癌症患者生存率的预测方法
1.本发明涉及确定癌症患者预后和/或预测治疗反应以指导癌症患者治疗方案选择和/或预测癌症患者生存率和/或生存风险和/或临床结果的方法和/或确定一种治疗是否适合特定癌症患者的方法和/或确定癌症患者疗程的方法(例如治疗方案分层方法),特别是那些患有肺癌诸如非小细胞肺癌的患者。
背景技术:
2.肺癌是全球癌症死亡的主要原因,非小细胞肺癌(nsclc)占全球确诊病例的85
‑
90%。如detterbeck等人在chest 15,193
‑
203,2017上发表的“lung cancer stage classification”所述,肿瘤分期有助于指导临床决定是否进行辅助化疗。然而,如vargas等人在nat rev cancer(2016)上发表的“biomarker development in the precision medicine era:lung cancer as a case study”中所述,tnm分期是不完善的生存风险预测因子,因为同一肿瘤分期的患者可能出现明显不同的临床结果。
3.有人建议,在当前的诊断标准中纳入分子生物标志物(例如基于基因表达的肿瘤侵袭性相关物),可以将癌症患者划分为更精确的疾病亚型。这方面的实例在van’t veer等人在nature452,564
–
570(2008)发表的“enabling personalized cancer medicine through analysis of gene
‑
expression patterns”、vargas等人在nat.rev.cancer 16,525
–
537(2016)上发表的“biomarker development in the precision medicine era;lung cancer as a case study”及kumar
‑
sinha等人在nat.biotechnol.36,46
‑
60(2018)发表的“precision oncology in the age of integrative genomics”中进行了描述。准确识别术后非小细胞肺癌(nsclc)复发高危患者可能具有相当高的临床价值,有助于指导决策,例如是否施用手术切除后的辅助化疗或患者随访的所需强度。
4.在过去的二十年中,人们进行了多次尝试,得出了肺腺癌(luad)患者的预后基因表达特征,肺腺癌是非小细胞肺癌(nsclc)最常见的组织学亚型。这方面的实例在beer等人在nat med 8,816
‑
824(2002)发表的“gene
‑
expression profiles predict survival of patients with lung adenocarcinoma”中,krystanek等人在biomark res 4,4(2016)发表的“a robust prognostic gene expression signature for early stage lung adenocarcinoma”中及wistuba等人在clin cancer res(2013)发表的“validation of a proliferation based expression signature as prognostic marker in early stage lung adenocarcinoma”中进行了描述。然而,这些努力由于可重现性差,或独立于现有临床病理风险因素的预后能力有限而受到阻碍,例如如subramanian等人在jncl j natl cancer inst 102,464
‑
474(2010)发表的“gene expression
‑
based prognostic signatures in lung cancer;ready for clinical use?”中所述。
5.图1a至1d阐明了与已知特征相关的一些问题。图1a示出了包含肿瘤12的肺10。多个区域r1、r2、r3和r4可以进行肺活检。然而,如红色和蓝色的示意图所示,采用人们已知的预后生物标志物,区域r1、r2和r3的活检将得出高风险分类,而区域r4的活检将得出低风险
分类。通常,在常规临床实践中,采用单次活检14进行诊断或开展预后评估。因此,图1a所示的假设预后特征将显示出不一致的肿瘤风险分类,因为区域r4的活检结果与不同区域所采集样本的结果不匹配。因此,特征的读出容易受到肿瘤采样偏差的影响。
6.图1b阐明了肿瘤采样偏差对患者群体的影响。图中示出了多个肺肿瘤20、22、24、26、28、30,每个肺肿瘤具有多个采样区域(例如r1至r5)。对其中一个区域活检应用预后生物标志物,根据估计的生存风险,将肺癌患者40、42、44、46、48、50分为更精确的疾病亚型,这可能有助于指导治疗决策。正确区分需要辅助化疗的高风险患者和仅通过手术即可治愈的低风险患者非常重要。
7.在肺肿瘤20、22的每个区域中,活检将正确地得出相关患者40、42为低风险分类,因此这些患者将被分类为适合仅通过手术切除进行治疗。类似地,在肺肿瘤28、30的每个区域中,活检将正确地得出相关患者48、50为高风险分类,因此这些患者将被分类为需要通过手术切除和辅助化疗进行治疗。然而,第三位患者44具有类似于图1a所示的肺肿瘤24。如图中所示,区域r4的活检得出该患者分类为低风险,这与肺肿瘤24中其他区域活检结果得出的分类不一致。这一点非常重要,因为根据该诊断,患者不可能接受辅助治疗,从而未接受充分的治疗。因此,该患者具有次优的治疗和随访。类似地,第四位患者46具有肺肿瘤26,根据活检取样位置将得出不同的结果。在说明中,活检得出高风险分类,这可能导致患者接受不必要的治疗,从而承受化疗的副作用。
