结合5
结合5
′‑
末端缀合物和中性/阳离子混合脂材包载的小干扰rna及其修饰方法
技术领域
1.本发明涉及一种结合5
′‑
末端缀合物和中性脂材/阳离子混合脂材包载修饰的小干扰rna(sirna)及其化学修饰方法和载体递送策略。本发明实现了对sirna反义链的5
′‑
末端缀合,缀合物的存在不影响sirna的基因沉默活性。经过本发明方法缀合修饰并包载后的sirna,具有稳定性好、递送效率高效、体内靶器官分布高、入胞能力强、毒性低且生物活性好等有点,能够广泛应用于抗肿瘤、抗病毒及代谢类疾病的药物研究中。本发明属于生物医药技术领域。
背景技术:
2.小干扰rna(sirna)是一类具有巨大临床前景的药物候选分子,它的靶标是细胞内的mrna,可以在基因水平上直接沉默靶标基因的表达,从根本上预防疾病的发生和发展。天然的sirna由dsrna在胞内被dicer剪切至21
‑
25mer,通常会有5
′‑
磷酸基和3
′‑
羟基。合成的sirna的5
′‑
羟基会在胞内被clp1细胞激酶迅速磷酸化,这也是sirna发挥活性的必要条件,因为sirna只有在5
′‑
磷酸化后,才可以进入risc发挥其基因沉默活性。与传统的小分子药物相比,sirna具有更高的特异性,更低的毒性和副作用,易于制备等优点。但是由于其血清稳定性差、跨膜困难、易脱靶、激发免疫应答和其他缺陷,使临床应用受到限制。
3.2018年,fda(美国食品药品监督管理局)批准了第一例sirna药物(patisiran),它是hattr(遗传性运甲状腺素蛋白淀粉样变性)的候选疗法,使用由dlin
‑
mc3
‑
dma、dspc、peg
‑
dmg和胆固醇组成的脂质体进行递送。体外实验表明在hepg2细胞中,10nm sirna可敲低95%ttr mrna表达,在啮齿动物模型中的ed
50
为0.03mg/kg。这一sirna药物的安全性优于hattr的反义核酸药物inotersen2019年,fda批准了第二例sirna药物givlaari
tm
(givosiran),它是除了静脉注射血红素以外唯一有效的急性肝卟啉症的治疗方法,且血红素极难获得。givosiran使用了galnac缀合进行递送,在啮齿动物模型中的有效剂量是1
‑
3mg/kg。随着基因组学的快速发展,rnai(rna干扰)药物的成功表明个体化治疗新时代的开始,但遗憾的是目前的上市药物使用靶向肝脏脱唾液酸糖蛋白受体的lnp递送体系和galnac缀合物而仅能进行肝脏递送。而且鉴于目前正处于临床iii期研究的sirna均未使用载体包载,依赖电荷作用的阳离子脂材的应用已经陷入了困境。
4.阳离子脂材依靠其与sirna的正负电荷间的库仑力作用结合并包载sirna。但阳离子脂质体的毒性较强,且易于在生理条件下带负电的血清蛋白结合,产生免疫原性、肝脏毒性并使sirna易在靶器官外释放。这些缺陷限制了阳离子脂质体在包载sirna成药的进一步应用。
5.本发明人前期设计合成了碱基乙酰胺甘油醚分子dnca(cn108059619a),因其具有碱基性质的头部,可以通过氢键作用和π
‑
π堆积作用结合并包载单链核酸药物和质粒(cn1084478807a)。结合dnca及发明人前期设计合成的以赖氨酸作为头部的阳离子脂材cld共同包载3
′
,3
″‑
双肽缀sirna已在细胞水平成功应用(mol pharm,2019,16,4920)。在此,我
们对包载方法进行优化,重新制定并探索了最优的dnca/cld转染处方,并加入dspe
‑
peg,以优化混合制剂体内应用的性质。结合使用磷酸二酯键作为sirna链末端与缀合基团的链接的缀合策略和化学修饰方法,获得了高效低毒的缀合物/中性/阳离子混合脂材sirna药物包载递送系统。
技术实现要素:
6.为了提高小干扰rna(sirna)体内运送过程中靶器官递送的特异性,入胞效率及胞内释放的有效性,本发明提供了一种结合5
′‑
末端缀合物和中性脂材/阳离子脂材混合载体包载的sirna的化学修饰方法和载体递送策略。本发明通过将缀合基团制备为亚磷酰胺单体,使用固相合成仪实现缀合物的快速合成。本发明利用中性脂材和阳离子混合脂材包载sirna缀合物,以实现更有效且安全无毒的体内sirna递送,从而提高sirna的成药性。
7.为了达到上述目的,本发明采用的技术方案如下:
8.本发明的一种对小干扰rna(sirna)的化学修饰方法,包括在sirna的正义链或/和反义链的5
′‑
末端缀合一个或多个链接基团作为缀合基团,并利用中性脂材和阳离子混合脂材进行包载,其中所述链接基团的结构式如化学式i或化学式ii所示,其中,化学式i所示的结构为一个六碳单元的链接基团,化学式ii所示的结构为一个三碳单元的链接基团;
[0009][0010]
其中,所述的混合脂材由化学式iii所示的中性胞苷脂材、化学式iv所示的阳离子脂材组成,或由化学式iii所示的中性胞苷脂材、化学式iv所示的阳离子脂材、化学式v所示的peg2000
‑
dspe组成:
[0011][0012]
其中,优选的,在sirna的反义链或/和正义链5
′‑
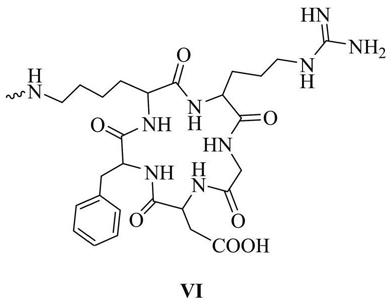
末端缀合一个或多个链接基团的同时,还包括在链接基团的末端活泼酯上通过缩合反应链接靶向基团,所述的靶向基团结构式如化学式vi所示:
[0013][0014]
其中,优选的,在对sirna进行缀合前,将化学式vii、化学式viii所示的链接基团
分别制备成化学式ix、化学式x所示的亚磷酰胺单体,然后通过磷酸二酯键与sirna进行缀合;随后在化学式ix或化学式x所示的亚磷酰胺单体的活泼酯上通过缩合反应连接化学式vi所示的靶向基团;
[0015][0016]
其中,优选的,使用固相合成技术,采用亚磷酰胺法合成天然的及缀合链接基团的sirna正义链及反义链序列,在rna合成仪上,序列从3
′‑
端至5
′‑
端进行合成,每偶联一个核苷为一个循环,在天然核苷亚磷酰胺单体偶联21次后,将一个或几个所述的亚磷酰胺单体ix或x在序列的5
′‑
