯度99.7%。
[0036]
实例2:6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的制备
[0037]
向2000毫升反应瓶中依次加入110克(0.42mol)2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,1100毫升回收乙腈,2.7克草酸,55g醋酸酐及89.7克(0.50mol)nbs,升温至60℃反应12小时,反应毕,降温至0~10℃,保温4h,过滤,100ml乙腈洗涤,烘干得白色固体139克,收率97.2%,hplc纯度98.6%。
[0038]
将反应母液常压蒸馏至近干,的紫色液体1130ml,加入50g氢氧化钠固体,常温搅拌2h,过滤,得无色滤液,常压蒸馏得1110ml乙腈,gc检测99.9%,水分(卡氏法)0.3%。
[0039]
剩余物加200ml水溶解,用氨水调ph值至8,减压蒸馏除水,得粘稠状固体。高真空蒸馏(5mmhg),收集170~20℃馏分,得39g固体,收率78.2%,为丁二酰亚胺,纯度99.7%。
[0040]
实例3:6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的制备
[0041]
向2000升反应釜中依次加入870千克乙腈,110千克2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,2.7千克草酸,55g醋酸酐及89.7千克nbs,升温至60℃反应12小时,反应毕,降温至0~10℃,保温4h,离心,80kg乙腈洗涤,,烘干得白色固体138.4千克,收率96.9%,hplc纯度98.4%。
[0042]
将反应母液转移至回收釜,常压蒸馏,共计得紫红色液体920kg,将45kg氢氧化钠加入回收乙腈中,常温搅拌4h,压滤,滤液压至二次蒸馏釜,滤渣经水洗涤,排入污水站。经二次常压蒸馏,得到885kg回收乙腈,纯度99.9%(gc),水分0.3%。
[0043]
向母液蒸馏残渣中,加入200kg纯化水,加入氨水调ph值至8,将料液转移至高真空蒸馏釜,常压蒸除水,再减压蒸馏(≤1000pa),收集170~200℃馏分,得42kg无色片状晶体,收率84.2%。
[0044]
实例4:6
‑
溴
‑2‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮的制备
[0045]
向2000升反应釜中依次加入870千克回收乙腈,110千克2
‑
氯
‑8‑
环戊基
‑5‑
甲基吡啶并[2,3
‑
d]嘧啶
‑
7(8h)
‑
酮,2.7千克草酸,55g醋酸酐及89.7千克nbs,升温至60℃反应12小时,反应毕,降温至0~10℃,保温4h,离心,80kg乙腈洗涤,,烘干得白色固体139.5千克,收率97.6%,hplc纯度98.5%。
[0046]
将反应母液转移至回收釜,常压蒸馏,共计得紫红色液体922kg,将45kg氢氧化钠加入回收乙腈中,常温搅拌4h,压滤,滤液压至二次蒸馏釜,滤渣经水洗涤,排入污水站。经二次常压蒸馏,得到883kg回收乙腈,纯度99.9%(gc),水分0.3%。
[0047]
向母液蒸馏残渣中,加入200kg纯化水,加入氨水调ph值至8,将料液转移至高真空蒸馏釜,常压蒸除水,再减压蒸馏(≤1000pa),收集170~200℃馏分,得43kg无色片状晶体,
收率86.2%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1