8.图1c示出了shukla等人在jncl j natl cancer inst 109(2017)发表的“development of arna
‑
seq based prognostic signature in lung adenocarcinoma”中描述的luad已知特征的分析结果。所述特征采用世界上最大的多区域测序研究tracerx肺试验提供的信息进行分析,从而能够对肿瘤进化进行详细探索。该项研究,例如在jamal
‑
hanjani等人在plos biol 12(2014)公开的“tracking genomic cancer evolution for precision medicine;the lung tracerx study”中进行了描述。在图1c中,对tracerx研究中28名患者的89个肿瘤区域进行了分析,如图所示,每个患者按预测生存“风险分数”排序,风险最低的患者位于图中左侧。为了计算本实施例中的“风险分数”,通过对特征中四个基因表达值的计算风险分数进行回归,拟合无截距的线性模型,从原始出版物中提供的补充数据重新推导回归系数。图1c上的每个点代表单个肿瘤区域,垂直线表示每个患者的风险分数范围。不管活检的位置如何,11名患者被归类为低风险,5名患者被归类为高风险。然而,有12名患者的分类不一致,其风险分数取决于活检的位置。
9.图1d将图1c中的数据以条形图展示,其中示出了低风险、高风险和不一致患者的百分数。图1e是基于免疫相关基因对的不同特征的类似条形图,如li等人在jama oncol(2017)发表的“development and validation of an individualized immune prognostic signature in early
‑
stage nonsquamous non
‑
small cell lung cancer”中所述。在这两种情况下,有很大比例的不一致患者
‑
43%或29%
‑
来自同一肿瘤的不同区域可能被归类为具有不同的分子风险特征。易受肿瘤取样偏差影响的患者比例很高,这可能限制了此类预后分析的临床应用。
10.迄今为止,luad中大多数基于基因表达的预后特征都是通过微阵列表达谱而不是rna测序来确定的。图1f示出了下表详述的9个已公开的luad预后特征的一致性结果。指明了每篇论文中的患者数n。按照gyanchandani等人在clin cancer res 22,5362
‑
5369
(2016)发表的“intratumour heterogenity affects gene expression profile test prognostic risk stratification in early breast cancer”中的描述,采用曼哈顿计量的ward法对每个预后特征进行分层聚类(hierarchical clustering)。对于给定数量的聚类(cluster),聚类一致性被量化为所有肿瘤区域为同一聚类的患者百分比。结果以肿瘤区域属于同一聚类的患者百分数对照聚类数绘图。垂直虚线标记聚类的范围(2、3、14和28):
[0011][0012][0013]
在28个聚类中,聚类不一致率的中位数为50%(15.5/28个luad肿瘤),表明一半的肿瘤区域会因采样偏差而存在被错误分类的风险。该范围在18
‑
82%之间,表明某些特征明显优于其它特征。综上所述,图1a至1f表明采样偏差会使分子生物标志物在几种癌症类型中的使用变得混乱。正如jamal
‑
hanjani等人在n engl j med 376,2109
‑
2121(2017)发表的“tracking the evolution of non
‑
small cell lung cancer”中所述,肿瘤内异质性(ith)和染色体不稳定性(cin)是nsclc和其他类型癌症的共同特征。此外,如burrell等人在nature 501,338
‑
345(2013)发表的“the causes and consequence of genetic heterogeneity in cancer evolution”中所述,遗传肿瘤内异质性(ith)在各种类型癌症
中普遍存在。
[0014]
有关先前肺癌预后特征的背景信息参见国际专利公开wo201/063121(描述了使用16
‑
基因预后特征将非小细胞肺癌(nsclc)患者分类为各个风险组);美国专利公开us2010/184063(描述了使用15
‑
基因的预后和预测特征将nsclc患者分类为各个风险组);和国际专利公开wo2015/138769(描述了使用9
‑
基因预后特征将nsclc患者分类为各个风险组)。
[0015]
本技术人已经认识到需要改进的基因特征来帮助临床医生改进预后准确性以帮助指导治疗决策,例如选择仅手术切除或手术切除后开展化疗或其他辅助治疗。
技术实现要素:
[0016]
根据本发明,提供了如所附权利要求中所述的装置和方法。本发明的其他特征将从从属权利要求和下面的描述中显而易见。