末端相应位置进行偶联,每个循环包括四个反应:脱dmt、偶联、封闭、氧化。
[0017]
其中,优选的,每个循环亚磷酰胺单体进样后的偶联时间为600秒/次,偶联3次。
[0018]
其中,优选的,按照摩尔百分比计算,所述的混合脂材由72.7%化学式iii所示的中性胞苷脂材、27.3%化学式iv所示的阳离子脂材组成,其中,优选的,总脂质与sirna的摩尔比为10:1。或所述的混合脂材由39.7%的化学式iii所示的中性胞苷脂材、59.6%化学式iv所示的阳离子脂材以及0.7%化学式v所示的peg2000
‑
dspe组成,其中,优选的,总脂质与sirna的摩尔比为5.3:1。
[0019]
其中,优选的,利用中性脂材和阳离子脂材的混合脂材进行包载的方法为:将所述的混合脂材溶于乙醇溶液中,将反义链或/和正义链的5
′‑
末端通过磷酸二酯键缀合一个或多个链接基团的sirna溶于depc水中,使用未缓冲溶液或缓冲溶液混合,超声处理,使用0.22μm滤膜过滤,滤液中包含修饰后的sirna,其中,优选的,所述的未缓冲溶液为生理盐水、水或任何转染优化溶液,所述的缓冲溶液包含乙酸盐、柠檬酸盐、碳酸盐、磷酸盐或其任何组合。
[0020]
其中,所述的化学修饰策略还包括与其它化学修饰策略的共同使用,包括sirna的2
′‑
o
‑
甲基(2
′‑
ome)、2
′‑
氟代(2
′‑
f)、2
′‑
o
‑
甲氧乙基(2
′‑
o
‑
moe)、锁核苷酸(lna),磷硫骨架修饰及其他末端缀合方式等。
[0021]
进一步的,本发明还提出了按照以上任一项所述的化学修饰方法合成的结合5
′‑
末端缀合物和中性/阳离子混合脂材载体包载修饰的小干扰rna。
[0022]
本发明所述的5
′‑
末端缀合方式与3
′‑
末端缀合相比具有以下特点:
[0023]
a.将缀合基团制备成相应的亚磷酰胺单体,使缀合部分可以直接作为单体使用固相合成仪进行合成,合成简单快速;
[0024]
b.链接基团x的末端保留活泼酯,作为活性反应位点,可利用缩合反应高效链接多种官能团,快速扩展缀合物规模;
[0025]
c.链接基团间以磷酸二酯键进行连接,可由细胞内的酶进行水解剪切,释放出带有磷酸末端的未缀合sirna单链,最终达到更易载入risc复合物、提高sirna基因沉默活性的能力。
[0026]
在本发明的具体实施例中,5
‑
末端缀合物修饰后的序列选自以下序列所组成的群
组:
[0027]
(1)在小干扰rna序列的正义链或反义链的5
′‑
末端缀合一个化学式ii所示的链接基团而得到的反义链或正义链;
[0028]
(2)在小干扰rna序列的正义链或反义链的5
′‑
末端依次延3
′‑5′
方向缀合一个或几个化学式i和一个化学式ii所示的链接基团而得到的反义链或正义链;
[0029]
(3)在小干扰rna序列的正义链或反义链的5
′‑
末端缀合一个化学式ii所示的链接基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式x,随后在化学式x所示的基团的活泼酯上通过缩合反应连接化学式vi所示的靶向基团,而得到的正义链或反义链;
[0030]
(4)在小干扰rna序列的反义链的正义链或5
′‑
末端依次延3
′‑5′
方向缀合一个或几个化学式i和一个化学式ii所示的链接基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式ix和x,随后在化学式x所示的基团的活泼酯上通过缩合反应连接化学式vi所示的靶向基团,而得到的反义链或正义链缀合物;
[0031]
上述(1)
‑
(4)中的反义链和正义链为自由组合的形式。
[0032]
本发明所述的sirna及其缀合物是按照需求,将缀合或未缀合修饰的反义链与正义链经退火后形成的rna双链复合物。
[0033]
以rl1as/rl0s
‑
sirna为例,在sirna的反义链(as表示)5
′‑
末端依次(3
′‑5′
方向)通过固相合成缀合一个化学式i(数字表示化学式i所示的链接基团的数量)和一个化学式ii(l表示)所示的链接基团,其中缀合基团间通过磷酸二酯键链接,随后通过缩合反应缀合一个化学式vi所示的靶向基团crgd(r表示),即rl1as。在正义链(s表示)5
′‑
末端通过固相合成缀合一个化学式ii(l表示),随后通过缩合反应缀合一个化学式vi所示的靶向基团crgd(r表示),即rl0s,随后将合成纯化完毕的反义链与正义链按照1:1.1的摩尔比进行退火,形成rl1as/rl0s
‑
sirna,其余缀合的sirna以此类推。
[0034]
在本发明的一个具体实施例中,待修饰的sirna为靶向erk通路中braf
v600e
突变的mrna的simb3序列。simb3修饰前的序列如下:
[0035]
simb3:正义链:5
′‑
gcuacagagaaaucucgaudtdt
‑3′
[0036]
反义链:5
′‑
aucgagauuucucuguagcdtdt
‑3′
[0037]
在本发明的一个具体实施例中,在以上所述simb3序列由化学式iii所示的中性脂材、化学式iv所示的化合物阳离子脂材以及化学式v所示的化合物组成的混合脂材包载时,能够以球形囊泡形态均匀分布,sirna脂质复合物粒径均一,在100nm左右,表面电性中性偏负。
[0038]
在本发明的一个具体实施例中,simb3及其反义链末端缀合一个化学式i和一个化学式vi所示的缀合基团,并使用商用阳离子转染试剂lipofectamine2000或由化学式iii所示的中性脂材、化学式iv所示的化合物阳离子脂材以及化学式v所示的化合物组成的混合脂材包载时,混合脂材具有更优的入胞效率和基因沉默活性。
[0039]
在本发明的一个具体实施例中,在以上所述的simb3序列的反义链5
‑
末端缀合依次缀合三个化学式i所示的缀合基团、一个化学式ii所示的缀合基团,并通过活性酯反应缀合一个化学式vi所示的靶向基团crgd,并由化学式iii所示的中性脂材、化学式iv所示的化合物阳离子脂材以及化学式v所示的化合物组成的混合脂材包载时,具有最优的细胞摄取能力。
