具有聚(甲基)丙烯酸酯基团的聚有机硅氧烷及其制备方法和用途与流程
具有聚(甲基)丙烯酸酯基团的聚有机硅氧烷及其制备方法和用途
1.相关申请的引用
2.本技术根据35 u.s.c.第119(e)条要求于2019年3月14日提交的美国临时专利申请第62/818131号的权益。美国临时专利申请第62/818131号通过引用并入本文。
技术领域
3.公开了一种具有硅键合的聚(甲基)丙烯酸酯基团的聚有机硅氧烷(以下简称“聚(甲基)丙烯酸酯接枝的聚有机硅氧烷”)。还公开了一种用于制备聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的方法。
背景技术:
4.丙烯酸链可以使用自由基聚合通过在巯基官能的聚二甲基硅氧烷的存在下进行聚合而结合到聚二甲基硅氧烷主链上。巯基基团将充当链转移剂并且使丙烯酸链能够作为侧基和/或端基接枝到聚二甲基硅氧烷链上。然而,如果巯基官能的聚二甲基硅氧烷还含有乙烯基或其它脂肪族不饱和单价烃基官能团,则该体系可能由于脂肪族不饱和基团在自由基聚合期间的反应而变得饱和或交联。此外,由于链转移是动力学控制的过程,由于巯基官能化的低水平,接枝效率可能很低。
技术实现要素:
5.一种具有聚(甲基)丙烯酸酯基团的聚有机硅氧烷(下文简称“聚(甲基)丙烯酸酯接枝的聚有机硅氧烷”)包含以下单元式:
6.[r
3w
(r5‑
s
‑
r
″
)(or4)
(2
‑
w)
si
‑
o
1/2
]
p
[r
3v
(r5‑
s
‑
r
″
)(or4)
(1
‑
v)
si
‑
o
2/2
]
q
[(r5‑
s
‑
r
″
)si
‑
o
3/2
]
k
(r6r
72
sio
1/2
)
r
(r
72
sio
2/2
)
s
(r6r7sio
2/2
)
t
(r
73
sio
1/2
)
u
,其中每个下标w独立地为0、1或2,每个下标v独立地为0或1,每个r3为独立选择的单价烃基;每个r4为独立选择的烷基;每个r5为独立选择的二价烃基,每个r
″
独立地为聚(甲基)丙烯酸酯聚合物或共聚物,每个r6为独立选择的脂肪族不饱和单价烃基,每个r7为独立选择的不含脂肪族不饱和基团的单价烃基,下标p≥0,下标q≥0,下标k≥0,数量(p+q+k)≥1,下标r≥0,下标s≥0,下标t≥0,下标u≥0,数量(r+t)≥2,并且数量(p+q+k+r+s+t+u)足以向聚(甲基)丙烯酸酯接枝的聚有机硅氧烷提供至少50kda的分子量。
[0007]
一种用于制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法,其包含:
[0008]
i)组合起始材料,其包含:
[0009]
烷氧基硅基官能的(甲基)丙烯酸酯大分子单体;
[0010]
聚二有机硅氧烷,其选自由以下组成的组:
[0011]
每分子具有至少一个硅键合的脂肪族不饱和基团的不饱和聚二有机硅氧烷,
[0012]
羟基官能的聚二有机硅氧烷,以及
[0013]
不饱和聚二有机硅氧烷和羟基官能的聚二有机硅氧烷两者的组合;以及
[0014]
缩合反应催化剂;
[0015]
从而制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷和包含水和/或醇的副产物的产物;以及
[0016]
ii)在步骤i)期间和/或之后除去副产物的全部或部分。
附图说明
[0017]
图1示出了方案1的代表性实例,其中(甲基)丙烯酸酯聚合物在从自由基引发剂片段或生长链中提取氢原子之后从硫醇官能团生长。然后用来自不同硫醇分子的氢原子封端/端封(甲基)丙烯酸酯低聚物,从而形成烷氧基硅基官能的(甲基)丙烯酸酯大分子单体。这在下面的参考实例a中进行了描述。
[0018]
图2示出了用于制备以下参考实例b中描述的聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的方案2的代表性实例。在图2中,双乙烯基封端的聚二甲基硅氧烷、双羟基封端的聚二甲基硅氧烷和在图1中的方案1中制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体在磷腈催化剂和溶剂(甲苯)的存在下反应以形成聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。
[0019]
图3示出了来自对比实例27的方案3,其中双乙烯基封端的聚二甲基硅氧烷、双硅烷醇封端的聚二甲基硅氧烷和巯基丙基甲基二甲氧基硅烷在磷腈催化剂1和甲苯的存在下组合,从而制备巯基官能化的双乙烯基封端的聚(二甲基/甲基巯基丙基)硅氧烷共聚物。
[0020]
图4示出了方案4,其中在实例17中合成了具有聚丙烯酸酯封端的聚(二甲基/甲基乙烯基)硅氧烷共聚物。
[0021]
图5示出了方案5,其中在实例18中合成了具有三甲基硅氧基末端嵌段的聚(二甲基/甲基,乙烯基/甲基,聚丙烯酸酯)硅氧烷共聚物。
具体实施方式
[0022]
公开了一种用于制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法。该方法包含:
[0023]
i)组合起始材料,其包含:
[0024]
a)烷氧基硅基官能的(甲基)丙烯酸酯大分子单体;
[0025]
b)聚二有机硅氧烷,其选自由以下组成的组:
[0026]
b1)每分子具有至少一个硅键合的脂肪族不饱和基团的不饱和聚二有机硅氧烷;
[0027]
b2)每分子具有至少两个硅键合的羟基的羟基官能的聚二有机硅氧烷,以及
[0028]
b3)b1)和b2)的组合;
[0029]
c)缩合反应催化剂;
[0030]
任选地d)聚二烷基硅氧烷;以及
[0031]
任选地e)溶剂;从而制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷和副产物的产物;
[0032]
ii)在步骤i)期间和/或之后除去副产物的全部或部分;
[0033]
任选地iii)中和产物;以及
[0034]
任选地iv)回收聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。
[0035]
该方法中的步骤i)可以通过任何方便的方式进行,如混合。步骤i)可以在惰性条件,例如氮气或其它惰性气体下进行。
[0036]
可以在升高的温度下进行组合起始材料,例如在80℃至120℃下加热。可以任选地添加溶剂,例如以促进起始材料的组合。起始材料的组合可以以任何添加顺序进行,例如组合a)烷氧基硅基官能的(甲基)丙烯酸酯大分子单体,b)聚二有机硅氧烷(例如,b1)不饱和聚二有机硅氧烷和b2)羟基官能的聚二有机硅氧烷中的一种或两种)和/或d)聚二烷基硅氧烷。然后可将所得混合物加热,然后可添加c)缩合反应催化剂,任选地溶于e)溶剂中。在不希望受到理论约束的情况下,据认为添加e)溶剂可有益于制备具有高mw的聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。当起始材料反应时,产生副产物。副产物可以包含水和/或醇(如甲醇)。可以在步骤i)期间和/或之后除去副产物的所有或部分。在不希望受到理论约束的情况下,据认为除去副产物可以驱动反应完成和/或促进mw增加。副产物可以通过任何方便的方式除去,如汽提。
[0037]
方法中的步骤iii)是中和产物。中和可以通过在步骤ii)期间或之后向产物中添加f)中和剂来进行。中和可以在环境温度或升高的温度下进行。方法中的步骤iv)是回收聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。回收聚(甲基)丙烯酸酯接枝的聚有机硅氧烷可通过任何方便的方式进行,如过滤、汽提和/或蒸馏。上述方法中使用的起始材料如下。
[0038]
起始材料a),在上述方法中使用的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体可具有式a
‑
1):其中每个r3为独立选择的单价烃基;每个r4为独立选择的烷基;r5为二价烃基;r
″
为(甲基)丙烯酸酯聚合物或共聚物,并且下标a为0、1或2。r
″
可以具有1至1,000,或者5至600的dp。替代地,烷氧基硅基官能的(甲基)丙烯酸酯大分子单体可具有式a
‑
2):其中每个r1独立地选自由氢和烷基组成的组;r2独立地选自由氢、烷基、芳基和芳烷基组成的组;下标n为1至1,000;并且r3、r4和r5如上所述。
[0039]
替代地,在式a
‑
2)中,下标n可为5至600。适合于r1的烷基可以是1至4个碳原子的烷基;替代地甲基或乙基。替代地,每个r1可以为甲基。适合于r2的烷基可具有1至18个碳原子,或者1至8个碳原子。适合于r2的烷基包括甲基、乙基、丙基和丁基。适合于r2的芳基具有6至18个碳原子并包括苯基,适合于r2的芳烷基具有6至18个碳原子并包括苯乙烯基。替代地,在式a
‑
2)中,每个r2可以为独立选择的1至18个碳原子,或者1至8个碳原子的烷基。
[0040]
替代地,在式a
‑
1)和a
‑
2)中,下标a可以为1或2,或者1,或者2。每个r3可以为独立选择的1至18个碳原子的单价烃基。适合于r3的单价烃基包括烷基(例如,甲基、乙基、丙基
和丁基)和烯基(例如,乙烯基、烯丙基和己烯基)、如苯基的芳基和如苄基、甲苯基、二甲苯基和苯乙基的芳烷基。替代地,每个r3可独立地选自由烷基和烯基组成的组。替代地,每个r3可为1至8个碳原子的烷基。替代地,每个r3可选自由甲基和乙烯基组成的组。
[0041]
每个r4可为独立选择的1至6个碳原子的烷基。适合于r4的烷基包括甲基、乙基、丙基和丁基;替代地甲基。
[0042]
每个r5可为1至18个碳原子的二价烃基。
[0043]
适合于r5的二价烃基包括如亚乙基(
‑
ch2‑
ch2‑
)的亚烷基、如
‑
ch2‑
ch2‑
ch2‑
或
‑
ch(ch3)ch2‑
)的亚丙基、亚丁基或亚己基;如亚苯基的亚芳基,或亚烷芳基,如:
[0044]
替代地,r5可为2至6个碳原子的亚烷基,如亚丙基。
[0045]
用于制备包含上述聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法中使用的起始材料a)的量取决于各种因素,然而,按该方法中起始材料a)和b)的组合重量计,起始材料a)的用量可以为4%至11%。替代地,在相同的基础上,起始材料a)的量可以为1%至50%,或者5%至10%,或者5%至9%。
[0046]
适合用作起始材料a)的烷氧基硅基官能的(甲基)丙烯酸酯单体可以通过已知方法制备,如在brown的美国专利6,733,884中公开的方法。替代地,烷氧基硅基官能的(甲基)丙烯酸酯可通过包含以下步骤的方法制备:
[0047]
1)组合起始材料,起始材料包含
[0048]
i)式的(甲基)丙烯酸酯单体,其中r1和r2如上所述;
[0049]
ii)式的巯基官能的烷氧基硅烷,其中r3、r4、r5和下标a如上所述;
[0050]
任选地iii)自由基引发剂;以及
[0051]
任选地iv)溶剂;从而制备包含烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的产物;以及
[0052]
任选地2)回收a)烷氧基硅基官能的(甲基)丙烯酸酯大分子单体。
[0053]
在这一方法中,i)(甲基)丙烯酸酯单体可在步骤1)之前与iv)溶剂(当存在时)组合。在与起始材料i)和/或任何其它起始材料组合之前,可任选地干燥溶剂。替代地,i)(甲基)丙烯酸酯单体),ii)巯基官能的烷氧基硅烷和iv)溶剂(当存在时)可在步骤1)之前组合。所得组合可以在反应器中加热至例如50℃至150℃。起始材料i)和ii)的反应可以在充