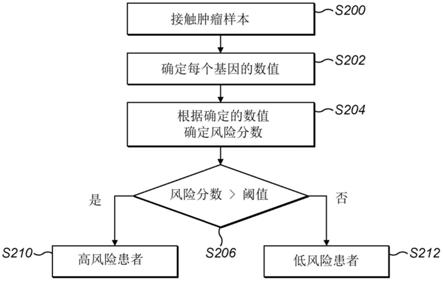
[0017]
我们描述了为肺癌受试者提供预后的方法,所述方法包括:(a)将来自受试者的生物样本与试剂接触,所述试剂和一组生物标志物中的每个成员特异性结合,所述生物标志物包括anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1;(b)根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;(c)根据受试者的风险分数,提供肺癌的预后。
[0018]
确定受试者的风险分数可以包括:对于每个生物标志物,确定指示组织样本中核酸表达水平的分数;根据确定的分数,计算风险分数,其中风险分数通过将加权的生物标志物的分数相加计算得出,其中生物标志物的分数基于确定的分数,并且每个生物标志物的分数都具有相关的权重;及将风险分数与阈值进行比较。这样,每个受试者都可能例如被分到高风险组(例如风险分数高于阈值)或低风险组(例如风险分数等于或低于阈值)。例如,当考虑所有类型的肺癌时,高风险组的生存率可能较低,而低风险组的生存率可能较高。或者,在考虑早期癌症时,高风险组可能比低风险组更容易复发。golga8a、scpep1、slc46a3和xbp1中的每个生物标志物分数的相关权重可能是负值,表明它们是有利的基因。anln、aspm、cdca4、errfi1、furin、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、snx7和tpbg中的生物标志物分数的相关权重可能是正值。
[0019]
风险分数的加权总和可以通过以下方式确定:
[0020]
风险分数=b1x
1i
+b2x
2i
+
…
+b
n
x
ni
[0021]
其中x
1i
、x
2i
、
……
、x
ni
是每个受试者i的四个选定生物标志物的生物标志物分数,b1、b2、
……
、b
n
是每个生物标志物分数的一组相关权重。
[0022]
所述方法可以进一步包括采用cox比例风险模型确定加权总和的权重,该模型采用包括有关一组受试者多个生物标志物的信息的训练数据进行训练。所述方法可包括识别在cox比例风险模型中使用的多个生物标志物,其中所述多个生物标志物选自anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。
[0023]
阈值可以是训练数据的中值风险分数。
[0024]
确定指示生物标志物水平的分数可以包括确定缩放(scaled)强度分数。生物标志物分数可以基于已通过减去调整因子而调整的缩放强度分数。确定指示生物标志物水平的
分数可以包括当水平高于阈值时赋予第一值,当水平低于阈值时赋予第二值。确定指示生物标志物水平的分数可以包括当水平高于上阈值时赋予第一值,当水平低于上阈值但高于下阈值时赋予第二值,以及当水平低于下阈值时赋予第三值。
[0025]
试剂可以是核酸。
[0026]
此处所述术语“核酸”、“核酸序列”、“核苷酸”、“核酸分子”或“多核苷酸”旨在包括dna分子(例如,cdna或基因组dna)、rna分子(例如,mrna、mirna、lncrna)、天然存在的、突变的、合成的dna或rna分子,以及使用核苷酸类似物生成的dna或rna类似物。核酸可以是单链或双链。所述核酸或多核苷酸包括但不限于结构基因的编码序列、反义序列及不编码mrna或蛋白质产物的非编码调控序列。这些术语还包括基因。术语“基因”、“等位基因(allele)”或“基因序列”广泛用于指与生物学功能相关的dna核酸。因此,基因可以包括基因组序列中的内含子和外显子,或者可以仅包括cdna中的编码序列,和/或可以包括与调控序列组合的cdna。因此,根据本发明的各个方面,可以使用基因组dna、cdna或编码dna。在一个实施方案中,核酸是cdna或编码dna。因此,基因可以包括基因组序列中的内含子和外显子,或者可以仅包括cdna中的编码序列,和/或可以包括与调控序列组合的cdna。
[0027]
核酸分析可采用合适的技术进行,例如用于测量基因表达的技术,包括但不限于数字pcr、qpcr、微阵列、rna
‑
seq或分析。在此处所述的某些实施方案中,基因表达通过定量rna来测定,包括rna
‑
seq或分析。应当理解的是,基因表达可以使用不止一种技术进行测定。
[0028]
rna测序(rna
‑
seq)是一种转录组分析技术,它利用基于下一代测序(ngs)的下一代测序平台。rna
‑
seq转录本被逆转录成cdna,且接头与cdna的每一端连接。测序可以单向(单端测序)或双向(双端测序)进行,然后与参考基因组数据库比对或组装以获得重新的转录(de novo transcript),证明全基因组表达谱。rna
‑
seq可以定性和定量研究任何类型的rna,包括信使rna(mrna)、微rna、小干扰rna和长链非编码rna。
[0029]
rna可以采用nanostring ncounter基因表达分析法进行分析。nanostring是一种相对较新的分子表达谱技术,可以从少量固定的患者组织得到准确的基因组信息。nanostring平台采用数字、彩色编码的条形码或标记到序列特异性探针的代码集,可以对mrna表达进行量化(geiss等人nat biotechnol.2008mar;26(3);317
‑