[0040]
在本发明的一个具体实施例中,在以上所述的simb3序列的反义链5
‑
末端缀合依次缀合三个化学式i所示的缀合基团、一个化学式ii所示的缀合基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式ix和x,其中缀合基团间通过磷酸二酯键链接,随后在化学式x所示的基团的活泼酯上通过缩合反应连接一个化学式vi所示的靶向基团crgd,并由化学式iii所示的中性脂材、化学式iv所示的化合物阳离子脂材以及化学式v所示的化合物组成的混合脂材包载时,具有最优的细胞水平基因沉默活性。
[0041]
在本发明的一个具体实施例中,在以上所述的simb3序列的反义链5
‑
末端缀合依次缀合三个化学式i所示的缀合基团、一个化学式ii所示的缀合基团,缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式ix和x,其中缀合基团间通过磷酸二酯键链接,随后在化学式x所示的基团的活泼酯上通过缩合反应连接一个化学式vi所示的靶向基团crgd,并由化学式iii所示的中性脂材、化学式iv所示的化合物阳离子脂材以及化学式v所示的化合物组成的混合脂材包载时,具有最优的动物体内抗肿瘤活性。
[0042]
在本发明的一个具体实施例中,在以上所述的simb3序列的反义链5
‑
末端缀合一个化学式ii所示的缀合基团。缀合使用固相合成仪完成,使用的亚磷酰胺单体为化学式x,随后在化学式x所示的基团的活泼酯上通过缩合反应连接一个化学式vi所示的靶向基团crgd,并由化学式iii所示的中性脂材、化学式iv所示的化合物阳离子脂材以及化学式v所示的化合物组成的混合脂材包载时,具有最优的动物体内肿瘤组织分布。
[0043]
相较于现有技术,本发明的优点在于:
[0044]
1.本发明提供的结合靶向基团缀合及中性/阳离子混合载体包载的化学修饰策略,所获得的sirna脂质复合物具有更好生物活性,细胞水平表现出优异的跨膜转运能力和靶mrna的沉默活性,动物水平表现出高效的靶器官分布能力和抑制肿瘤生长的能力,而且在细胞水平和动物水平应用的安全性好,无明显毒性,为sirna技术在临床的广泛应用奠定了基础;
[0045]
2.中性核苷脂材具有碱基头部,可以与sirna通过氢键作用和π
‑
π堆积作用结合,并且与阳离子脂材和sirna间的电荷作用相比,在体内应用时更为稳定,在循环过程中不易吸附带电的颗粒或蛋白,避免了脂质复合物在靶器官外的解体与sirna的释放。并且基于非电性作用的结合减少了阳离子脂材在制剂处方中的用量,阳离子脂材与sirna的氮磷比仅为3/1,远低于其余报道中的阳离子脂材用量。
[0046]
3.5
′‑
缀合具有合成简单快速,可作为底物高效扩展缀合物组合,并且在胞内降解代谢时可释放出5
′‑
末端为磷酸的sirna反义链,更易进入risc复合物发挥基因沉默活性。本发明证明了反义链5
′‑
末端缀合不影响sirna的基因沉默活性,扩展了sirna缀合位点,丰富了sirna的修饰策略。
附图说明
[0047]
图1为sirna/dnca/cld/peg
‑
dspe脂质复合物和空载体的透射电镜(tem)照片;
[0048]
图2为反义链5
′‑
末端单缀合的sirna中性/阳离子脂质复合物的细胞摄取(10nm,4h);
[0049]
图3为反义链5
′‑
末端单缀合的sirna中性/阳离子脂质复合物的胞内分布情况(50nm,4h),其中蓝色表示细胞核,红色表示cy3标记的sirna,绿色表示溶酶体,图中标尺为
10μm;
[0050]
图4为5
′‑
末端单缀合和5
′
,5
″‑
双缀合的sirna中性/阳离子脂质复合物的靶基因沉默活性(50nm,48h);
[0051]
图5为5
′‑
末端单缀合和5
′
,5
″‑
双缀合的sirna中性/阳离子脂质复合物的抗肿瘤活性和载体细胞毒性(100nm,72h);
[0052]
图6为反义链5
′‑
末端单缀合的sirna中性/阳离子脂质复合物的动物体内(a)以及离体后(b)的分布情况,使用小动物活体成像系统拍摄,激发光为745nm,发射光为800nm;
[0053]
图7为反义链5
′‑
末端单缀合的sirna(rl0as/s)的动物体内抗肿瘤活性(balb/c
‑
裸鼠,腋下接种a375);
[0054]
a.当肿瘤体积达到约50mm3时为第0天,制剂在第1、3、5、8天通过静脉注射给药,每天测量并计算肿瘤体积;b.整个治疗过程中小鼠的体重变化曲线;c.最后一次给药后48h安乐死小鼠,收集肿瘤并对肿瘤称重记录;d.取出的肿瘤内braf
v600e
mrna表达;e.免疫组化考察取出的肿瘤组织内braf
v600e
表达,黄褐色斑点代表braf
v600e
蛋白表达量,蓝色代表细胞核,黑色标尺为50μm。数据使用mean
±
sd表示,n=5,***p<0.001;
[0055]
图8为反义链5
′‑
末端单缀合的sirna(rl2as/s)的动物体内抗肿瘤活性(balb/c
‑
裸鼠,腋下接种a375);
[0056]
a.当肿瘤体积达到约50mm3时为第0天,制剂在第1、3、5、7、9天通过静脉注射给药,每天测量并计算肿瘤体积,每只小鼠以第0天的肿瘤体积为1进行归一化处理;b.最后一次给药后48h安乐死小鼠,收集肿瘤并对肿瘤称重记录;c.对取出的肿瘤拍照观察;d.取出的肿瘤内braf
v600e
mrna表达;e.免疫组化考察取出的肿瘤组织内braf
v600e
表达,黄褐色斑点代表braf
v600e
蛋白表达量,蓝色代表细胞核,黑色标尺为50μm;f.整个治疗过程中小鼠的体重变化曲线。数据使用mean
±
sd表示,n=5,*p<0.05,**p<0.01,***p<0.001,****p<0.0001;
[0057]
图9为反义链5
′‑
末端单缀合的sirna(rl0as/s)在动物体内应用时的肝肾生化指标;
[0058]
图10为反义链5
′‑
末端单缀合的sirna(rl2as/s)单次给药48h后瘤内的靶基因沉默活性,sirna给药剂量为1.5mg/kg,每只小鼠2nmol sirna,数据使用mean