分加热下进行以生成自由基。替代地,iii)自由基引发剂,任选地溶于iv)溶剂中,可以添加到反应容器中。步骤1)可以在惰性条件下进行,例如,通过用氮气吹扫反应器。步骤1)中的起始材料可以通过混合、加热或两者进行组合。例如,混合和加热可以通过混合同时从50℃加热至起始材料的回流温度,或者50℃至150℃,或者50℃至110℃;或者50℃至100℃;或者75℃至85℃进行1至6小时。起始材料可以以任何顺序添加,然而,iii)自由基引发剂可以溶于iv)溶剂中并任选地与i)(甲基)丙烯酸酯单体组合,然后可以将所得组合添加到含有ii)巯基官能的烷氧基硅烷的反应器中。替代地,i)(甲基)丙烯酸酯单体和ii)巯基官能的烷氧基硅烷组合以形成混合物,然后可将iii)自由基引发剂加入到混合物中。在不希望受到理论约束的情况下,据认为所得烷氧基硅基官能的(甲基)丙烯酸酯大分子单体(以这一添加顺序制备)将具有与当使用不同添加顺序时不同的分子量分布。
[0054]
步骤2),回收烷氧基硅基官能的(甲基)丙烯酸酯大分子单体可以通过任何方便的方式进行,如将步骤1)中制备的反应产物冷却至室温,并在将使烷氧基硅基官能的(甲基)丙烯酸酯大分子单体沉淀的非溶剂(例如,如己烷的烷烃或如甲醇的醇)中沉淀。回收可以任选地进一步包含干燥沉淀物,如通过在环境压力或减压下加热,例如80℃至100℃,以除去残留单体、溶剂或两者。
[0055]
在制备烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的方法中,起始材料i)是(甲基)丙烯酸酯单体。合适的(甲基)丙烯酸酯单体具有式i
‑
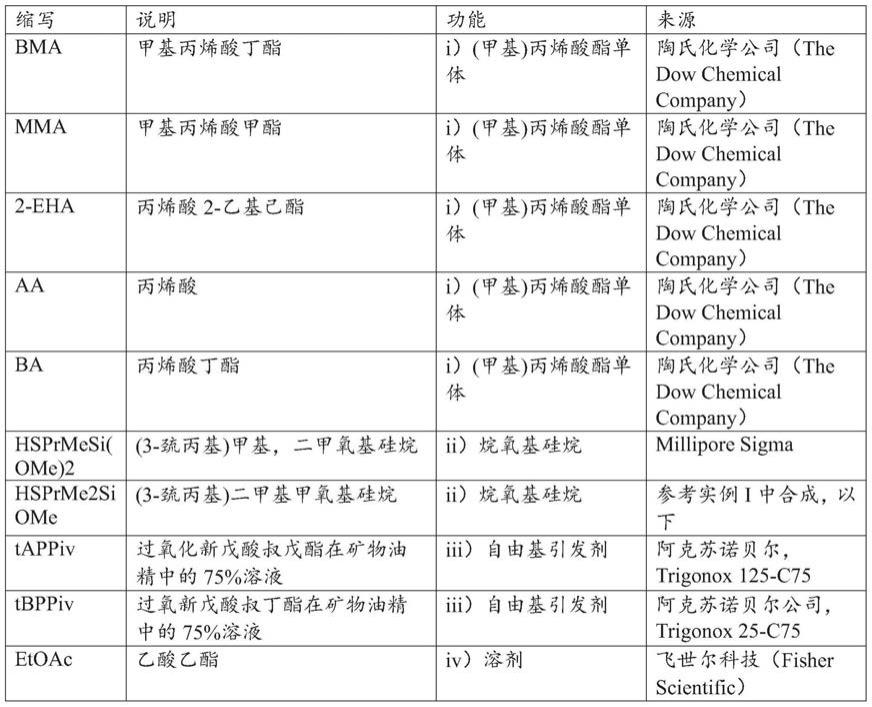
1):其中r1和r2如上所述。合适的(甲基)丙烯酸酯单体是本领域已知的并且可商购获得,并且一些实例示于表1中。按起始材料i)、ii)、iii)和iv)的组合重量计,起始材料i)的量可为20%至99.8%,或者30%至90%,或者70%至75%。
[0056]
表1
‑
示范性(甲基)丙烯酸酯单体
[0057]
缩写(甲基)丙烯酸酯单体商业来源aa丙烯酸millipore sigmaba丙烯酸丁酯millipore sigmabma甲基丙烯酸丁酯millipore sigmaeha丙烯酸2
‑
乙基己酯millipore sigmaehma甲基丙烯酸2
‑
乙基己酯millipore sigmaiba丙烯酸异丁酯millipore sigmaibma甲基丙烯酸异丁酯millipore sigmamma甲基丙烯酸甲酯millipore sigmamaa甲基丙烯酸millipore sigmabzma甲基丙烯酸苄酯millipore sigmapma甲基丙烯酸苯酯millipore sigma
[0058]
在制备烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的方法中,起始材料ii)是
巯基官能的烷氧基硅烷。巯基官能的烷氧基硅烷可具有式ii
‑
1):其中下标a和r3、r4和r5如上所述。合适的巯基官能的烷氧基硅烷是本领域已知的并且是可商购的。这些包括3
‑
巯基丙基三甲氧基硅烷、3
‑
巯基丙基三乙氧基硅烷、11
‑
巯基十一烷基三甲氧基硅烷、(巯基甲基)甲基二乙氧基硅烷、3
‑
巯基丙基甲基二甲氧基硅烷,它们均可购自美国宾夕法尼亚州莫里斯维尔的gelest公司。替代地,如上述那些巯基官能的烷氧基硅烷以及巯基官能的单烷氧基硅烷,例如,3
‑
巯基丙基二甲基甲氧基硅烷和3
‑
巯基丙基二甲基乙氧基硅烷,可通过如美国专利申请2005/0124821和agina,e.v,《美国化学学会应用材料与界面(acs applied materials&interfaces)》,2015,22,11755
‑
11764中公开的那些已知方法合成。按起始材料i)、ii)、iii)和iv)的组合重量计,起始材料ii)的量可为0.1%至50%,或者1%至10%,或者1%至8%。
[0059]
用于制备烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的起始材料iii)是自由基引发剂。自由基引发剂可选自由以下组成的组:iii
‑
1)偶氮化合物,iii
‑
2)过氧化物(例如,羟基过氧化物、过酸和过酸酯,如过氧新戊酸叔烷基酯),和iii
‑
3)其组合。合适的自由基引发剂是本领域已知的,参见例如美国专利8,258,243第2栏第9
‑
34行。替代地,合适的自由基引发剂是可商购的。例如,过氧新戊酸叔烷基酯可从阿克苏诺贝尔公司(akzo nobel)商购获得,例如,过氧新戊酸叔戊基酯可作为trigonox 125
‑
c75获得,过氧新戊酸叔丁基酯可作为trigonox 25
‑
c75获得。按起始材料i)、ii)、iii)和iv)的组合重量计,起始材料iii)的量可为0至5%,或者0.1%至2%,或者1%至2%。
[0060]
溶剂可用于上述方法中。用于制备烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的一种或多种起始材料可在与其它起始材料组合之前溶解于iv)溶剂中。例如,自由基引发剂可以溶解在矿物油精中。替代地,该溶剂可以选自由以下组成的组:iv
‑
1)沸点高于100℃的烃(例如,芳香族烃,如甲苯或二甲苯),iv
‑
2)极性溶剂(如丙酮、甲基乙基酮、乙酸甲酯、乙酸乙酯、乙腈、甲醇、异丙醇或叔丁醇),iv
‑
3)硅油(例如,该硅油可以是聚二烷基硅氧烷,如下文所述),以及iv
‑
4)它们中的两种或更多种的组合。替代地,溶剂可以是例如甲苯,以促进起始材料a)与其它起始材料的组合,从而制备聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。当使用聚二烷基硅氧烷作为溶剂时,聚二烷基硅氧烷可用作制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法中的起始材料。当使用甲苯时,在制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法中可以不包括溶剂交换。按起始材料i)、ii)、iii)和iv)的组合重量计,起始材料iv)的量可为0至70%,或者0至25%。
[0061]
在用于制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法中,起始材料b)为聚二有机硅氧烷。聚二有机硅氧烷选自由以下组成的组:b1)每分子具有至少一个硅键合的脂肪族不饱和基团的不饱和聚二有机硅氧烷,b2)每分子具有至少两个硅键合的羟基的羟基官能的聚二有机硅氧烷,以及b3)b1)和b2)两者的组合。当使用起始材料b1)时,聚(甲基)丙烯酸酯接枝的聚有机硅氧烷具有硅键合的脂肪族不饱和基团和硅键合的聚(甲基)丙烯酸酯基团两者。
[0062]
起始材料b1)为每分子具有至少一个硅键合的脂肪族不饱和基团的不饱和聚二有
机硅氧烷。脂肪族不饱和基团可在末端位置、侧链位置或末端和侧链位置两者中。
[0063]
起始材料b1),即不饱和聚二有机硅氧烷,可以包含单元式b1
‑
1):(r6r
72
sio
1/2
)
b
(r
72
sio
2/2
)
c
(r6r7sio
2/2
)
d
(r
73
sio
1/2
)
e
(r
′
or
72
sio
1/2
)f(r
′
or7sio
2/2
)
g
,其中每个r6为独立选择的脂肪族不饱和烃基,每个r7为独立选择的不含脂肪族不饱和基团的单价烃基,每个r
′
独立地选自由h和r7组成的组,下标b为0、1或2,下标c≥1,下标d≥0,下标e为0、1或2,下标f为0、1或2,并且下标g≥0,条件是数量(b+d)≥1,数量(b+e+f)=2,并且数量(b+c+d+e+f+g)至少为3,或者3至250。
[0064]
r6的脂肪族不饱和烃基可以具有2至18个碳原子并以烯基为例,如乙烯基、烯丙基或己烯基;和炔基,如丙炔基、丁炔基或己炔基。替代地,每个r6可为烯基。替代地,每个r6可为乙烯基。
[0065]
r7的不含脂肪族不饱和基团的单价烃基可以具有1至18个碳原子,并以烷基、芳基和芳烷基为例;或者烷基和芳基。合适的烷基包括甲基、乙基和丙基;替代地甲基。合适的芳基包括苯基。替代地,每个r7可为烷基,如甲基。
[0066]
替代地,在单元式b1
‑
1)中,下标b可为0或2,下标e可为0或2,并且下标g可为0。替代地,下标c可为1至250,下标d可为0至1,下标g可为0至1,并且数量(c+d+g)可为1至250。替代地,数量(b+e)可为2。替代地,下标c可为1至100,或者10至75,或者25至75,或者30至60。替代地,下标d可为0至50,或者0至25,或者0至10,或者0至5。替代地,下标g可为0至50,或者0至25,或者0至10,或者0至5。下标b为0至2,或者下标b可为0,或者下标b可为2。下标e为0至2,或者下标e可为0,或者下标e可为2。下标f为0至2,或者下标f可为0,或者下标f可为2。
[0067]
起始材料b1)可以含有硅键合的脂肪族不饱和烃基和硅键合的羟基。含有硅键合的脂肪族不饱和基团和硅键合的羟基的起始材料b1)的实例包括oh封端的聚甲基乙烯基硅氧烷和oh封端的聚(二甲基/甲基乙烯基)硅氧烷共聚物,其可从gelest商购获得。参见例如《gelest活性硅酮:锻造新的聚合物链(gelest reactive silicones:forging new polymer links)》,2016,https://www.gelest.com/wp
‑
content/uploads/reactive
‑
silicones
‑
no
‑
price
‑
2016.pdf,第11页。替代地,起始材料b1)可以具有硅键合的脂肪族不饱和烃基、硅键合的羟基和硅键合的烷氧基。此类材料的实例包括dowsil
tm 4
‑
7042,其为可从美国密歇根州米德兰市的陶氏硅树脂有限公司(dow silicones corporation)商购获得的羟基封端的聚(二甲基,甲基乙烯基硅氧烷)和α
‑
羟基封端的、ω
‑
甲氧基封端的聚(二甲基,甲基乙烯基硅氧烷)的混合物。当起始材料b1)含有硅键合的脂肪族不饱和烃基和足够的硅键合的羟基时,则起始材料b2)的羟基官能的聚二有机硅氧烷是任选的。
[0068]
替代地,在上述单元式b1
‑
1)中,数量(f+g)可小于2(使得起始材料b1)每分子可具有小于2个硅键合的羟基)。合适的不饱和聚二有机硅氧烷的实例包括
[0069]
b
‑
i)二甲基乙烯基硅氧基封端的聚二甲基硅氧烷,
[0070]
b
‑
ii)二甲基乙烯基硅氧基封端的聚(二甲基硅氧烷/甲基乙烯基硅氧烷),
[0071]
b
‑
iii)二甲基乙烯基硅氧基封端的聚甲基乙烯基硅氧烷,
[0072]
b
‑
iv)三甲基硅氧基封端的聚(二甲基硅氧烷/甲基乙烯基硅氧烷),
[0073]
b
‑
v)三甲基硅氧基封端的聚甲基乙烯基硅氧烷,
[0074]
b
‑
vi)二甲基乙烯基硅氧基封端的聚(二甲基硅氧烷/甲基乙烯基硅氧烷),
[0075]
b
‑
vii)二甲基乙烯基硅氧基封端的聚(二甲基硅氧烷/甲基苯基硅氧烷),
[0076]
b
‑
viii)二甲基乙烯基硅氧基封端的聚(二甲基硅氧烷/二苯基硅氧烷),
[0077]
b
‑
ix)苯基、甲基、乙烯基
‑
硅氧基封端的聚二甲基硅氧烷,
[0078]
b
‑
x)二甲基己烯基硅氧基封端的聚二甲基硅氧烷,
[0079]
b
‑
xi)二甲基己烯基硅氧基封端的聚(二甲基硅氧烷/甲基己烯基硅氧烷),
[0080]
b
‑
xii)二甲基己烯基硅氧基封端的聚甲基己烯基硅氧烷,
[0081]
b
‑
xiii)三甲基硅氧基封端的聚(二甲基硅氧烷/甲基己烯基硅氧烷),
[0082]
b
‑
xiv)三甲基硅氧基封端的聚甲基己烯基硅氧烷
[0083]
b
‑
xv)二甲基己烯基
‑
硅氧基封端的聚(二甲基硅氧烷/甲基己烯基硅氧烷),
[0084]
b
‑
xvi)二甲基乙烯基硅氧基封端的聚(二甲基硅氧烷/甲基己烯基硅氧烷)
[0085]
b
‑
xvii)其组合。乙烯基官能的聚二有机硅氧烷是可用的,参见例如《gelest反应性硅酮:锻造新的聚合物链》,2016,https://www.gelest.com/wp
‑
content/uploads/reactive
‑
silicones