25,das等人nanostring expression profiling identifies candidate biomarkers of rad001 response in metastatic gastric cancer,esmo open2016,1
‑
9)。nanostring系统将两个探针与每个目标转录物杂交:生物素标记的捕获探针和荧光条形码标记的报告探针。报告探针与样本中的特定rna杂交,捕获探针通过亲和素将它们锁定在静态表面上。nanostring ncounter分析系统使用其条形码对固定化rna进行计数。
[0030]
肺癌可以是非小细胞肺癌(nsclc)。nsclc可以选自浸润性腺癌(luad)、鳞状细胞癌(lusc)、大细胞癌、腺鳞癌、癌肉瘤、大细胞神经内分泌癌、未分化非小细胞肺癌或细支气管肺泡癌。luad和lusc占nsclc病例的大部分,其他类型往往被归为一组。nsclc可以是i期、ii期、iii期或iv期。
[0031]
样本可以来自手术切除的肿瘤。样本可以来自肺组织或肺肿瘤活检。
[0032]
预后可提供风险评估。
[0033]
所述方法可以进一步包括确定治疗方案。因此,我们还描述了用于确定受试者治
疗方案的方法,所述方法包括上述方法及进一步包括确定治疗方案的进一步步骤。所述治疗方案可以选自外科治疗、化疗、手术、放疗、免疫疗法或car
‑
t疗法。所述治疗方案是本领域熟悉的。应当理解的是,存在多种类型的免疫疗法,例如免疫检查点抑制剂、溶瘤病毒疗法、t细胞疗法和癌症疫苗。可以选择合适的疗法。
[0034]
我们还描述了包含一组试剂的组合物,所述试剂与一组生物标志物的每个成员特异性结合,所述生物标志物包括以下生物标志物或由以下生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1。
[0035]
我们还描述了包含试剂的试剂盒,所述试剂与一组生物标志物的每个成员特异性结合,所述生物标志物包括anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1。
[0036]
上述组合物或试剂盒中的所述试剂可以是核酸。我们还描述了组合物或试剂盒在为如上所述肺癌受试者提供预后的方法中的用途。我们还描述了组合物或试剂盒在为如上所述肺癌受试者提供治疗的方法中的用途。
[0037]
我们还描述了anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1在为如上所述肺癌受试者提供预后的方法中的用途。我们还描述了anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1在为如上所述肺癌受试者提供治疗的方法中的用途。
[0038]
我们还描述了治疗肺癌受试者的方法,包括预测肺癌受试者的死亡风险水平的步骤,所述方法包括(a)将来自受试者的生物样本与试剂接触,所述试剂与一组生物标志物中的每个成员特异性结合,所述生物标志物包括anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1;(b)根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;(c)将风险分数与阈值进行比较,以预测受试者是否存在高死亡风险;(d)选择治疗方案;(e)实施所述治疗方案。
[0039]
我们还描述了为癌症受试者生成生物标志物特征的方法,所述方法包括:从多个癌症受试者生成训练数据,所述训练数据包括多个受试者中每一个受试者多个基因的基因表达数据;根据基因表达数据,计算多个基因中每个基因的肿瘤内异质性度量和肿瘤间异质性度量;以及应用异质性过滤器,选择肿瘤内异质性低于肿瘤内异质性阈值且肿瘤间异质性高于肿瘤间异质性阈值的的基因;其中所述生物标志物特征包括所选基因中的至少一些基因。所述方法可适用于各种不同的癌症,特别是与ith相关的癌症。
[0040]
所述方法可以进一步包括:计算每个基因的一致性分数;并应用一致性过滤器选择一致性分数低于一致性阈值的基因。一致性过滤器可以被认为是一种去除混杂基因的异质性过滤器。所选基因的一致性分数可在应用异质性过滤器之后计算。或者,可以在计算肿瘤内异质性度量和肿瘤间异质性度量之前应用一致性过滤器。
[0041]
每个基因的肿瘤内异质性度量可以通过以下方式计算:获得每个基因在同一肿瘤
内多个位置的基因表达值,计算每个肿瘤指示每个基因所得基因表达值的度量,并获得作为每个肿瘤中每个基因指示性度量平均值的肿瘤内异质性度量。指示基因表达值的度量可以选自标准偏差、中值绝对偏差和变异系数。
[0042]
肿瘤间异质性度量可以通过以下方式计算:获得每个受试者每个基因在肿瘤多个区域其中一个区域的基因表达值;并对所得值取标准偏差。所述方法可以进一步包括多次迭代获得和取值步骤,并且平均迭代的标准偏差,获得肿瘤间异质性度量。应当理解的是,也可以使用除标准偏差之外的其他度量,例如变异系数和中值绝对偏差。
[0043]
生物标志物特征可以是预后的。所述方法可以进一步包括:为多个受试者中的每一个受试者产生包括相关生存数据的训练数据;根据生存数据计算多个基因中每个基因的预后度量;并且应用预后过滤器选择预后度量高于预后阈值的基因。预后度量可采用cox单变量回归分析计算。
[0044]