±
sd表示,n=4,**p<0.01,***p<0.001。
具体实施方式
[0059]
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随具体实施例的描述而更为清楚。但实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
[0060]
实施例1 5
‑
末端缀合的sirna的单链固相合成
[0061]
rna的合成采用appllied biosystems model 394rna固相合成仪。
[0062]
正常的核苷酸亚磷酰化单体(dt,rgibu,rabz,rcac,ru)从安徽芜湖华仁科技有限公司购买;cpg(cpg
‑
dt),cap
‑
a和cap
‑
b,氧化i2液,cl3ccooh从北京奥科生物科技公司购买;0.25m的5
‑
乙巯基1h
‑
四氮唑溶液从上海智研科技有限公司(上海)购买。
[0063]
按照文献(biomacromolecules 2018,19,7,2526
‑
2534)的方法,将化学式i/化学
式ii所示的链接基团分别制备成化学式ix、化学式x所示的亚磷酰胺单体,即:
[0064]
化学式vii所示的化合物(8.4g,4.2mmol)和三乙胺(0.43g,4.2mmol)溶解在10ml无水dcm中。然后将溶解在dcm中的dmtrcl(1.41g,4.2mmol)缓慢加入上述混合物中。将反应混合物在室温下搅拌4小时。减压除去溶剂,通过柱色谱(pe:ea=5:1)纯化,得产物(1.45g,41%)。在氩气保护下,将双(二异丙基氨基)(2
‑
氰基乙氧基)膦(0.64g,2.1mmol)加入到产物(0.45g,1.1mmol)和四氮唑(0.18g,2.6mmol)的混合物中,用无水dcm(5ml)溶解。将反应混合物在室温下搅拌2小时。通过柱色谱(pe:ea=10:1)纯化,得化学式ix所示的化合物(0.40g,60.6%)。
[0065]
化学式viii所示的化合物(1.5g,12.7mmol)和1
‑
乙基
‑3‑
(3
‑
二甲基氨基丙基)碳二亚胺盐酸盐(edc hcl)(2.7g,15.1mmol)溶解在10ml无水dcm(ch2cl2)中。然后加入n
‑
羟基琥珀酰亚胺(nhs)(1.6g,14.3mmol)并将混合物搅拌过夜。减压除去溶剂,通过柱色谱(pe:ea=1:1)纯化,得产物(1.9g,69.6%)。氩气保护下,将双(二异丙基氨基)(2
‑
氰基乙氧基)膦(0.8g,2.6mmol加入到产物(0.3g,1.4mmol)和四氮唑(0.21g,3mmol)的混合物中用无水dcm(5ml)溶解。将反应混合物在室温下搅拌2小时。通过柱色谱(pe:ea=10:1)纯化,得到化学式x所示的化合物(0.38g,66%)。
[0066][0067]
合成规模:~1.0μmol
[0068]
核苷亚磷酰化单体溶液的配制:氩气保护下称量,加无水乙腈,1g单体对应20ml乙腈;
[0069]
连接臂亚磷酰化单体溶液的配制:氩气保护下称量化学式ix、x所示的化合物,加无水乙腈,1g单体对应20ml乙腈。
[0070]
采用固相合成的方法合成了天然的及缀合的sirna反义链及正义链序列。在rna合成仪上,序列从3
′‑
端至5
′‑
端进行合成,每偶联一个核苷为一个循环,在天然核苷亚磷酰胺单体偶联21次后,将一个或几个所述的链接臂亚磷酰胺单体在序列的5
′‑
末端相应位置进行偶联,每个循环包括四个反应:脱dmt、偶联、封闭、氧化。
[0071]
合成序列:
[0072]
在本实施例中使用待修饰的sirna为靶向erk通路中braf
v600e mrna的simb3,修饰前的序列如下:
[0073]
simb3:正义链:5
′‑
gcuacagagaaaucu cgaudtdt
‑3′
[0074]
反义链:5
′‑
auc gagauu ucu cug uag c dtdt
‑3′
[0075]
修饰策略选择以下任意一种:
[0076]
(1)在以上所述的simb3序列反义链或正义链的5
′‑
末端缀合化学式ii的链接基团;
[0077][0078]
(2)在以上所述的simb3序列反义链或正义链的5
′‑
末端依次(3
′‑5′
方向)缀合一个或多个化学式i和一个化学式ii的链接基团,其中缀合基团间通过磷酸二酯键链接;
[0079]
(3)在以上所述的simb3序列反义链或正义链的5
′‑
末端缀合一个化学式ii所示的链接基团,缀合使用固相合成仪完成,使用的亚磷单体为化学式x,在化学式x所示的链接基团的活泼酯上通过缩合反应连接化学式vi所示的靶向基团crgd;
[0080][0081]
(4)在以上所述的simb3序列反义链或正义链的5
′‑
末端依次(3
′‑5′
方向)缀合一个或几个化学式i和一个化学式ii所示的链接基团,缀合使用固相合成仪完成,使用的亚磷单体为化学式ix和x,其中缀合基团间通过磷酸二酯键链接,随后在化学式x的活泼酯上通过缩合反应连接化学式vi所示的靶向基团crgd。
[0082]
经过上述修饰策略后获得的sirna如下表1所示:
[0083]
表1
[0084]
[0085][0086]
合成步骤:每次称量约33mg cpg
‑
dt装入合成柱中,按照abi394核酸合成仪的rna标准步骤,采用dmt
‑
on的合成策略。以合成rl2as为例,在合成仪序列面板上设定好合成序列。其中,将化学式ix和x所示的连接臂也视为亚磷单体编入序列中,即在as链5
′‑
末端再编入2个化学式ix和1个化学式x所示的连接臂。crgd官能团在合成完毕后手动单独反应,合成完毕后,将合成柱中的粉末转移到1.5ml玻璃反应瓶中,加入1mg化学式xi所示的化合物、100μl dipea、1ml dmf室温搅拌反应24小时,后弃去上清液,使用1ml乙醇洗涤两次,1ml乙醚洗涤一次,后晾干。
[0087][0088]