‑
no
‑
price
‑
2016.pdf,第8
‑
11页和第15
‑
16页。当起始材料b1)不含有足够的硅键合的羟基时,在上述方法中使用起始材料b2)。用于该方法中的起始材料b1)的量取决于各种因素,包括b1)是否具有末端脂肪族不饱和基团、侧链脂肪族不饱和基团,或末端脂肪族不饱和基团和侧链脂肪族不饱和基团两者,然而,起始材料b1)的量足以向用于制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法的步骤i)中的所有起始材料提供0.1%至10%,或者0.1%至2%的脂肪族不饱和基团。替代地,按起始材料a)和b)的组合重量计,起始材料b1)的量可为0.5%至5%,或者1%至4%,或者1%至3%。
[0086]
替代地,当b1)具有羟基官能团且不使用起始材料b2)时,起始材料b1)可以以较高量存在,例如高达90%。
[0087]
在制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法中,起始材料b2)为每分子具有至少两个硅键合的羟基的羟基官能聚二有机硅氧烷。羟基可以在末端位置、在侧链位置或两者中。起始材料b2)可以包含单元式b2
‑
1):(r
82
sio
2/2
)
h
(r
83
sio
1/2
)
i
(hor
82
sio
1/2
)
j
,其中每个r8为独立选择的不含脂肪族不饱和基团的单价烃基,下标j为1或2,下标i为0或1,数量(j+i)=2,下标h≥1,并且数量(h+i+j)至少为3,或者3至250,或者3至100。替代地,下标h可为1至250,或者1至100。替代地,i可为0,且j可为2。r8的单价烃基包括烷基、芳基和芳烷基;或者烷基和芳基。合适的烷基包括甲基、乙基和丙基;替代地甲基。合适的芳基包括苯基。替代地,每个r8可为烷基,如甲基。起始材料b2)的实例包括羟基封端的聚二甲基硅氧烷、羟基封端的聚(二甲基/二苯基)硅氧烷共聚物、羟基封端的聚(二甲基/甲基苯基)硅氧烷共聚物。替代地,合适的双羟基封端的聚二甲基硅氧烷可购自美国密歇根州米德兰的陶氏硅树脂有限公司。示范性羟基官能的聚二有机硅氧烷是可商购的,其包括在《gelest反应性硅酮:锻造新的聚合物链》中的硅烷醇官能的聚合物,2016,https://www.gelest.com/wp
‑
content/uploads/reactive
‑
silicones
‑
no
‑
price
‑
2016.pdf,第22页和第24
‑
25页。按起始材料a)和b)的组合重量计,起始材料b2)的用量可为80%至95%,或者85%至95%;或者87%至94%,或者89%至94%,基于相同的基准。
[0088]
在用于制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法中,起始材料c)为磷腈缩合反应催化剂,如磷腈卤化物。在不希望受到理论约束的情况下,据认为当在该方法中使用磷腈缩合反应催化剂时,可以最小化环状硅氧烷副产物(如八甲基环四硅氧烷)的形成。
[0089]
磷腈缩合反应催化剂以美国专利9,051,428中公开的催化剂为例。示范性磷腈缩合反应催化剂可以每分子含有至少一个
‑
(n=p<)
‑
单元,并且可以是具有至多10个这样的磷腈单元的低聚物,例如具有平均1.5至5个磷腈单元。磷腈缩合反应催化剂可以例如是卤代磷腈,如氯磷腈(磷腈氯化物)、含氧卤代磷腈,或磷腈的离子衍生物,如磷腈盐,如磷腈卤化物的离子衍生物,例如全氯低磷腈盐。
[0090]
一种合适类型的磷腈缩合反应催化剂是含氧卤代磷腈,如含氧氯代磷腈。这样的含氧氯磷腈可以例如具有式c
‑
1):或c
‑
2):在式c
‑
1)和c
‑
2)中,下标p可以具有1至10,或者1至5的平均值。催化剂还可以包含式c
‑
2)的催化剂的互变异构体。另一种类型的合适的含氧氯磷腈具有式c
‑
3):其中r9表示经由氧键合到磷的有机硅部分,例如式c
‑
4):的磷腈催化剂,其中每个r
10
表示具有1至18个碳原子的单价烃基或具有1至18个碳原子的单价卤代烃基,并且下标q具有1至10,或者1至5的平均值。催化剂还可以包含这种含有机硅的磷腈的缩合产物。任何上述含氧磷腈中的所有或一些氯原子可以被基团q替代,其中q表示选自由以下组成的组的部分:羟基、如烷氧基或芳氧基的单价有机基团、除氯之外的卤素原子、有机硅基团和含磷基团。
[0091]
另一种合适类型的磷腈催化剂是式c
‑
5):的全氯低聚磷腈盐,其中下标o具有1至10的平均值,并且z
‑
表示阴离子。替代地,下标o可以具有1至6的平均值,并且替代地,下标o可以具有2的平均值。阴离子可以是络合阴离子并且可以例如具有式mx
(v+1)
,其中m是在鲍林标度上具有1.0至2.0的电负性和化合价v的元素,并且
为独立选择的二价烃基,每个r
″
独立地为(甲基)丙烯酸酯聚合物或共聚物,每个r6为独立选择的脂肪族不饱和单价烃基,每个r7为独立选择的不含脂肪族不饱和基团的单价烃基,下标p≥0,下标q≥0,下标k≥0,数量(p+q+k)≥1,下标r≥0,下标s≥0,下标t≥0,下标u≥0,数量(r+t)≥2,并且数量(p+q+k+r+s+t+u)足以向聚(甲基)丙烯酸酯接枝的聚有机硅氧烷提供至少50kda的分子量。
[0099]
替代地,下标w为1或2。替代地,下标p为0至2,或者1或2。替代地,下标q为0至100。替代地,下标k<5;替代地,k可为0、1或2;并且替代地,k=0。替代地,数量(p+q+k)为1至100。替代地,下标r为0至2。替代地,下标s为0至100。替代地,下标t为0至100。替代地,下标u为0至2。替代地,数量(p+q+r+s+t+u)足以向聚(甲基)丙烯酸酯接枝的聚有机硅氧烷提供50kda至1,000kda,或者60kda至1,000kda,或者50kda至600kda,或者60kda至300kda的分子量。
[0100]
替代地,在上述聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的式中,每个r3为1至6个碳原子的烷基。替代地,每个r4为1至6个碳原子的烷基。替代地,每个r5为2至8个碳原子的亚烷基。替代地,每个r6为选自乙烯基、烯丙基和己烯基的烯基。替代地,每个r7为1至6个碳原子的烷基。
[0101]
具有硅键合的聚(甲基)丙烯酸酯基团和硅键合的脂肪族不饱和基团的聚(甲基)丙烯酸酯接枝的聚有机硅氧烷可用于氢化硅烷化反应可固化组合物中。在不希望受到理论约束的情况下,据认为聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的硅键合的脂肪族不饱和基团中的一些或全部可以与硅键合的氢原子反应。一种氢化硅烷化反应可固化组合物包含:
[0102]
i)具有硅键合的聚(甲基)丙烯酸酯基团和硅键合的脂肪族不饱和基团的聚(甲基)丙烯酸酯接枝的聚有机硅氧烷(如上所述),
[0103]
ii)每分子具有至少3个硅键合的氢原子的有机氢硅交联剂,以及
[0104]
iii)氢化硅烷化反应催化剂。氢化硅烷化反应可固化组合物可任选地进一步包含一种或多种附加成分,如iv)氢化硅烷化反应催化剂抑制剂,v)聚有机硅酸盐树脂,vi)间隔基;vii)增量剂、增塑剂或其组合;viii)填料;ix)填料处理剂;x)杀生物剂;xi)阻燃剂;xii)表面改性剂;xiii)增链剂;xiv)封端剂;xv)助熔剂;xvi)抗老化添加剂;xvii)颜料;xviii)酸受体;xix)流变添加剂;xx)媒剂(例如,溶剂或稀释剂);xxi)表面活性剂;xxii)腐蚀抑制剂,及其组合。合适的有机氢硅交联剂、氢化硅烷化反应催化剂和额外的起始材料是本领域已知的,例如参见美国专利申请2014/0228570,第[0096]段至第[0173]段,其通过引用并入本文。
[0105]
在氢化硅烷化反应组合物中,起始材料ii)为有机氢硅交联剂,矽每分子平均具有3个或更多个硅键合氢原子的化合物。起始材料ii)可以包含硅烷和/或聚有机氢硅氧烷化合物。起始材料ii)在组合物中的量取决于各种因素,包括起始材料ii)的sih含量、i)聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的不饱和基团含量和所需组合物的反应产物的性质,然而,起始材料ii)的量可以足以提供起始材料ii)中的sih基团与起始材料i)中的脂肪族不饱和基团的摩尔比(通常称为sih∶vi比率)范围为0.3∶1至40∶1,或者0.1∶1至35∶1,或者0.1∶10至10∶1。起始材料ii)可以具有直链、支链、环状或树脂状结构。当起始材料ii)是聚合物时,则起始材料ii)可以是均聚物或共聚物。起始材料ii)中的硅键合的氢原子可位于末端、侧链或位于末端和侧链位置。起始材料ii)可以是一种sih官能化合物。替代地,起始材料
ii)可以包含两种或更多种sih官能化合物的组合。起始材料ii)可以是两种或更多种有机氢聚硅氧烷,其在以下特性中的至少一个方面不同:结构、平均分子量、粘度、硅氧烷单元和序列。
[0106]
起始材料ii)的有机氢硅化合物可以包含聚有机氢硅氧烷,聚有机氢硅氧烷包含硅氧烷单元,其包括但不限于hr
132
sio
1/2
、r
133
sio
1/2
、hr
13
sio
2/2
、r
132
sio
2/2
、r
13
sio
3/2
、hsio
3/2
和sio
4/2
单元。在前述式中,每个r
13
为独立选择的单价烃基。适合于r
13
的单价烃基包括上文针对r7所述的不含脂肪族不饱和基团的基团。
[0107]
起始材料ii)可以包含以下的聚有机氢硅氧烷:
[0108]
式ii
‑
1):r
133
sio(r
132
sio)
aa
(r
13
hsio)
bb
sir
133
,
[0109]
式ii
‑
2):r
132
hsio(r
132
sio)
cc
(r
13
hsio)
dd
sir
132
h,或
[0110]
其组合。
[0111]
在上式ii
‑
1)和ii
‑
2)中,下标aa具有0至2000的平均值范围,下标bb具有2至2000的平均值范围,下标cc具有0至2000的平均值范围,并且下标dd具有0至2000的平均值范围。每个r
13
独立地为单价烃基,如不含脂肪族不饱和基团的单价烃基,如上文针对r7所述的单价烃基。r
13
可以具有1至18个碳原子。r
13
可为烷基或芳基。合适的烷基包括甲基和乙基,或者甲基。合适的芳基包括苯基。
[0112]
用于起始材料ii)的聚有机氢硅氧烷以以下为例:
[0113]
a)二甲基氢硅氧基封端的聚二甲基硅氧烷,
[0114]
b)二甲基氢硅氧基封端的聚(二甲基硅氧烷/甲基氢硅氧烷),
[0115]
c)二甲基氢硅氧基封端的聚甲基氢硅氧烷,
[0116]
d)三甲基硅氧基封端的聚(二甲基硅氧烷/甲基氢硅氧烷),
[0117]
e)三甲基硅氧基封端的聚甲基氢硅氧烷,
[0118]
f)基本上由h(ch3)2sio
1/2
单元和sio
4/2
单元组成的树脂,以及
[0119]
g)其组合。
[0120]
制备适合用作起始材料ii)的直链、支链和环状有机氢聚硅氧烷的方法,如有机卤代硅烷的水解和缩合,是本领域公知的。制备适用作起始材料ii)的有机氢聚硅氧烷树脂的方法也是众所周知的,以美国专利5,310,843;4,370,358;和4,707,531为例。
[0121]
此类有机氢硅化合物可商购获得,并且包括syl
‑
off
tm sl2 crosslinker和syl
‑
off
tm sl12 crosslinker,两者均可从美国密歇根州米德兰的陶氏硅树脂有限公司商购获得。
[0122]
组合物中起始材料ii)的确切量取决于各种因素,包括i)聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的反应性,起始材料ii)的类型和量,以及任何额外的起始材料(除起始材料ii)之外)(如果存在)的类型和量。然而,按组合物中所有起始材料的总重量计,组合物中起始材料ii)的量可以在>0%至25%,或者0.1%至15%,或者1%至5%的范围内。