生物标志物特征可以预测受试者对特定治疗(例如免疫疗法)的反应。所述方法可以进一步包括:为多个受试者中的每一个受试者产生包括相关反应数据(例如,特定治疗的结果)的训练数据;根据反应数据计算多个基因中每个基因的预测度量;并且应用预测过滤器选择预测度量高于预测阈值的基因。预测度量可以采用回归分析,将基因表达与治疗反应相关联或治疗反应的代理度量来计算。所述方法可用于建立治疗反应的预测特征,帮助对患者进行分类,获得最合适的治疗方案。因此,如上所述产生的生物标志物特征有可能区分各种癌症亚型,并根据癌症亚型确定治疗策略。应当理解的是,上述提供预后的方法、确定受试者治疗方案的方法、组合物、试剂盒、治疗方法和用途可以适用于如上所述产生的任何特征。
[0045]
我们还描述了为癌症受试者提供预后的方法,所述方法包括:将来自受试者的生物样本与试剂接触,所述试剂与如上所述产生的特征中的一组生物标志物的每个成员特异性结合;根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;及根据受试者的风险分数提供癌症的预后。我们还描述了用于确定受试者治疗方案的方法,所述方法包括提供预后的方法及进一步包括确定治疗方案的进一步步骤。我们还描述了包含一组试剂的组合物,所述试剂与如上所述产生的特征中的一组生物标志物的每个成员特异性结合。我们还描述了包含试剂的试剂盒,所述试剂与如上所述产生的特征中的一组生物标志物的每个成员特异性结合。
[0046]
我们还描述了如上所述产生的特征中的生物标志物在为癌症受试者提供预后的方法中的用途。我们还描述了如上所述产生的特征中的生物标志物在为癌症受试者提供治疗方案的方法中的用途。我们还描述了治疗癌症受试者的方法,包括预测癌症受试者的死亡风险水平的步骤,所述方法包括将来自受试者的生物样本与试剂接触,所述试剂与如上所述产生的特征中的一组生物标志物的每个成员特异性结合;根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;将风险分数与阈值进行比较,以预测受试者是否存在高死亡风险;选择治疗方案;实施所述治疗方案。
[0047]
还可能提供计算机装置,包括至少一个处理器;以及指令,当所述指令由至少一个处理器执行时,使所述计算机装置执行上述方法的确定、计算和比较步骤中的任一个步骤。还可能提供其上记录指令的有形非瞬态计算机可读存储介质,当所述指令由计算机装置执行时,使计算机装置按上述方式布置和/或使计算机装置执行如上所述方法相关步骤的任
一个步骤。还可能提供试剂盒,包括计算机装置和组织样本的微阵列和/或一种或多种确定生物标志物存在的试剂。
[0048]
到目前为止,我们已经描述了采用包括或由23个特定生物标志物组成的一组生物标志物。我们现在描述使用一组生物标志物的实施方案,所述一组生物标志物包括选自23个特定生物标志物的两个或更多个生物标志物。
[0049]
我们还描述了为肺癌受试者提供预后的方法,所述方法包括:(a)将来自受试者的生物样本与和一组生物标志物中的每个成员特异性结合的试剂接触,所述一组生物标志物包括至少两个选自anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1的生物标志物;(b)根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;及(c)根据受试者的风险分数,提供肺癌的预后。
[0050]
确定受试者的风险分数可以包括:对于每个选择的生物标志物,确定指示组织样本中核酸表达水平的分数;根据确定的分数,计算风险分数,其中风险分数通过将加权的生物标志物的分数相加计算得出,其中生物标志物的分数基于确定的分数,并且每个生物标志物的分数都具有相关的权重;及将风险分数与阈值进行比较。这样,每个受试者都可能例如被分到高风险组(例如风险分数高于阈值)或低风险组(例如风险分数等于或低于阈值)。例如,当考虑所有类型的肺癌时,高风险组的生存率可能较低,而低风险组的生存率可能较高。或者,在考虑早期癌症时,高风险组可能比低风险组更容易复发。golga8a、scpep1、slc46a3和xbp1中的每个生物标志物分数的相关权重可能是负值,表明它们是有利的基因。anln、aspm、cdca4、errfi1、furin、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、snx7和tpbg中的生物标志物分数的相关权重可能是正值。
[0051]
风险分数的加权总和可以通过以下方式确定:
[0052]
风险分数=b1x
1i
+b2x
2i
+
…
+b
n
x
ni
[0053]
其中x
1i
、x
2i
、
……
、x
ni
是每个受试者i的四个选定生物标志物的生物标志物分数,b1、b2、
……
、b
n
是每个生物标志物分数的一组相关权重。
[0054]
所述方法还可以包括采用cox比例风险模型确定加权总和的权重,该模型采用包括有关一组受试者多个生物标志物的信息的训练数据进行训练。所述方法可包括识别在cox比例风险模型中使用的多个生物标志物,其中所述多个生物标志物选自anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。
[0055]
阈值可以是训练数据的中值风险分数。
[0056]