rna的切割、脱保护:向晾干的粉末中加入750μl甲胺醇和750μl甲氨水切割,置于摇床上,60℃,80rpm,震摇90min。之后将上清液取出,用depc水洗涤三次,均匀分装至两个1.5ml ep管中,最后用冻干机冻干。在每管中加入100μl dmso和125μl三乙胺三氢氟酸,封口膜缠住,80rpm,65℃,震摇90min。冷却后,加入100μl 3m ch3coona溶液,涡旋,加入1ml正丁醇溶液,涡旋后放入
‑
80℃静置30min。后取出12500rpm离心10min,弃去上清液,留下白色沉淀,加入0.75ml无水乙醇涡旋,放入
‑
80℃静置30min,12500rpm离心10min,弃去上清,再重复一次该操作,后冻干。
[0089]
rna的分离纯化:将样品用400μl depc水稀释溶解,用0.22μm滤头过滤,在用200μl depc水洗涤两次,最终总体积大约为800μl。用进样针吸取,每针体积120μl进样。使用xbridge
tm oligonucleotide beh c18 obd
tm prep column10x50mm 2.5μm核酸分离柱梯度洗脱(0min,98%teab,2%ch3cn;25min,85%teab,15%ch3cn,v=1ml/min)。馏分收集完毕后,冻干成粉末。再用700μl depc水溶解,用葡聚糖凝胶柱(hitrap 5ml desalting)进行脱盐(depc水,v=1.5ml/min)出峰时间在2min左右,收集馏分,再次冻干,得纯产物,
‑
80℃保存。
[0090]
实施例2中性/阳离子混合制材包载sirna形成脂质复合物(sirna/dnca/cld/peg2000
‑
dspe)
[0091]
将sirna与混合制材混合,混合制材由39.7%的化学式iii所示的化合物(dnca,中性型)、59.6%的化学式iv所示的化合物(cld,阳离子型)、0.7%的化学式v所示的化合物(peg2000
‑
dspe)组成。这些比例指的是所有脂质总量的摩尔%。脂质与sirna的摩尔比为5.3:1。按文献(cn108059619a)的方法合成dnca,按文献(new jchem,2014,38(10),4952
‑
4962)的方法合成cld,peg2000
‑
dspe购自源叶公司。
[0092][0093]
简要来说,将化学式iii所示的化合物(dnca,中性型)、化学式iv所示的化合物(cld,阳离子型)以及化学式v所示的化合物溶于乙醇溶液中,将sirna溶于depc水中,使用genopti(北京迈晨)混合,然后于70℃超声10min,超声频率150w,40khz。超声结束恢复至室温后,使用0.22μm滤膜过滤,滤液中包含脂质复合物。
[0094]
例如,在一个具体的方法中,在乙醇中配置新鲜的脂质储液:称取156.5mg dnca、
315mg cld、10.5mg peg2000
‑
dspe,并在20ml乙醇中溶解以形成脂质储备液,将其在37℃超声4分钟以形成均匀的脂质混合物。随后,将20μl上述储备液加入480μl genopti中形成500μl工作脂质储液。将这个用量的脂质用于形成具有10nmol的sirna脂质复合物。在sirna干粉溶解在depc水中形成200μm的sirna储备液。将50μl上述储备液加入450μl genopti中形成500μl工作sirna储液,将500μl的工作脂质储液与500μl的工作sirna储液混合,短暂低速离心后,于70℃超声10min形成脂质复合物。体内和体外实验所用制剂使用0.22μm滤膜过滤(默克密理博,merck millipore),使用前使用pbs溶液稀释至1
×
sirna脂质复合物溶液。
[0095]
图1显示通过这些方法制备的sirna的脂质复合物和不含sirna的空载体的示例性电性显微镜照片(tem)。这些脂质体包含的sirna为未缀合的靶向erk通路中braf
v600e
mrna的simb3。不加入sirna的空载体的颗粒较为松散,且颗粒表面皱缩。加入sirna后的脂质复合物可以形成颗粒均一的球型纳米颗粒,而且边缘光滑形态较好。
[0096]
动态光散射显示包载有sirna的脂质复合物的平均直径是138.7
±
6.6(根据密度),多分散系数是0.188
±
0.051,其表面电性是
‑
16.9
±
0.4mv;空载体的平均直径是363.7
±
20.1(根据密度),多分散系数是0.635
±
0.045,其表面电性是24.3
±
2.8mv。由于tem拍摄前需对样品脱水干燥处理,颗粒会因水分的流失产生皱缩,而dls测定的是纳米颗粒的水和粒径,因此tem法所测得的粒径小于dls法。
[0097]
实施例3利用流式细胞术考察反义链5
‑
末端单缀合的sirna中性/阳离子脂质复合物的细胞摄取
[0098]
1.样品:
[0099]
as/cy3
‑
s
‑
simb3、rl0as/cy3
‑
s
‑
simb3、rl1as/cy3
‑
s
‑
simb3、rl2as/cy3
‑
s
‑
simb3、rl3as/cy3
‑
s
‑
simb3、rl4as/cy3
‑
s
‑
simb3。simb3的正义链5
‑
末端使用荧光染料cy3标记。反义链缀合物的合成参见实施例1,sirna脂质复合物sirna/dnca/cld的制备参见实施例2,区别在于混合制材由72.7%化学式iii所示的中性胞苷脂材、27.3%化学式iv所示的阳离子脂材组成。这些比例指的是所有脂质总量的摩尔%。总脂质与sirna的摩尔比为10:1。
[0100]
2.方法:
[0101]
将a375以6万/孔铺24孔板,培养18h后进行转染。simb3及其缀合物的给药浓度为10nm,转染使用的lipofectamine2000用量为0.5ul,按照其使用说明书与sirna混合制配置转染复合物。将复合物加入至培养板中,培养4h后,使用dmem润洗细胞表面,胰酶消化收集细胞,流式细胞仪pe通道测定细胞内荧光强度,考察simb3及其缀合物的细胞摄取能力,实验结果见图2。
[0102]
3.结果:
[0103]