[0123]
用于起始材料iii)的氢化硅烷化反应催化剂是本领域已知的并且是可商购的。氢化硅烷化反应催化剂包括铂族金属催化剂。此类氢化硅烷化催化剂可以是选自铂、铑、钌、钯、锇和铱的金属。可替代地,氢化硅烷化催化剂可以是例如以下等此类金属的化合物:氯三(三苯基膦)铑(i)(威尔金森氏催化剂(wilkinson
′
s catalyst))、如[1,2
‑
双(二苯基膦基)乙烷]二氯二铑或[1,2
‑
双(二乙基膦基)乙烷]二氯二铑的二膦铑螯合物、氯铂酸(斯皮
尔氏催化剂(speier
′
s catalyst))、六水合氯铂酸、二氯化铂以及所述化合物与低分子量有机聚硅氧烷的络合物或在基质或核壳型结构中微囊化的铂族金属化合物。铂与低分子量有机聚硅氧烷的络合物包含1,3
‑
二乙烯基
‑
1,1,3,3
‑
四甲基二硅氧烷与铂的络合物(卡斯特催化剂(karstedt
′
s catalyst))。这些络合物可以被微囊化在树脂基质中。可替代地,氢化硅烷化催化剂可以包括1,3
‑
二乙烯基
‑
1,1,3,3
‑
四甲基二硅氧烷与铂的络合物。示范性氢化硅烷化催化剂描述于美国专利3,159,601;3,220,972;3,296,291;3,419,593;3,516,946;3,814,730;3,989,668;4,784,879;5,036,117;和5,175,325;和ep 0 347 895 b。微胶囊化的氢化硅烷化催化剂及其制备方法是本领域已知的,如美国专利4,766,176和5,017,654中所示例的。
[0124]
本文所用的催化剂的量将取决于各种因素,包括起始材料a)和b)的选择以及它们各自的硅键合氢原子和脂肪族不饱和基团的含量,以及是否存在抑制剂,然而,催化剂的量足以催化硅和脂肪族不饱和基团的氢化sih化反应,或者催化剂的量足以按所有起始材料的组合重量计提供1ppm至1000ppm的铂族金属,或者基于相同基准提供5ppm至100ppm的铂族金属。
[0125]
用于起始材料iv的氢化硅烷化反应可固化组合物的抑制剂以炔醇为例,如甲基丁炔醇、乙炔基环己醇、二甲基己炔醇和3,5
‑
二甲基
‑1‑
己炔
‑3‑
醇、1
‑
丁炔
‑3‑
醇、1
‑
丙炔
‑3‑
醇、2
‑
甲基
‑3‑
丁炔
‑2‑
醇、3
‑
甲基
‑1‑
丁炔
‑3‑
醇、3
‑
甲基
‑1‑
戊炔
‑3‑
醇、3
‑
苯基
‑1‑
丁炔
‑3‑
醇、4
‑
乙基
‑1‑
辛炔
‑3‑
醇、3,5
‑
二甲基
‑1‑
己炔
‑3‑
醇和1
‑
乙炔基
‑1‑
环己醇,及其组合;环烯基硅氧烷,如甲基乙烯基环硅氧烷,以1,3,5,7
‑
四甲基
‑
1,3,5,7
‑
四乙烯基环四硅氧烷、1,3,5,7
‑
四甲基
‑
1,3,5,7
‑
四己烯基环四硅氧烷为例,及其组合;烯
‑
炔化合物,如3
‑
甲基
‑3‑
戊烯
‑1‑
炔、3,5
‑
二甲基
‑3‑
己烯
‑1‑
炔;三唑类,如苯并三唑;膦;硫醇;肼;胺化合物,如四甲基乙二胺;富马酸酯,如富马酸二烷基酯、富马酸二烯基酯、富马酸二烷氧基烷基酯;马来酸二烯丙酯,如马来酸二烯丙酯;腈;醚类;一氧化碳;烯烃,如环辛二烯、二乙烯基四甲基二硅氧烷;醇,如苯甲醇;及其组合。
[0126]
替代地,抑制剂可以是硅烷化的炔属化合物。在不希望受到理论约束的情况下,据认为与来自不含有硅烷化炔属化合物或含有有机炔属醇稳定剂(例如上述那些)的组合物的氢化硅烷化的反应产物相比,添加硅烷化炔属化合物降低由组合物的氢化硅烷化反应制备的反应产物的黄化。
[0127]
硅烷化炔属化合物的实例为(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)三甲基硅烷、((1,1
‑
二甲基
‑2‑
丙炔基)氧基)三甲基硅烷、双(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)二甲基硅烷、双(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)硅烷甲基乙烯基硅烷、双((1,1
‑
二甲基
‑2‑
丙炔基)氧基)二甲基硅烷、甲基(三(1,1
‑
二甲基
‑2‑
丙炔基氧基))硅烷、甲基(三(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基))硅烷、(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)二甲基苯基硅烷、(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)二甲基己烯基硅烷、(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)三乙基硅烷、双(3
‑
甲基
‑1‑
丁炔
‑3‑
氧基)甲基三氟丙基硅烷、(3,5
‑
二甲基
‑1‑
己炔
‑3‑
氧基)三甲基硅烷、(3
‑
苯基
‑1‑
丁炔
‑3‑
氧基)二苯基甲基硅烷、(3
‑
苯基
‑1‑
丁炔
‑3‑
氧基)二甲基苯基硅烷、(3
‑
苯基
‑1‑
丁炔
‑3‑
氧基)二甲基乙烯基硅烷、(3
‑
苯基
‑1‑
丁炔
‑3‑
氧基)二甲基己烯基硅烷、(环己基
‑1‑
乙炔
‑1‑
氧基)二甲基己烯基硅烷、(环己基
‑1‑
乙炔
‑1‑
氧基)二甲基乙烯基硅烷、(环己基
‑1‑
乙炔
‑1‑
氧基)二苯基甲基硅烷、(环己基
‑1‑
乙炔
‑1‑
氧基)三甲基硅烷及其组合。替代地,成分(iv)示例为甲基(三(1,1
‑
二甲基
‑2‑
丙炔基
氧基))硅烷、((1,1
‑
二甲基
‑2‑
丙炔基)氧基)三甲基硅烷或其组合。硅烷化炔属化合物可以通过本领域已知的方法制备,如通过使上述炔属醇与氯硅烷在酸受体存在下反应而使其硅烷化。
[0128]
添加到氢化硅烷化反应可固化组合物中的抑制剂的量将取决于各种因素,包括组合物的所需适用期、组合物是否为单组分组合物或多组分组合物、所使用的具体抑制剂。然而,当存在时,按氢化硅烷化反应可固化组合物中所有起始材料的重量计,抑制剂的量可以在0%至1%,或者0%至5%,或者0.001%至1%,或者0.01%至0.5%,或者0.0025%至0.025%的范围内。
[0129]
氢化硅烷化反应可固化组合物可任选地进一步包含起始材料v)聚有机硅酸盐树脂。聚有机硅酸盐树脂是基本上由r
143
sio
1/2
单元和sio
4/2
单元组成的mq树脂,其中每个r
14
独立地选自由羟基和单价烃基组成的组。r
14
的单价烃基可以选自由以下组成的组:1至18个碳原子的烷基、2至18个碳原子的烯基,和6至18个碳原子的芳基或芳烷基。合适的烷基、烯基、芳基和芳烷基如下文所定义。替代地,每个r
14
可为羟基或烷基,如甲基。
[0130]
替代地,聚有机硅酸盐树脂可以包含单元式(ii):(r
152
r
16
sio
1/2
)
x
(r
153
sio
1/2
)
y
(sio
4/2
)
z
,其中r
15
为烷基、芳基或芳烷基,并且r
16
为2至18个碳原子的烯基,如乙烯基、烯丙基或己烯基,下标x≥0,下标y≥0,下标z为>0,数量(x+y)为>0,并且下标x、y和z具有使得0.9≤(x+y)/z≤1.3的值。
[0131]
树脂可以含有平均3至30摩尔百分比,或者0.1至30摩尔百分比,或者0.1至5摩尔百分比,或者3至10摩尔百分比的烯基。树脂中烯基的摩尔百分比是树脂中含烯基的硅氧烷单元的摩尔数与树脂中硅氧烷单元的总摩尔数之比乘以100。
[0132]
聚有机硅酸盐树脂的mn通常大于3,000da,或者>3,000da至8,000da,或者4,500至7,500da。可采用下述方法通过gpc测量mn。
[0133]
制备树脂的方法是本领域公知的。例如,树脂可以通过使用至少一种含烯基的封端试剂处理由daudt等人的二氧化硅水溶胶封端方法生产的树脂共聚物来制备。在美国专利2,676,182中公开了daudt等人的方法。
[0134]
daudt等人的方法涉及使二氧化硅水溶胶在酸性条件下与如三甲基氯硅烷的可水解的三有机硅烷、如六甲基二硅氧烷的硅氧烷或其混合物反应,并回收具有m
‑
单元和q
‑
单元的共聚物。所得的共聚物通常含有2重量%到5重量%的羟基。
[0135]
通常含有小于2%的硅键合的羟基的树脂可通过使daudt等天的产物与含烯基的封端剂和/或不含脂肪族不饱和度的封端剂反应来制备,其量足以在最终产物中提供3至30摩尔%的不饱和有机基团。封端剂的实例包含但不限于硅氮烷、硅氧烷和硅烷。合适的封端剂在本领域中是已知的并在美国专利4,584,355;4,591,622;和4,585,836中例示。可使用单一封端剂或此类封端剂的混合物来制备树脂。
[0136]
各种合适的聚有机硅酸盐树脂可商购自如美国密歇根州米德兰市的陶氏硅树脂有限公司,美国纽约奥尔巴尼的迈图高新材料公司(momentive performance materials)和美国新泽西州东布朗士维克的美国蓝星硅树脂公司(bluestar silicones usa corp.)等来源。例如,dowsil
tm mq
‑
1600固体树脂、dowsil
tm mq
‑
1601固体树脂,和dowsil
tm 1250表面活性剂,dowsil
tm 7466树脂,和dowsil
tm 7366树脂适用于本文,所有这些都可商购自美国密歇根州米德兰的陶氏硅树脂有限公司。这样的树脂可以在有机溶剂中提供。
[0137]
实例
[0138]
这些实例旨在说明本发明,而不应被解释为限制权利要求中阐述的范围。实例中使用的起始材料示于下表2中。
[0139]
表2.起始材料
[0140]
[0141][0142]
在这一参考实例a中,提供了用于制备大分子单体实例1(37bma/63mma,命名法指重量%,bma=甲基丙烯酸丁酯,mma=甲基丙烯酸甲酯)的方法的描述,其中基于单体的(3
‑
巯基丙基)甲基二甲氧基硅烷为1.9重量%。该方法用于通过改变合适的起始材料及其量来制备实例mm
‑
2至mm
‑
5中的其它烷氧基硅基官能的(甲基)丙烯酸酯大分子单体,如下表3所示。乙酸乙酯(etoac)经分子筛干燥,其它成分按供应使用。制备含有etoac(75g)、mma(233g)、bma(136.9g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(7g)的单体混合物。将etoac
(100g)和12.5%的单体混合物(56.5g)添加到配备有冷凝器和顶部混合器的4颈1升玻璃反应器中。将混合物加热至77℃并用氮气鼓泡30分钟。将在etoac(10g)中的trigonox 125
‑
c75(tappiv,1g)添加到反应器中并保持5分钟。将温度缓慢升至85℃,然后以0.1ml/min的速度进料单体混合物180分钟,并将引发剂溶液(20g的etoac;2g的trigonox 125
‑
c75)进料240分钟(180分钟加上在单体进料结束后60分钟过量进料)。在引发剂进料结束后,将反应在85℃下保持180分钟,然后使所得混合物冷却至室温。将所得混合物在大量过量的己烷中沉淀三次并在每个沉淀步骤后在真空烘箱中干燥。最后的真空干燥进行至少24小时并加热至80至100℃以除去残留的单体和溶剂。实例4偏离了这一纯化方案,因为bma低聚物可溶于己烷,因此使用旋转蒸发器对其进行干燥。表3总结了每个实例使用这一方法制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的组成和特征。
[0143]
在这一参考实例b中,提供了用于生产具有0.2重量%基于单体的(3
‑
巯基丙基)甲基二甲氧基硅烷的大分子单体实例mm
‑
6(100bma)的方法的描述。该方法用于通过改变适当的起始材料及其量来制备另一种烷氧基硅基官能的(甲基)丙烯酸酯大分子单体(实例mm
‑