确定指示生物标志物水平的分数可以包括确定缩放强度分数。生物标志物分数可以基于已通过减去调整因子而调整的缩放强度分数。确定指示生物标志物水平的分数可以包括当水平高于阈值时赋予第一值,当水平低于阈值时赋予第二值。确定指示生物标志物水平的分数可以包括当水平高于上阈值时赋予第一值,当水平低于上阈值但高于下阈值时赋予第二值,以及当水平低于下阈值时赋予第三值。
[0057]
试剂可以是核酸。
[0058]
肺癌可以是非小细胞肺癌(nsclc)。nsclc可以选自浸润性腺癌(luad)、鳞状细胞癌(lusc)、大细胞癌、腺鳞癌、癌肉瘤、大细胞神经内分泌癌、未分化非小细胞肺癌或细支气
管肺泡癌。luad和lusc占nsclc病例的大部分,其他类型往往被归为一组。nsclc可以是i期、ii期、iii期或iv期。
[0059]
样本可以来自手术切除的肿瘤。样本可以来自肺组织或肺肿瘤活检。
[0060]
预后可提供风险评估。
[0061]
所述方法还可以包括确定治疗方案。因此,我们还描述了用于确定受试者治疗方案的方法,所述方法包括上述方法及进一步包括确定治疗方案的进一步步骤。所述治疗方案可以选自外科治疗、化疗、手术、放疗、免疫疗法或car
‑
t疗法。所述治疗方案是本领域熟悉的。应当理解的是,存在多种类型的免疫疗法,例如免疫检查点抑制剂、溶瘤病毒疗法、t细胞疗法和癌症疫苗。可以选择合适的治疗。
[0062]
我们还描述了包含一组试剂的组合物,所述试剂与一组生物标志物的每个成员特异性结合,所述一组生物标志物包括选自以下生物标志物的至少两个生物标志物或由选自以下生物标志物的两个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1。
[0063]
我们还描述了包含试剂的试剂盒,所述试剂与一组生物标志物的每个成员特异性结合,所述一组生物标志物包括选自以下生物标志物的至少两个生物标志物:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1。上述组合物或试剂盒中的所述试剂可以是核酸。我们还描述了组合物或试剂盒在为如上所述肺癌受试者提供预后的方法中的用途。我们还描述了组合物或试剂盒在为如上所述肺癌受试者提供治疗方案的方法中的用途。
[0064]
我们还描述了选自anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1的至少两个生物标志物在为肺癌受试者提供预后的方法中的用途。我们还描述了选自anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg和xbp1的至少两个生物标志物在为如上所述肺癌受试者提供治疗方案的方法中的用途。
[0065]
我们还描述了治疗肺癌受试者的方法,包括预测肺癌受试者的死亡风险水平的步骤,所述方法包括:(a)将来自受试者的生物样本与和一组生物标志物中的每个成员特异性结合的试剂接触,所述一组生物标志物包括选自anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1的至少两个生物标志物;(b)根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;(c)将风险分数与阈值进行比较,以预测受试者是否存在高死亡风险;(d)选择治疗方案;(e)实施所述治疗方案。
[0066]
在本发明的每个实施方案中,一组生物标志物包括生物标志物的选择,本领域技术人员将理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少3个生物标志物或由选自以下生物标志物的至少3个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可
以包括选自以下生物标志物的至少4个生物标志物或由选自以下生物标志物的至少4个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少5个生物标志物或由选自以下生物标志物的至少5个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少6个生物标志物或由选自以下生物标志物的至少6个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少7个生物标志物或由选自以下生物标志物的至少7个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少8个生物标志物或由选自以下生物标志物的至少8个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少9个生物标志物或由选自以下生物标志物的至少9个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少10个生物标志物或由选自以下生物标志物的至少10个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少11个生物标志物或由选自以下生物标志物的至少11个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少12个生物标志物或由选自以下生物标志物的至少12个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少13个生物标志物或由选自以下生物标志物的至少13个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少14个生物标志物或由选自以下生物标志物的至少14个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少15