与lipofectamine2000相比,混合载体dnca/cld具有更高效的递送sirna入胞的能力,说明血清蛋白的存在对dnca/cld/simb3纳米复合物的摄取影响不大,即使血清中含有潜在的转染竞争者,例如玻连蛋白和纤连蛋白。
[0104]
对比不同长度链接臂连接的crgd缀合物的摄取情况发现,随链接臂延长(rl0as/s至rl3as/s),摄取逐渐增加。细胞摄取rl4as/s的能力较低,提示继续延长链接臂长度已经不能增强simb3缀合物的细胞摄取能力,这有可能是由于增加链接臂长度的同时会引入更多的带负电荷的磷酸二酯键,其可能影响simb3缀合物/dnca/cld脂质复合物的形成。
[0105]
实施例4利用共聚焦激光显微镜考察反义链5
‑
末端单缀合的sirna中性/阳离子脂
质复合物的细胞内分布
[0106]
1.样品:
[0107]
as/cy3
‑
s
‑
simb3、rl0as/cy3
‑
s
‑
simb3、rl1as/cy3
‑
s
‑
simb3。simb3的正义链5
‑
末端使用荧光染料cy3标记。反义链缀合物的合成参见实施例1,sirna脂质复合物sirna/dnca/cld的制备参见实施例3。
[0108]
2.方法:
[0109]
将a375以6万/孔铺于共聚焦皿中,培养18h后进行转染。simb3及其缀合物的给药浓度为50nm,转染使用的lipofectamine2000用量为1μl,按照其使用说明书与sirna混合制配置转染复合物。将复合物加入至培养板中,培养4h后,使用hoechst、lysobrite nir分别对细胞核和溶酶体进行染色。加入染料后孵育30分钟。使用dmem润洗细胞表面去除游离的染料,使用共聚焦显微镜拍照。考察simb3及其缀合物的细胞内分布情况,实验结果见图3。
[0110]
3.结果:
[0111]
共聚焦结果与流式细胞术结果相吻合,lipofectamine2000递送sirna入胞的效率远低于dnca/cld(mix)。crgd缀合于反义链5
′‑
末端后,细胞摄取有明显增加,这表明部分缀合的crgd可能不在dnca/cld/simb3纳米复合物内部,从而通过靶向αvβ3整合素进入细胞内,增加αvβ3阳性细胞a375摄取脂质复合物的能力。cy3标记的sirna(红色)在细胞膜上及细胞内呈点状聚集,可能分布于细胞膜转运囊泡和内体中,从而进行胞内转运。sirna经dnca/cld混合制剂包载后几乎不与溶酶体(绿色)共定位,提示这一脂质复合物可有效避免进入溶酶体或可快速从溶酶体中逃逸,从而最大程度减少sirna在溶酶体内的酸化降解。
[0112]
实施例5利用rt
‑
qpcr技术考察5
‑
末端单/双缀合的sirna中性/阳离子脂质复合物的基因沉默活性
[0113]
1.样品:
[0114]
as/s
‑
simb3、l0as/s
‑
simb3、l1as/s
‑
simb3、rl0as/s
‑
simb3、rl1as/s
‑
simb3、rl2as/s
‑
simb3、rl3as/s
‑
simb3、rl4as/s
‑
simb3、l1as/l0s
‑
simb3、as/rl0s
‑
simb3、rl0as/rl0s
‑
simb3、rl1as/rl0s
‑
simb3、l0as/rl0s
‑
simb3、l1as/rl0s
‑
simb3。以上5
‑
末端单双缀合物的合成参见实施例1。
[0115]
2.rt
‑
qpcr实验:
[0116]
将a375以8万/孔铺12孔板,培养18h后进行转染。simb3及其缀合物的给药浓度为50nm,转染使用的lipofectamine2000用量为1ul,按照其使用说明书与sirna混合制配置转染复合物。脂质复合物sirna/dnca/cld按照实施例3的方法制备。将复合物加入至培养板中,培养48h后,每孔加入0.5ml trizol试剂提取总rna,使用逆转录试剂盒将rna逆转录为cdna,进行实时荧光定量pcr,考察simb3及其缀合物的靶mrna沉默能力,实验结果见图4。
[0117]
3.结果
[0118]
首先比较使用商用转染试剂和dnca/cld混合制剂转染sirna的效率,发现混合脂材dnca/cld包载的sirna基因沉默活性优于使用商用转染试剂lipofectamine2000包载(lipo组),且具有显著性差异(p<0.0001)。
[0119]
对反义链5
′‑
单缀合物基因沉默活性考察发现,末端缀合短的连接臂(l0as/s和l1as/s)与未缀合的simb3(as/s)的基因沉默能力没有显著性差异(p>0.05)。继续考察了rl0
‑
rl4一这系列末端缀合了较大体积基团crgd的缀合物的基因沉默活性,结果表明基因
沉默活性存在一定差异且与链接臂长度相关,具有更长链接臂(rl2as/s,rl3as/s和rl4as/s)的5
′‑
单缀合物的活性优于较短链接臂(rl1as/s和rl0as/s)连接的缀合物,这一结果可能是由于在反义链5
′‑
末端和crgd之间插入更长的链接臂会使缀合部分的结构更加松散,减少了sirna缀合物载入risc时的空间阻碍。rl2as/s
‑
simb3与simb3的活性差异较小,可能是因其可以采用适当的构象避免与risc的空间冲突,因此与未缀合物具有相似的基因沉默活性。
[0120]
正义链5
′‑
末端单缀合crgd对基因沉默活性无明显影响(as/rl0s
‑
simb3),即使是在链接臂最短的情况下。而双缀合物与未缀合的simb3相比,基因沉默活性略有下降,但在此浓度条件下活性差异不明显,当在正义链的5
′‑
末端通过较短连接基团缀合crgd后,反义链5
′‑
缀合大基团的活性优于缀合较小基团。
[0121]
实施例6考察5
‑
末端单/双缀合的sirna中性/阳离子脂质复合物的抗肿瘤活性及空载体细胞毒性
[0122]
1.样品:
[0123]
as/s
‑
simb3、l0as/s
‑
simb3、l1as/s
‑
simb3、rl0as/s
‑
simb3、rl1as/s
‑
simb3、l1as/l0s