7),如下表3所示。制备溶剂(5g)、bma(400g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(0.7g)的单体混合物。将溶剂(80g)添加到配备有冷凝器和顶部混合器的4颈1升玻璃反应器中。将溶剂加热至85℃,然后在2小时的时间段内进料单体混合物(400.7g),5分钟后开始添加引发剂溶液(20g的溶剂和11g的trigonex 25
‑
c75 tbppiv),并在4小时的时间段内进料。在3小时后,添加ba(丙烯酸丁酯,4g)丸。在引发剂进料结束后,将反应在85℃下保持80分钟,然后使混合物冷却至室温并收集。将反应混合物用50g的甲苯稀释,然后收集。
[0144]
在这一参考实例c中,提供了用于生产具有8.2重量%基于单体的(3
‑
巯基丙基)甲基二甲氧基硅烷的实例mm
‑
8(37bma/63mma)的方法的描述。该方法用于通过改变适当的起始材料及其量来制备另一种烷氧基硅基官能的(甲基)丙烯酸酯大分子单体(实例mm
‑
9),如下表3所示。制备甲苯(5g)、mma(252g)、bma(148g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(36g)的单体混合物。将甲苯(80g)添加到配备有冷凝器和顶部混合器的4颈1升玻璃反应器中。将溶剂加热至85℃,然后在1小时的时间段内进料单体混合物(441g),在3小时的时间段内进料引发剂溶液(30g的甲苯和16.5g的trigonex 25
‑
c75 tbppiv)。在2小时后,在30分钟的时间段内添加ba(丙烯酸丁酯,10g)。在引发剂进料结束后,将反应在85℃下保持80分钟,然后使混合物冷却至室温并收集。对于x1,在收集之前用160g的甲苯稀释反应混合物,对于x2,不需要额外的稀释。
[0145]
表3.在参考实例a
‑
c中制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的概述
[0146]
[0147]
*bom=基于单体
[0148]
用于生产具有8.3重量%基于单体的(3
‑
巯基丙基)甲基二甲氧基硅烷的实例mm
‑
10(90bma/10aa)的方法。制备甲苯(10g)、bma(350g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(0.7g)的单体混合物。将溶剂(80g)添加到配备有冷凝器和顶部混合器的4颈1升玻璃反应器中。将溶剂加热至85℃。一旦达到该温度,作为bma(10g)、aa(10g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(1.8g)的初始进料,然后将单体混合物(384.2g)添加到反应器中。5分钟后,在2小时的时间段内进料单体混合物,并在4小时的时间段内进料引发剂溶液(20g的溶剂和11g的trigonex 25
‑
c75 tbppiv)。在引发剂进料结束后,将反应在85℃下保持80分钟,然后使混合物冷却至室温并收集。将反应混合物用100g的甲苯稀释,然后收集。
[0149]
用于生产具有10.1重量%基于单体的(3
‑
巯基丙基)甲基二甲氧基硅烷的实例mm
‑
11(100eha)的方法。制备甲苯(5g)、eha(350)和(3
‑
巯基丙基)甲基二甲氧基硅烷(34.2g)的单体混合物。将溶剂(80g)添加到配备有冷凝器和顶部混合器的4颈1升玻璃反应器中。将溶剂加热至85℃。一旦达到该温度,作为eha(20g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(1.8g)的初始进料,然后将单体混合物(384.2g)添加到反应器中。5分钟后,在2小时的时间段内进料单体混合物,并在4小时的时间段内进料引发剂溶液(20g的溶剂和11g的trigonex 25
‑
c75 tbppiv)。在引发剂进料结束后,将反应在85℃下保持80分钟,然后使混合物冷却至室温并收集。
[0150]
用于生产具有8.3重量%基于单体的(3
‑
巯基丙基)甲基二甲氧基硅烷的实例mm
‑
12(90eha/10aa)的方法。制备eha(360g)、aa(40g)和(3
‑
巯基丙基)甲基二甲氧基硅烷(36g)的单体混合物。将溶剂(80g)添加到配备有冷凝器和顶部混合器的4颈1升玻璃反应器中。将溶剂加热至85℃。一旦达到该温度,在2小时的时间段内进料单体混合物,并在4小时的时间段内进料引发剂溶液(20g的溶剂和11g的trigonex 25
‑
c75 tbppiv)。在引发剂进料结束后,将反应在85℃下保持80分钟,然后使混合物冷却至室温并收集。
[0151]
表4.实例mm
‑
10至mm
‑
12的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的概述
[0152][0153]
表5.烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的表征结果
[0154]
实例idmn/mwa(kda)pdia残留单体(ppm)b%硅酮缩合物ctg(℃)emm
‑
17.7/15.12.075mma/341bma072mm
‑
21.9/3.01.6392mma/1880bma933mm
‑
38.0/15.62.0382bma021mm
‑
43.2/4.81.5139mma/5660bma0
‑
23mm
‑
57.9/12.01.5751bma046mm
‑
614/815.8nddndndmm
‑
70.62/6.510.5ndndnd
mm
‑
82.3/4.21.8153bma&mmandndmm
‑
92.3/4.21.8184bmandndmm
‑
102.4/5.02.1160bma/0aand2.9mm
‑
111.9/3.21.7501ehand
‑
77mm
‑
122.4/4.51.9130ehand
‑
62
[0155]
a.使用thf作为洗脱液在gpc上测量的分子量分布。
[0156]
b.通过hs
‑
gc测量的残留单体浓度。除了内标物(~20mg)之外,将反应混合物的样品(~20mg)添加到小瓶中,并将小瓶压紧。通过加热小瓶并对顶部空间取样以测定残留单体的ppm水平来进行顶部空间气相色谱法。
[0157]
c.通过
29
si nmr测量的硅氧烷部分的缩合百分比。
[0158]
d.nd=未确定
[0159]
e.t
g
,玻璃化转变温度,用dsc在第2个加热周期上测量。
[0160]
在这一参考实例d中,上述实例mm
‑
1至mm
‑
12的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体用于制备聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。程序如下:在实验开始之前,将具有搅拌棒和叶片的1升4颈圆底烧瓶预称重并记录。向这个烧瓶中添加181.0克oh封端的pdms 1,5.65克vi封端的pdms 1,11.41克实例mm
‑
5(其具有mn=7.9kda且bma=70%),和238.5克甲苯,装入配备有顶置式机械搅拌器、热电偶和具有附接至氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的圆底烧瓶中。将加热套插入温度控制器中以防止反应混合物加热至大于120℃的温度。当釜温度达到80℃时,添加0.47ml磷腈催化剂1。随着反应混合物继续加热,在迪安斯塔克设备中收集水、甲醇和甲苯馏出物。在蒸馏足够的甲苯以获得60%固体溶液的最终浓度之后中断加热,通常通过除去100g馏出物。当达到60%固体的最终浓度时,在搅拌下向反应烧瓶中添加0.20ml三辛胺以中和反应混合物,并使所得混合物冷却至室温。冷却至室温后,称重并记录具有搅拌棒和叶片的圆底烧瓶。然后基于质量平衡(预重量与最终重量之间的差)计算nvc,假定所有聚合物含量保留在烧瓶中。另外,计算溶液的乙烯基含量。
[0161]
使用上述参考实例d中的程序重复实例gp
‑
2至gp
‑
20,不同之处在于用不同的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体代替实例5中制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体并改变烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的wt%。这些实例列于表6中。
[0162]
表6.合成的pdms和所选性质的概述。
[0163][0164]
a
通过gpc分析测定的mw和pdi,
b
nd=未确定
[0165]
*可能存在超高mw材料,但在gpc注射中滤出
[0166]
除了上述聚丙烯酸酯接枝的pdms实例之外,下面示出了具有各种结构变化的另外几个实例。
[0167]
实例gp
‑
24
‑
具有聚丙烯酸酯末端的聚(二甲基,甲基乙烯基)硅氧烷共聚物的合成
[0168]
将甲基丙烯酸丁酯(200g)和hsprme2siome(5g)的单体混合物添加到广口瓶中。引发剂溶液由tbppiv(5.5g)和甲苯(10g)制成。向四颈圆底烧瓶中加入40克甲苯。将甲苯加热至85℃,此时分别在2和4小时内进料单体和引发剂混合物。单体混合物进料开始后3小时向烧瓶中添加丙烯酸丁酯(2g)。在引发剂进料结束后,将反应在85℃下保持80分钟。这产生了具有6.6kda100bma的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体。
[0169]
在实验之前称量一个具有搅拌棒和叶片的1l 4颈圆底烧瓶(571.50g)。在图4的方案4所示的典型合成中,将181.0g的oh封端的pdms 1、2.46g的4
‑
7042、10.098g的上述制备的具有100bma的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体和238g的甲苯装入烧瓶中,该烧瓶配备有热电偶和具有适用于氮气鼓泡器的水冷冷凝器的迪安斯塔克设备。在氮气层下将釜温加热至80至100℃,并添加0.47ml的磷腈催化剂1。然后当釜温升高至103℃时,在迪安斯塔克设备中收集水、甲醇和甲苯。在蒸馏出总共67.0g馏出物之后停止加热,其中釜温升高到113℃。反应运行1小时14分钟,从在80℃下添加催化剂的时间直到反应淬灭。由于聚合物的粘度,在仅67.0克馏出物之后停止反应。接下来,在搅拌下将0.2ml三辛胺(中和剂)添加到烧瓶中,并使所得混合物冷却至室温。在将釜温冷却至室温后,再次称量具有搅拌棒和叶片的烧瓶(930.0g)。基于质量平衡计算的非挥发物含量(nvc)为54.0%,假定所有聚合物内容物保留在烧瓶中。所得聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的乙烯基含量为
0.069%。
[0170]
实例gp
‑
25——具有甲基末端的聚丙烯酸酯接枝的乙烯基pdms侧链。
[0171]
在实验之前称量一个具有搅拌棒和叶片的1l 4颈圆底烧瓶(571.49g)。在图5的方案5所示的典型合成中,将181.0g的oh封端的pdms 1、2.46g的4
‑
7042、2.70g的md
22
m、18.62g的实例4中制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体和238g的甲苯装入烧瓶中,该烧瓶还配备有热电偶和具有适用于氮气鼓泡器的水冷冷凝器的迪安斯塔克设备。在氮气层下将釜温加热至80至100℃并添加0.47ml磷腈催化剂1。然后当釜温升高至103℃时,在迪安斯塔克设备中收集水、甲醇和甲苯。在蒸馏出总共96.8g馏出物之后停止加热,其中釜温升高到113℃。反应运行1小时20分钟,从在80℃下添加催化剂的时间直到反应淬灭。接着在搅拌下将0.2ml的三辛胺(中和剂)添加到烧瓶中,并使混合物冷却至室温。在将釜温冷却至室温后,再次称量具有搅拌棒和叶片的烧瓶(929.3g)。基于质量平衡计算的非挥发物含量(nvc)为57.2%,假定所有聚合物内容物保留在烧瓶中。将样品收集在广口瓶中。所得聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的乙烯基含量为0.069%。
[0172]
实例gp
‑
26
[0173]
在这一实例gp
‑
26中,如下合成具有侧链聚甲基丙烯酸丁酯基团的双羟基封端的聚二甲基硅氧烷。在实验开始之前,将具有搅拌棒和叶片的1升4颈圆底烧瓶预称重并记录。向这个烧瓶中添加181.3克oh封端的pdms 1、18.17克实例mm
‑
3的大分子单体(其具有mn=8,000g/mol且bma=100%),和239.61克甲苯,装入配备有顶置式机械搅拌器、热电偶和具有连接到氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的圆底烧瓶中。当在氮气层下釜温达到80至100℃时,加入0.47ml磷腈催化剂1。随着反应混合物继续加热,在迪安斯塔克设备中收集水、甲醇和甲苯馏出物。在蒸馏足够的甲苯以获得50%固体溶液的最终浓度之后,停止加热。当达到50%固体的最终浓度时,在搅拌下向反应烧瓶中添加0.20ml三辛胺以中和反应混合物,并使所得混合物冷却至室温。冷却至室温后,称重并记录具有搅拌棒和叶片的圆底烧瓶。然后基于质量平衡(预重量与最终重量之间的差)计算nvc,假定所有聚合物含量保留在烧瓶中。
[0174]
参考实例e
‑
gpc实验细节
[0175]
通过在waters 2695型gpc上的分析测定分子量数据。将聚(甲基)丙烯酸酯接枝的聚有机硅氧烷以5mg固体/ml的浓度溶解于thf中,并且在注射100μl样品等分试样之前通过0.45μm的ptfe注射器式过滤器过滤。gpc配备有两根聚合物实验室plgel 5μm混合c柱(300mm x 7.5mm),前面是plgel 5μm保护柱(50mm x 7.5mm),在35℃下的流速为1.0ml/min。使用waters 2410差示折光率检测器进行检测。覆盖580g/mole至2,300,000g/mole范围的16个窄聚苯乙烯标准物的常规校准并拟合为三阶多项式曲线。
[0176]
参考实例
‑
dsc
[0177]
t
g
:ta instruments q1000dsc用于玻璃化转变温度(t
g
)和固化研究。在密封dsc盘中称出样品(各5至15mg),首先记录盘重量,然后记录样品重量。然后将其置于90℃的真空烘箱中8小时。一旦从烘箱中取出,再次称重样品以获得实际样品重量,然后置于ta instruments q1000 dsc上。对于t
g
测量,样品以10℃/min的速度从
‑
90℃到150℃进行2次循环,并且由第2次加热升温确定t
g
。
[0178]
参考实例f——制备和固化氢化硅烷化反应可固化组合物的一般程序
[0179]
基于44.1∶2.7∶53.2∶0.4∶0.05的典型重量比,将实例gp
‑
1至gp
‑
4和gp
‑
8至gp
‑
10中制备的样品与sih交联剂、树脂1、卡斯特催化剂和etch混合,然后将10mg混合物置于密封dsc盘中并密封。记录样品重量。dsc以10℃/min的速度从rt运行至200℃。对于乙烯基与硅烷的氢化硅烷化,通常观察到在100℃与120℃之间的放热峰。
[0180]
对于基准,使用具有搅拌棒和叶片的1升4颈圆底烧瓶制备乙烯基封端的乙烯基硅氧烷,在实验开始之前预称重并记录。将169.4克oh封端的pdms 1、5.56克vi封端的pdms 1和219.5克甲苯装入配备有顶置式机械搅拌器、热电偶和具有连接到氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的烧瓶中。将加热套插入温度控制器中以防止反应混合物加热至大于120℃的温度。当釜温度达到84℃时,添加0.47ml磷腈催化剂1。随着反应混合物继续加热,在迪安斯塔克设备中收集水、甲醇和甲苯馏出物。蒸馏96.1g共沸物后停止加热。然后在搅拌下将0.20ml三辛胺添加到反应烧瓶中以中和反应混合物,并使所得混合物冷却至室温。将具有搅拌棒和叶片的圆底烧瓶称重并记录。然后基于质量平衡(预重量与最终重量之间的差)计算nvc为59%,假定所有聚合物含量保留在烧瓶中。所得聚合物的m
w
为153kda且pdi为1.98。根据参考实例f中概述的方法制备psa。
[0181]
参考实例g——膜样品制备
[0182]
制作膜:将一张纸放置在真空板上并抽真空,然后将一张防粘衬垫放置在纸上。将如上文针对参考实例f所述制备的所需组合物倾倒到防粘衬垫上并且使用4密耳鸟棒来制备膜。将膜置于150℃的despatch
tm
烘箱中5分钟。在样品冷却后,切割2英寸x 81/2条并通过使用~5磅辊转移到玻璃基板上。
[0183]
参考实例h——雾度和清晰度
[0184]
用来自byk gardner的haze
‑
gard plus仪器在玻璃基板上测量根据参考实例f制备的膜样品的光学性质(雾度和透明度)。
[0185]
参考实例i——nmr
[0186]
在配备有16mm无硅autox探针的agilent 500mhz dd2(mi
‑
mr
‑
06)系统上获得
29
si nmr光谱。用cdcl3+0.02m cr(acac)3在无si的teflon nmr管中制备样品。应用标准参数,nt=1024除外。在配备有5mm tci h c/si冷冻探针(mi
‑
mr
‑
07)的bruker avance iii hd nmr光谱仪上获得1h nmr光谱。在5mm nmr管中用cdcl3制备样品。应用标准参数。
[0187]
参考实例j——hsprme2siome的合成
[0188]
将ch3mgbr在乙醚(50ml,0.15mol)中的3m溶液滴加到3
‑
巯基丙基三甲氧基硅烷(8.5g,0.043mol)在50ml的thf中的溶液中,同时将温度保持在0至10℃。将反应混合物在0℃搅拌1小时,然后用ch3oh(40ml)逐滴处理。过滤固体。将粗物质直接用于下一反应。收集5.4g,得到75%产率的纯产物。nmr与参考文献agina,e.v,《美国化学学会应用材料与界面(acs applied materials&interfaces)》,2015,22,11755
‑
11764一致。
[0189]
在这一参考实例d中,上述实例mm
‑
1至mm
‑
12的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体用于制备聚(甲基)丙烯酸酯接枝的聚有机硅氧烷。程序如下:在实验开始之前,将具有搅拌棒和叶片的1升4颈圆底烧瓶预称重并记录。向这个烧瓶中添加181.0克oh封端的pdms 1,5.65克vi封端的pdms 1,11.41克实例mm
‑
5(其具有mn=7.9kda且bma=70%),和238.5克甲苯,装入配备有顶置式机械搅拌器、热电偶和具有附接至氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的圆底烧瓶中。将加热套插入温度控制器中以防止反应混合物加热至
大于120℃的温度。当釜温度达到80℃时,添加0.47ml磷腈催化剂1。随着反应混合物继续加热,在迪安斯塔克设备中收集水、甲醇和甲苯馏出物。在蒸馏足够的甲苯以获得60%固体溶液的最终浓度之后中断加热,通常通过除去100g馏出物。当达到60%固体的最终浓度时,在搅拌下向反应烧瓶中添加0.20ml三辛胺以中和反应混合物,并使所得混合物冷却至室温。冷却至室温后,称重并记录具有搅拌棒和叶片的圆底烧瓶。然后基于质量平衡(预重量与最终重量之间的差)计算nvc,假定所有聚合物含量保留在烧瓶中。另外,计算溶液的乙烯基含量。
[0190]
使用上述参考实例d中的程序重复实例gp
‑
2至gp
‑
20,不同之处在于用不同的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体代替实例5中制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体并改变烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的wt%。这些实例列于表6中。
[0191]
表6.合成的pdms和所选性质的概述。
[0192][0193]
a
通过gpc分析测定的mw和pdi,
b
nd=未确定
[0194]
*可能存在超高mw材料,但在gpc注射中滤出
[0195]
除了上述聚丙烯酸酯接枝的pdms实例之外,下面示出了具有各种结构变化的另外几个实例。
[0196]
实例gp
‑
24
‑
具有聚丙烯酸酯末端的聚(二甲基,甲基乙烯基)硅氧烷共聚物的合成
[0197]
将甲基丙烯酸丁酯(200g)和hsprme2siome(5g)的单体混合物添加到广口瓶中。引发剂溶液由tbppiv(5.5g)和甲苯(10g)制成。向四颈圆底烧瓶中加入40克甲苯。将甲苯加热至85℃,此时分别在2和4小时内进料单体和引发剂混合物。单体混合物进料开始后3小时向烧瓶中添加丙烯酸丁酯(2g)。在引发剂进料结束后,将反应在85℃下保持80分钟。这产生了具有6.6kda和100bma的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体。
[0198]
在实验之前称量一个具有搅拌棒和叶片的1l 4颈圆底烧瓶(571.50g)。在图4的方案4所示的典型合成中,将181.0g的oh封端的pdms 1、2.46g的4
‑
7042、10.098g的上述制备的具有100bma的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体和238g的甲苯装入烧瓶中,该烧瓶配备有热电偶和具有适用于氮气鼓泡器的水冷冷凝器的迪安斯塔克设备。在氮气层下将釜温加热至80至100℃,并添加0.47ml的磷腈催化剂1。然后当釜温升高至103℃时,在迪安斯塔克设备中收集水、甲醇和甲苯。在蒸馏出总共67.0g馏出物之后停止加热,其中釜温升高到113℃。反应运行1小时14分钟,从在80℃下添加催化剂的时间直到反应淬灭。由于聚合物的粘度,在仅67.0克馏出物之后停止反应。接下来,在搅拌下将0.2ml三辛胺(中和剂)添加到烧瓶中,并使所得混合物冷却至室温。在将釜温冷却至室温后,再次称量具有搅拌棒和叶片的烧瓶(930.0g)。基于质量平衡计算的非挥发物含量(nvc)为54.0%,假定所有聚合物内容物保留在烧瓶中。所得聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的乙烯基含量为0.069%。
[0199]
实例gp
‑
25——具有甲基末端的聚丙烯酸酯接枝的乙烯基pdms侧链。
[0200]
在实验之前称量一个具有搅拌棒和叶片的1l 4颈圆底烧瓶(571.49g)。在图5的方案5所示的典型合成中,将181.0g的oh封端的pdms 1、2.46g的4
‑
7042、2.70g的md
22
m、18.62g的实例4中制备的烷氧基硅基官能的(甲基)丙烯酸酯大分子单体和238g的甲苯装入烧瓶中,该烧瓶还配备有热电偶和具有适用于氮气鼓泡器的水冷冷凝器的迪安斯塔克设备。在氮气层下将釜温加热至80至100℃并添加0.47ml磷腈催化剂1。然后当釜温升高至103℃时,在迪安斯塔克设备中收集水、甲醇和甲苯。在蒸馏出总共96.8g馏出物之后停止加热,其中釜温升高到113℃。反应运行1小时20分钟,从在80℃下添加催化剂的时间直到反应淬灭。接着在搅拌下将0.2ml的三辛胺(中和剂)添加到烧瓶中,并使混合物冷却至室温。在将釜温冷却至室温后,再次称量具有搅拌棒和叶片的烧瓶(929.3g)。基于质量平衡计算的非挥发物含量(nvc)为57.2%,假定所有聚合物内容物保留在烧瓶中。将样品收集在广口瓶中。所得聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的乙烯基含量为0.069%。
[0201]
实例gp
‑
26
[0202]
在这一实例gp
‑