‑
simb3、rl0as/rl0s
‑
simb3、rl1as/rl0s
‑
simb3、as/rl0s
‑
simb3、l0as/rl0s
‑
simb3、l1as/rl0s
‑
simb3。以上5
‑
末端单双缀合物的制备参见实施例1,sirna脂质复合物sirna/dnca/cld的制备参见实施例3。
[0124]
2.cck
‑
8实验
[0125]
将a375以1万/孔铺96孔板,培养18h后进行转染。simb3及其缀合物的给药浓度为100nm,将sirna脂质复合物加入至培养板中,培养72h后,弃去培养基,加入10%cck
‑
8试剂,37℃孵育2h,使用酶标仪测定450nm的吸光度。结果以control组的吸光度为1进行均一化处理。考察simb3及其缀合物的抗肿瘤活性和空载体的细胞毒性,实验结果见图5。
[0126]
3.结果
[0127]
对单缀合simb3/dnca/cld脂质复合物的细胞水平抗肿瘤活性进行评价发现,对反义链5
′‑
末端进行缀合会使其体外抗肿瘤活性降低,且与缀合基团的大小无关。正义链5
′‑
末端缀合后,抗肿瘤活性也随之下降,双缀合物中,仅双缀合物l1as/l0s的基因沉默活性与非缀合物as/s相似,l0as/rl0s同样具有较好的抗肿瘤活性。dnca/cld空载体(mix组)的细胞毒性较低,提示体外应用脂质复合物安全低毒。
[0128]
实施例7考察5
‑
末端单缀合的sirna中性/阳离子脂质复合物的动物体内分布
[0129]
1.样品:
[0130]
as/cy7
‑
s
‑
simb3、rl0as/cy7
‑
s
‑
simb3,simb3的正义链5
′‑
末端使用荧光染料cy7标记。反义链缀合物的合成参见实施例1,sirna脂质复合物sirna/dnca/cld/peg2000
‑
dspe的制备参见实施例2。
[0131]
2.方法:
[0132]
在6周龄的裸鼠腋下接种a375细胞,在10天后肿瘤体积达到约500mm3。随后,通过尾静脉注射dnca/cld/peg2000
‑
dspe包载的cy7标记的simb3(as/s)或crgd缀合物(rl0as/s),sirna给药剂量1.5mg/kg。给药后1.5h至36h通过体内成像系统测量sirna的荧光信号。给药后36h,安乐死动物,取出肿瘤组织和器官并拍照。实验结果见图6。
[0133]
3.结果:
[0134]
整个拍照过程中可以明显观察到rl0as/s在瘤内蓄积,但未缀合crgd的as/s仅有微弱的荧光信号(图6a)。对瘤内荧光信号进行定量,结果显示crgd
‑
缀合的simb3的肿瘤蓄积比未缀合的simb3高约90%(p<0.05),混合载体可有效递送sirna至肿瘤,as/s组的瘤内荧光信号显著高于blank组(p<0.01)。然而,由于胃肠道中食物的自发荧光,难以观察到肝脏和肾脏中的sirna信号,但其与肿瘤内的荧光信号无关。
[0135]
离体结果可以发现(图6b),cy7标记的simb3主要聚集在肿瘤和肝脏中,肿瘤中rl0as/s的荧光信号比as/s强,离体肿瘤内荧光定量结果与体内分布的肿瘤定量结果相对应,crgd缀合的simb3(rl0as/s)瘤内蓄积能力强于未缀合simb3(p<0.05)。以上结果表明,dnca/cld/peg2000
‑
dspe混合载体可高效递送simb3及其缀合物至肿瘤组织,并且给药后36小时仍有明显的瘤内和肝脏分布,这种组合的缀合和递送策略为全身递送sirna体内治疗肿瘤提供了平台。
[0136]
实施例8考察5
‑
末端单缀合的sirna中性/阳离子脂质复合物的动物体内抗肿瘤活性及其机制
[0137]
1.样品:
[0138]
as/s
‑
simb3、rl0as/s
‑
simb3、rl2as/s
‑
simb3。sirna及缀合物的合成参见实施例1,sirna脂质复合物sirna/dnca/cld/peg2000
‑
dspe的制备参见实施例2。
[0139]
2.方法:
[0140]
将160w黑色素瘤细胞a375接种至3周龄裸鼠腋下,待7天后肿瘤成型后将裸鼠分为5组,此时平均肿瘤体积约为50mm3(第0天),每组5只。尾静脉注射simb3及缀合物混合制剂进行治疗,空白对照为blank(尾静脉注射制剂溶剂genopti),阴性对照为simb3(未经包载的simb3)、mix(不含有sirna的dnca/cld/peg2000
‑
dspe空载体)、mix
‑
nc(经mix包载的靶向firefly萤光素酶mrna的sifl)。在第1、3、5、8天(rl0as/s)或第1、3、5、7、9天(rl2as/s)通过尾静脉注射给药,sirna的给药剂量为2mg/kg(rl0as/s)或1.48mg/kg(rl2as/s)。给药治疗期间每日或每两日量取肿瘤大小及小鼠体重,并按照公式肿瘤体积v=长
×
宽2×
0.5计算肿瘤体积。最后一次给药48h后,取1ml血液,随后安乐死小鼠,分离肿瘤组织称重并拍照记录。取部分瘤组织(50
‑
100mg)加入trizol后匀浆提取总rna,使用逆转录试剂盒将rna逆转录为cdna,进行实时荧光定量pcr,考察simb3及其缀合物的瘤内mrna沉默能力。另取部分瘤组织(50
‑
100mg)于多聚甲醛中固定过夜,随后使用石蜡包埋,并制成切片,对细胞核和braf
v600e
蛋白标记染色处理。小鼠血液4℃静止一夜,低速离心15分钟分离出血清,随后对血清中的肝肾生化指标进行检测。实验结果见图7、图8、图9、图10。
[0141]
3.结果:
[0142]
首先考察较短链接臂链接的rl0as/s
‑
simb3的体内抗肿瘤活性(图7)。结果表明,simb3及其缀合物包载后在整个治疗过程中可明显抑制肿瘤生长(图7a,p<0.001),但缀合物rl0as/s抑制肿瘤生长的能力的弱于非缀合物as/s,尽管p值为0.25,无显著性差异。整个给药治疗过程中,小鼠体重稳定增长,各组间无差异,说明缀合不影响sirna脂质复合物的安全性(图7b)。最后一次给药后48h安乐死小鼠并取出肿瘤称重(图7c),mix