26中,如下合成具有侧链聚甲基丙烯酸丁酯基团的双羟基封端的聚二甲基硅氧烷。在实验开始之前,将具有搅拌棒和叶片的1升4颈圆底烧瓶预称重并记录。向这个烧瓶中添加181.3克oh封端的pdms 1、18.17克实例mm
‑
3的大分子单体(其具有mn=8,000g/mol且bma=100%),和239.61克甲苯,装入配备有顶置式机械搅拌器、热电偶和具有连接到氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的圆底烧瓶中。当在氮气层下釜温达到80至100℃时,加入0.47ml磷腈催化剂1。随着反应混合物继续加热,在迪安斯塔克设备中收集水、甲醇和甲苯馏出物。在蒸馏足够的甲苯以获得50%固体溶液的最终浓度之后,停止加热。当达到50%固体的最终浓度时,在搅拌下向反应烧瓶中添加0.20ml三辛胺以中和反应混合物,并使所得混合物冷却至室温。冷却至室温后,称重并记录具有搅拌棒和叶片的圆底烧瓶。然后基于质量平衡(预重量与最终重量之间的差)计算nvc,假定所有聚合物含量保留在烧瓶中。
[0203]
对比实例27——通过巯基官能化的乙烯基pdms接枝聚丙烯酸酯。
[0204]
方案3:按照以下图3中所示的方案3,在实验之前称重具有搅拌棒和叶片的1l的4颈圆底烧瓶(571.49g)。在典型的合成中,将181.0g的oh封端的pdms 1、5.65g的vi封端的
pdms 1、0.132ml的(3
‑
巯基丙基)甲基二甲氧基硅烷和238g的甲苯装入配备有机械搅拌器、热电偶和具有适用于氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的烧瓶中。在氮气层下将釜温加热至80至100℃并加入0.47ml磷腈催化剂1,然后当釜温升高至103℃时在迪安斯塔克设备中收集水、甲醇和甲苯。在蒸馏出总计100.0g馏出物之后停止加热(釜温升至113℃)。(反应运行1小时6分钟,从在80℃下添加催化剂的时间开始,直到反应混合物猝灭。)接下来,在搅拌下将0.2ml三辛胺(中和剂)添加到烧瓶中,并使所得混合物冷却至室温。在将釜温冷却至室温后,再次称量具有搅拌棒和叶片的烧瓶(901.10g)。基于质量平衡计算的nvc为56.7%,假定所有聚合物内容物保留在烧瓶中。聚合物的乙烯基含量为0.024%。硫醇含量计算为0.00732%。这种聚合物是双乙烯基封端的、巯丙基官能化的聚二甲基硅氧烷,pdms(sh)。
[0205]
接枝bma:将如上所述制备的pdms(sh)在甲苯中的溶液(30.04g的pdms(sh),22.66g的甲苯)添加到4颈圆底烧瓶中。添加甲基丙烯酸丁酯(10g)。将溶液加热至85℃。在60分钟内将引发剂tbppiv于甲苯中的溶液(0.6g的tbppiv于5g的甲苯中)进料至反应混合物中。引发剂进料4分钟后,溶液变成混浊混合物。10分钟后,反应混合物呈现乳白色。随着反应进行,反应混合物的粘度增加。在引发剂进料结束后,将反应混合物在85℃下保持80分钟。在反应结束时加入30g甲苯以稀释所得混合物。
[0206]
方案3,如上所述,产生具有mw/mn 229kda/133kda和pdi 1.72的pdms(sh)。然而,随后在引发剂存在下接枝bma的尝试导致所得产物中mw/mn 76kda/6.5kda和pdi 11.82降低。
[0207]
对比实例28——尝试通过巯基官能化的乙烯基pdms接枝聚丙烯酸酯
[0208]
如对比实例27中所述通过方案3制备的相同pdms(sh)用于在这一实例中经由不同的反应条件接枝丙烯酸酯。将pdms(sh)在甲苯中的溶液(114g的pdms(sh),86g的甲苯)添加到4颈圆底烧瓶中。添加甲基丙烯酸丁酯(4.5g)和甲苯(50g)。将溶液加热至85℃。在60分钟内将引发剂tbppiv于甲苯中的溶液(0.125g的tbppiv于10g的甲苯中)进料至反应混合物中。在引发剂进料结束后,将反应在85℃下保持80分钟。在反应结束时添加60g的甲苯以稀释聚合物混合物。1h nmr光谱分析显示bma的转化率仅为19%。gpc分析显示有大量的嫁接证据。表6示出了对比实例27和28之间的比较。
[0209]
表6.对比实例27和28之间的比较
[0210]
条件实例27实例28pdms(sh)(克)22.7114单体(克)bma(10)bma(4.5)引发剂tbppivtbppiv引发剂(克)0.450.125引发剂(毫摩尔)2.580.54硫醇(毫摩尔)0.120.44单体(毫摩尔)70.331.65接枝xn(目标)60171.2接枝mn(目标)85460da10121da单体/引发剂(摩尔比)27.258.6
硫醇/引发剂(摩尔比)0.050.81
[0211]
参考实例e
‑
gpc实验细节
[0212]
通过在waters 2695型gpc上的分析测定分子量数据。将聚(甲基)丙烯酸酯接枝的聚有机硅氧烷以5mg固体/ml的浓度溶解于thf中,并且在注射100μl样品等分试样之前通过0.45μm的ptfe注射器式过滤器过滤。gpc配备有两根聚合物实验室plgel 5μm混合c柱(300mm x 7.5mm),前面是plgel 5μm保护柱(50mm x 7.5mm),在35℃下的流速为1.0ml/min。使用waters 2410差示折光率检测器进行检测。覆盖580g/mole至2,300,000g/mole范围的16个窄聚苯乙烯标准物的常规校准并拟合为三阶多项式曲线。
[0213]
参考实例f——制备和固化氢化硅烷化反应可固化组合物的一般程序
[0214]
基于44.1∶2.7∶53.2∶0.4∶0.05的典型重量比,将实例gp
‑
1至gp
‑
4和gp
‑
8至gp
‑
10中制备的样品与sih交联剂、树脂1、卡斯特催化剂和etch混合,然后将10mg混合物置于密封dsc盘中并密封。记录样品重量。dsc以10℃/min的速度从rt运行至200℃。对于乙烯基与硅烷的氢化硅烷化,通常观察到在100℃与120℃之间的放热峰。
[0215]
对于基准,使用具有搅拌棒和叶片的1升4颈圆底烧瓶制备乙烯基封端的乙烯基硅氧烷,在实验开始之前预称重并记录。将169.4克oh封端的pdms 1、5.56克vi封端的pdms 1和219.5克甲苯装入配备有顶置式机械搅拌器、热电偶和具有连接到氮气鼓泡器的水冷冷凝器的迪安斯塔克设备的烧瓶中。将加热套插入温度控制器中以防止反应混合物加热至大于120℃的温度。当釜温度达到84℃时,添加0.47ml磷腈催化剂1。随着反应混合物继续加热,在迪安斯塔克设备中收集水、甲醇和甲苯馏出物。蒸馏96.1g共沸物后停止加热。然后在搅拌下将0.20ml三辛胺添加到反应烧瓶中以中和反应混合物,并使所得混合物冷却至室温。将具有搅拌棒和叶片的圆底烧瓶称重并记录。然后基于质量平衡(预重量与最终重量之间的差)计算nvc为59%,假定所有聚合物含量保留在烧瓶中。所得聚合物的m
w
为153kda且pdi为1.98。根据参考实例f中概述的方法制备组合物。
[0216]
参考实例g——膜样品制备
[0217]
制作膜:将一张纸放置在真空板上并抽真空,然后将一张防粘衬垫放置在纸上。将如上文针对参考实例f所述制备的所需组合物倾倒到防粘衬垫上并且使用4密耳鸟棒来制备膜。将膜置于150℃的despatch
tm
烘箱中5分钟。在样品冷却后,切割2英寸x 81/2条并通过使用~5磅辊转移到玻璃基板上。
[0218]
参考实例h——雾度和清晰度
[0219]
用来自byk gardner的haze
‑
gard plus仪器在玻璃基板上测量根据参考实例f制备的膜样品的光学性质(雾度和透明度)。
[0220]
参考实例i——nmr
[0221]
在配备有16mm无硅autox探针的agilent 500mhz dd2(mi
‑
mr
‑
06)系统上获得
29
si nmr光谱。用cdcl3+0.02m cr(acac)3在无si的teflon nmr管中制备样品。应用标准参数,nt=1024除外。在配备有5mm tci h c/si冷冻探针(mi
‑
mr
‑
07)的bruker avance iii hd nmr光谱仪上获得1h nmr光谱。在5mm nmr管中用cdcl3制备样品。应用标准参数。
[0222]
参考实例j——hsprme2siome的合成
[0223]
将ch3mgbr在乙醚(50ml,0.15mol)中的3m溶液滴加到3
‑
巯基丙基三甲氧基硅烷(8.5g,0.043mol)在50ml的thf中的溶液中,同时将温度保持在0至10℃。将反应混合物在0
℃搅拌1小时,然后用ch3oh(40ml)逐滴处理。过滤固体。将粗物质直接用于下一反应。收集5.4g,得到75%产率的纯产物。nmr与参考文献agina,e.v,《美国化学学会应用材料与界面(acs applied materials&interfaces)》,2015,22,11755
‑
11764一致。
[0224]
工业适用性
[0225]
工业上需要将(甲基)丙烯酸类聚合物和共聚物掺入聚二有机硅氧烷(如聚二甲基硅氧烷)中以增加与基底表面的相互作用,同时保留聚二有机硅氧烷上的乙烯基或其它脂肪族不饱和反应性官能团,官能团将经由丙烯酸类聚合物在聚二有机硅氧烷上的自由基聚合而被破坏。脂肪族不饱和官能团然后可用于随后的反应(例如,氢化硅烷化固化)。在不希望受到理论约束的情况下,据认为本文所述的聚(甲基)丙烯酸酯接枝的聚有机硅氧烷可用于有机硅压敏粘合剂组合物中。上述氢化硅烷化反应可固化组合物可用作压敏粘合剂组合物。
[0226]
术语的定义和使用
[0227]
表7示出了本文使用的缩写。
[0228]
表7缩写
[0229]
缩写定义aa丙烯酸bma甲基丙烯酸丁酯da道尔顿dsc差示扫描量热法2
‑
eha或eha丙烯酸2
‑
乙基己酯g克hs
‑
gc如上所述测量的顶空气相色谱法gpc凝胶渗透色谱kda千道尔顿l升mg毫克mhz兆赫min分钟ml毫升mm毫米mma甲基丙烯酸甲酯mmole毫摩尔mn通过gpc测定的数均分子量mw通过gpc测定的重均分子量mw分子量nmr核磁共振nvc非挥发物含量pdi通过gpc测定的多分散性pdms聚二甲基硅氧烷
ppm百万分之重量份rt20℃至25℃的室温μm微米
[0230]
除非另有说明,否则所有量、比率和百分比均按重量计。除非说明书的上下文另有说明,否则冠词“一(a)”、“一个(an)”和“该(the)”均指一个或多个。范围的公开内容包括范围本身以及其中包含的任何内容,以及端点。例如,2.0至4.0的范围的公开不仅包括2.0至4.0的范围,而且还单独地包括2.1、2.3、3.4、3.5和4.0,以及包含在该范围内的任何其它数字。此外,例如2.0至4.0的范围的公开包括例如2.1至3.5、2.3至3.4、2.6至3.7和3.8至4.0的子集,以及包含在该范围内的任何其它子集。类似地,马库什(markush)组的公开内容包括整个组以及其中包含的任何单个成员和子组。例如,公开的马库什组、氢原子、烷基、芳基或芳烷基单独地包括成员烷基;子组烷基和芳基;以及其中包含的任何其它单个成员和子组。
[0231]“烷基”是指无环的、支链的或无支链的、饱和的单价烃基。烷基示例为但不限于甲基、乙基、丙基(例如,异丙基和/或正丙基)、丁基(例如,异丁基、正丁基、叔丁基和/或仲丁基)、戊基(例如,异戊基、新戊基和/或叔戊基)、己基、庚基、辛基、壬基和癸基以及6个或更多个碳原子的支链饱和单价烃基。
[0232]“烯基”是指在两个碳原子之间具有双键的无环、支链或非支链的单价烃基。烯基示例为但不限于乙烯基、烯丙基、丁烯基、戊烯基和己烯基,包括支链和直链物质。
[0233]“芳基”是指环状的完全不饱和的烃基。芳基由但不限于环戊二烯基、苯基、蒽基和萘基来示例。单环芳基可以具有5到9个碳原子,可替代地6到7个碳原子,并且可替代地5到6个碳原子。多环芳基可以具有10到17个碳原子,可替代地10到14个碳原子,并且可替代地12到14个碳原子。
[0234]“芳烷基”是指具有侧链和/或末端芳基的烷基或具有侧链烷基的芳基。示范性芳烷基包括甲苯基、二甲苯基、苄基、苯乙基、苯基丙基和苯基丁基。
[0235]“碳环”和“碳环的”各自表示烃环。碳环可以是单环,或者可以是稠合、桥连或螺环的多环。单环碳环可以具有3至9个碳原子,或者4至7个碳原子,或者5至6个碳原子。多环碳环可以具有7至17个碳原子,或者7至14个碳原子,或者9至10个碳原子。碳环可以是饱和或部分不饱和的。
[0236]“环烷基”是指饱和碳环。单环环烷基由环丁基、环戊基和环己基来示例。
[0237]“卤化烃”是指其中与碳原子键合的一个或多个氢原子已经形式上被卤素原子替代的烃。卤代烃基包括卤代烷基、卤代碳环基和卤代烯基。卤代烷基包括氟化烷基,如三氟甲基(cf3)、氟甲基、三氟乙基、2
‑
氟丙基、3,3,3
‑
三氟丙基、4,4,4
‑
三氟丁基、4,4,4,3,3
‑
五氟丁基、5,5,5,4,4,3,3
‑
七氟戊基、6,6,6,5,5,4,4,3,3
‑
九氟己基和8,8,8,7,7
‑
五氟辛基;和氯化烷基,如氯甲基和3
‑
氯丙基。卤代碳环基团包括氟化环烷基,如2,2
‑
二氟环丙基、2,3
‑
二氟环丁基、3,4
‑
二氟环己基和3,4
‑
二氟
‑5‑
甲基环庚基;和氯化环烷基,如2,2
‑
二氯环丙基、2,3
‑
二氯环戊基。卤代烯基包括氯烯丙基。
[0238]
如本文和所附权利要求中所用的术语“(甲基)丙烯酸”旨在用作涵盖丙烯酸和甲基丙烯酸中的任一者或两者的一般表达。
[0239]
如本文和所附权利要求中所用的术语“(甲基)丙烯酸酯”旨在用作涵盖丙烯酸酯
和甲基丙烯酸酯中的任一者或两者的一般表达。
[0240]“m
‑
单元”是指具有式r3sio
1/2
的硅氧烷单元,其中每个r独立地表示单价原子或有机基团。“d
‑
单元”是指具有式r2sio
2/2
的硅氧烷单元,其中每个r独立地表示单价原子或基团。“t
‑
单元”是指具有式rsio
3/2
的硅氧烷单元,其中每个r独立地表示单价原子或基团。“q
‑
单元”是指具有式sio
4/2
的硅氧烷单元。
[0241]
本发明的实施例
[0242]
在第一实施例中,一种具有脂肪族不饱和基团和聚(甲基)丙烯酸酯基团两者的聚有机硅氧烷包含以下单元式:
[0243]
[r
3w
(r5‑
s
‑
r
″
)(or4)
(2
‑
w)
si
‑
o
1/2
]
p
[r
3v
(r5‑
s
‑
r
″
)(or4)
(1
‑
v)
si
‑
o
2/2
]
q
[(r5‑
s
‑
r
″
)si
‑
o
3/2
]
k
(r6r
72
sio
1/2
)
r
(r
72
sio
2/2
)
s
(r6r7sio
2/2
)
t
(r
73
sio
1/2
)
u
,其中每个下标w独立地为0、1或2,每个下标v独立地为0或1,每个r3为独立选择的单价烃基;每个r4为独立选择的烷基;每个r5为独立选择的二价烃基,每个r
″
独立地为(甲基)丙烯酸酯聚合物或共聚物,每个r6为独立选择的脂肪族不饱和单价烃基,每个r7为独立选择的不含脂肪族不饱和基团的单价烃基,下标p≥0,下标q≥0,下标k≥0,数量(p+q+k)≥1,下标r≥0,下标s≥0,下标t≥0,下标u≥0,数量(r+t)≥1,并且数量(p+q+k+r+s+t+u)足以向聚有机硅氧烷提供至少50kda的分子量。
[0244]
在第二实施例中,在第一实施例的聚有机硅氧烷中,r3具有1至18个碳原子。
[0245]
在第三实施例中,在第一实施例或第二实施例的聚有机硅氧烷中,每个r3为1至6个碳原子的烷基。
[0246]
在第四实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r3为甲基。
[0247]
在第五实施例中,在前述实施例中任一项的聚有机硅氧烷中,r4具有1至6个碳原子。
[0248]
在第六实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r4为甲基。
[0249]
在第七实施例中,在前述实施例中任一项的聚有机硅氧烷中,r5具有1至18个碳原子。
[0250]
在第八实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r5为独立选择的具有2至8个碳原子的亚烷基。
[0251]
在第九实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r5为亚丙基。
[0252]
在第十实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r6具有2至18个碳原子。
[0253]
在第十一实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r6为选自乙烯基、烯丙基和己烯基的烯基。
[0254]
在第十二实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r6为乙烯基。
[0255]
在第十三实施例中,在前述实施例中任一项的聚有机硅氧烷中,r7具有1至18个碳原子。
[0256]
在第十四实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r7为1至6个碳原子的烷基。
[0257]
在第十五实施例中,在前述实施例中任一项的聚有机硅氧烷中,每个r7为甲基。
[0258]
在第十六实施例中,在前述实施例中任一项的聚有机硅氧烷中,r
″
具有1至1,000
的dp。
[0259]
在第十七实施例中,在前述实施例中任一项的聚有机硅氧烷中,r
″
具有5kda至600kda的dp。
[0260]
在第十八实施例中,在前述实施例中任一项的聚有机硅氧烷中,下标(p+r+u)=2,下标k=0,数量(p+q)为1至100,数量(r+t)为1至100,数量(p+q+r+s+t+u)足以向聚有机硅氧烷提供50kda至1,000kda的分子量。
[0261]
在第十九实施例中,在前述实施例中任一项的聚有机硅氧烷中,下标p为1或2。
[0262]
在第二十实施例中,在前述第一至第十八实施例中任一项的聚有机硅氧烷中,下标p=0,下标k=0,下标t=0,并且下标u=0。
[0263]
在第二十一实施例中,一种用于制备包含聚(甲基)丙烯酸酯接枝的聚有机硅氧烷的产物的方法包含以下步骤:
[0264]
i)组合起始材料,其包含:
[0265]
烷氧基硅基官能的(甲基)丙烯酸酯大分子单体;
[0266]
聚二有机硅氧烷,其选自由以下组成的组:
[0267]
每分子具有至少一个硅键合的脂肪族不饱和基团的不饱和聚二有机硅氧烷;
[0268]
每分子具有至少两个硅键合的羟基的羟基官能的聚二有机硅氧烷;以及
[0269]
不饱和聚二有机硅氧烷和羟基官能的聚二有机硅氧烷两者的组合;
[0270]
磷腈缩合反应催化剂;
[0271]
任选地聚二烷基硅氧烷;以及
[0272]
任选地溶剂;
[0273]
从而制备包含聚有机硅氧烷和副产物的产物;
[0274]
ii)在步骤i)期间和/或之后除去副产物的全部或部分;
[0275]
任选地iii)中和产物;以及
[0276]
任选地iv)回收聚有机硅氧烷。
[0277]
在第二十二实施例中,在第二十一实施例的方法中,烷氧基硅基官能的(甲基)丙烯酸酯大分子单体通过包括以下步骤的方法制备:
[0278]
1)组合起始材料,起始材料包含
[0279]
i)式的(甲基)丙烯酸酯单体,其中r1为氢或甲基,并且r2选自由氢、烷基、芳基和芳烷基组成的组;
[0280]
ii)式的巯基官能的烷氧基硅烷,其中下标a为0至2,每个r3为独立选择的单价烃基,r4为独立选择的烷基,并且r5为二价烃基;
[0281]
任选地iii)自由基引发剂;以及
[0282]
任选地iv)溶剂;从而制备包含烷氧基硅基官能的(甲基)丙烯酸酯大分子单体的产物;以及
[0283]
任选地2)回收a)烷氧基硅基官能的(甲基)丙烯酸酯大分子单体。
[0284]
在第二十三实施例中,在第二十二实施例的方法中,存在以下条件中的至少一个:
[0285]
在起始材料i)中,每个r1是甲基,并且每个r2是氢或1至8个碳原子的烷基;
[0286]
在起始材料ii)中,下标a为1至2,每个r3为1至6个碳原子的烷基,每个r4为1至6个碳原子的烷基,并且每个r5为1至8个碳原子的亚烷基;
[0287]
iii)存在自由基引发剂,并且自由基引发剂选自由以下组成的组:iii
‑
1)偶氮化合物,iii
‑
2)过氧化物,和iii
‑
3)其组合;
[0288]
iv)存在溶剂,并且溶剂选自由以下组成的组:iv
‑
1)沸点高于100℃的烃,iv
‑
2)极性溶剂,iv
‑
3)硅油,和iv
‑
4)iv
‑
1)、iv
‑
2)和iv
‑
3)中的两种或更多种的组合。
[0289]
在第二十四实施例中,在第二十一实施例的方法中,烷氧基硅基官能的(甲基)丙烯酸酯大分子单体具有式
[0290]
其中下标a为0至2,每个r3为独立选择的单价烃基,r4为独立选择的烷基,r5为二价烃基,并且r
″
为dp为1至1,000的(甲基)丙烯酸酯聚合物或共聚物。
[0291]
在第二十五实施例中,在第二十一实施例的方法中,存在不饱和聚二有机硅氧烷,并且不饱和聚二有机硅氧烷具有以下单元式:(r6r
72
sio
1/2
)
b
(r
72
sio
2/2
)
c
(r6r7sio
2/2
)
d
(r
73
sio
1/2
)
e
[(r
′
o)r
72
sio
1/2
]
f
[(r
′
o)r7sio
2/2
]
g
,其中每个r6为独立选择的脂肪族不饱和烃基,每个r7为独立选择的不含脂肪族不饱和基团的单价烃基,每个r
′
独立地选自由h和r7组成的组,下标b为0、1或2,下标e为0、1或2,下标f为0、1或2,数量(b+e+f)=2,下标c≥0,下标d≥0,下标g≥0,数量(c+d+g)为1至250,数量(b+d)≥1,并且数量(b+c+d+e+f+g)至少为3。
[0292]
在第二十六实施例中,在第二十五实施例的方法中,每个r6具有2至18个碳原子。
[0293]
在第二十七实施例中,在第二十五或第二十六实施例的方法中,每个r6为选自乙烯基、烯丙基和己烯基的烯基。
[0294]
在第二十八实施例中,在第二十五至第二十七实施例中任一项的方法中,每个r6为乙烯基。
[0295]
在第二十九实施例中,在第二十五至第二十八实施例中任一项的方法中,r7具有1至18个碳原子。
[0296]
在第三十实施例中,在第二十五至第二十九实施例中任一项的方法中,每个r7为甲基。
[0297]
在第三十一实施例中,在第二十一实施例的方法中,存在羟基官能的聚二有机硅氧烷并且其具有式(r
82
sio
2/2
)
h
(r
83
sio
1/2
)
i
(hor
82
sio
1/2
)
j
,其中每个r8为独立选择的不含脂肪族不饱和基团的单价烃基,下标j为0、1或2,下标i为0、1或2,数量(j+i)=2,下标h≥1,并且数量(h+i+j)为3至250。
[0298]
在第三十二实施例中,在第二十一实施例的方法中,磷腈缩合反应催化剂为磷腈
卤化物。
[0299]
在第三十三实施例中,在第二十一实施例的方法中,存在步骤iii),并且步骤iii)包含添加包含烷基胺的中和剂。
[0300]
在第三十四实施例中,在第二十一实施例的方法中,存在步骤iv),并且步骤iv)包含过滤、汽提和/或蒸馏。
[0301]
在第三十五实施例中,一种可固化组合物包含:
[0302]
i)第一至第二十实施例中任一项的聚有机硅氧烷,
[0303]
ii)每分子具有至少3个硅键合的氢原子的有机硅交联剂,
[0304]
iii)氢化硅烷化反应催化剂,
[0305]
任选地iv)氢化硅烷化反应抑制剂,
[0306]
任选地v)聚有机硅酸盐树脂,以及
[0307]
任选地xx)媒剂。
[0308]
在第三十六实施例中,一种用于制备涂层基底的方法包含:
[0309]
a)在基底的表面上形成根据第三十五实施例所述的组合物的膜,以及
[0310]
b)固化所述组合物以形成涂层。
[0311]
在第三十七实施例中,通过第三十六实施例的方法制备的涂层基底是压敏粘合剂制品。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1