‑
as/s肿瘤体积最小,mix
‑
rl0as/s的肿瘤体积相比空白和阴性对照也有一定降低,但整组内的治愈失败率为2/5,五只小鼠中有两只肿瘤体积与对照组相近。rt
‑
qpcr测定瘤内braf
v600e
mrna水平发现,as/s相较于rl0as/s可以更有效的沉默靶标mrna(图7d)。免疫组化结果与qpcr结果一致
(图7e),在小鼠的肿瘤石蜡切片中,simb3及缀合物治疗组均明显降低了braf
v600e
蛋白的表达(褐色斑点)。
[0143]
体内抗肿瘤实验说明,尽管具有crgd靶向基团的rl0as/s具有更优的肿瘤的蓄积能力,但由于其缀合基团与反义链间的空间距离过近,极大的影响了基因沉默活性,因此rl0as/s缀合物的治疗效果弱于未缀合的simb3。结合细胞实验,对rl2as/s体内抗肿瘤活性进行考察(图8)。
[0144]
经过5次尾静脉给药后,mix
‑
simb3及其缀合物mix
‑
rl2as/s的肿瘤生长速度显着低于blank(p<0.0001),也低于未包载的sirna(p<0.001)及mix
‑
nc组(p<0.001)(图8a)。空白及阴性对照组的的肿瘤体积是第0天的25倍以上,mix
‑
as治疗组增加了12倍,而mix
‑
rl2as/s组的肿瘤生长速度比mix
‑
as/s组慢得多(p<0.05),mix
‑
rl2as/s治疗组的肿瘤体积仅增加了约6倍。反义链5
′‑
末端经较长链接臂连接crgd缀合基团的simb3脂质复合物相比未缀合的simb3脂质复合物可更有效的治疗肿瘤。最后一次给药48h后,安乐死小鼠,分离肿瘤组织称重(图8b)并拍照记录(图8c)。如图8b所示,mix
‑
as/s组肿瘤重量明显小于空白及阴性对照组(p<0.05),缀合物mix
‑
rl2as/s组的肿瘤重量与空白及阴性对照组相比也显著减小(p<0.01)。缀合物与未缀合的肿瘤肿瘤没有显著性差异(p>0.5),但mix
‑
rl2as/s组的肿瘤重量均值小于mix
‑
as/s组。各组离体肿瘤体积的大小与肿瘤生长曲线一致(图8c)。rt
‑
qpcr测定瘤内braf
v600e mrna水平发现,as/s与rl2as/s可以更有效的沉默约40%靶标mrna表达(图8d),且与空白及阴性对照组有显著性差异(p<0.001)。通过免疫组化实验同步评估了瘤内braf
v600e
的蛋白水平,黄色斑点代表braf
v600e
蛋白的表达(图8e)。与nc相比,simb3及rl2as/s均下调了braf
v600e
,而rl2as/s与as/s的沉默效率相似。rl2as/s出色的抗肿瘤能力既与靶向基团的缀合,使肿瘤组织simb3的蓄积量增加有关,也与缀合基团与反义链5
′‑
末端的链接臂延长,使sirna的基因沉默活性恢复有关。因为在体内实验结束时,经rl0as/s治疗的肿瘤生长速度大于未缀合的simb3(图7a),尽管二者的肿瘤体积没有显著性差异(p=0.25)。这些结果表明,肿瘤靶向缀合基团crgd增加了simb3缀合物的纳米颗粒在肿瘤组织中的积累,并且crgd与5
′‑
末端之间的延长的空间距离(l2与l0相比)是反义链5
′‑
末端缀合物优异抗肿瘤效率的关键点。此外,稳定变化的体重和治疗期间无小鼠死亡均表明所有的缀合物在体内应用是足够安全的(图8f)。
[0145]
为验证缀合物及制剂的肝肾毒性,最后一次给药后48小时,收集全血,测量小鼠血液生化指标(图9)。各组的代表肾功能的crea
‑
j、ua、urea和肝功能的alt、ast、alp、tp、alb血生化指标与未给药组无明显差别,说明缀合和dnca/cld/peg2000
‑
dspe混合脂材制剂的安全性佳,几乎不存在肝肾毒性,simb3/dnca/cld/peg2000
‑
dspe脂质复合物可作为安全有效的小鼠肿瘤疗法。
[0146]
为验证simb3缀合物治疗肿瘤的机制,测定单次给药后瘤内braf
v600e
mrna的表达水平。使用balb/c
‑
nude 4周龄鼠,右侧腋下接种a375细胞160w,接种后20日,肿瘤体积约为500
‑
800mm3时随机分成四组,分别经尾静脉注射给药。给药后48h,安乐死动物,取肿瘤组织50
‑
100mg,定量mrna的表达量。如图10所示,一次给药后,simb3可沉默瘤内braf
v600e
mrna的表达。未缀合的as/s可沉默14%靶mrna,反义链5
′‑
末端缀合crgd的rl2as/s沉默22%的靶mrna表达,且与mix
‑
nc组相比有显著性差异(nc vs.as/s,p<0.05;nc vs.rl2as/s,p<0.001)。sirna瘤内靶基因抑制水平略低可能是因为给药时肿瘤体积较大,sirna不能完全
转染进入肿瘤组织中的所有细胞,多次给药后的基因沉默活性更为明显(图8d)。rl2as/s在动物水平抑制肿瘤生长与braf
v600e
mrna的沉默有关。
[0147]
本文显示并详细描述的信息足以实现本发明的上述目的,因此本发明的优选实施方案代表本发明的主题,该主题为本发明所广泛涵盖。本发明的范围完全涵盖其它对本领域技术人员来说显而易见的实施方案,因此,本发明的范围不被除所附权利要求之外的任何内容所限制,其中除了明确说明外,所用元素的单数形式并不是指“一个和唯一”,而是指“一个或更多”。对本领域一般技术人员来说,所有公知的上述优选的实施方案和附加实施方案部分的结构、组成和功能上的等价物因此引入本文作参考,而且试图被本发明的权利要求所涵盖。
[0148]
此外,不需要某种设备或方法来表达本发明所解决的每个问题,因为它们都已包括在本发明的权利要求之内。另外,无论本发明公开事实中的所有部分、成分,或者方法步骤是否在权利要求中被明确叙述,它们都没有贡献给公众。但是,对本领域普通技术人员来说,很明显在不背离如所附权利要求中所阐明的本发明的实质和范围的前提下,可以在形式、试剂和合成细节上做出各种改变和修饰。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1