个生物标志物或由选自以下生物标志物的至少15个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少16个生物标志物或由选自以下生物标志物的至少16个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少17个生物标志物或由选自以下生物标志物的至少17个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少18个生物标志物或由选自以下生物标志物的至少18个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少19个生物标志物或由选自以下生物标志物的至少19个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少20个生物标志物或由选自以下生物标志物的至少20个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少21个生物标志物或由选自以下生物标志物的至少21个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括选自以下生物标志物的至少22个生物标志物或由选自以下生物标志物的至少22个生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。
[0067]
在本发明的每个实施方案中,一组生物标志物包括生物标志物的选择,本领域技术人员将理解的是,所述一组生物标志物可以包括anln以及以下生物标志物的至少一个或由anln以及以下生物标志物的至少一个组成:aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括aspm以及以下生物标志物的至少一个或由aspm以及以下生物标志物的至少一个组成:anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括cdca4以及以下生物标志物的至少一个或由cdca4以及以下生物标志物的至少一个组成:aspm、anln、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解
的是,所述一组生物标志物可以包括errfi1以及以下生物标志物的至少一个或由errfi1以及以下生物标志物的至少一个组成:aspm、anln、cdca4、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括furin以及以下生物标志物的至少一个或由furin以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括golga8a以及以下生物标志物的至少一个或由golga8a以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括itga6以及以下生物标志物的至少一个或由itga6以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括jag1以及以下生物标志物的至少一个或由jag1以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括lrp12以及以下生物标志物的至少一个或由lrp12以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括maff以及以下生物标志物的至少一个或由maff以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括mrps17以及以下生物标志物的至少一个或由mrps17以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括plk1以及以下生物标志物的至少一个或由plk1以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括pnp以及以下生物标志物的至少一个或由pnp以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括ppp1r13l以及以下生物标志物的至少一个或由ppp1r13l以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括prkca以及以下生物标志物的至少一个或由prkca以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、pttg1、pygb、
rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括pttg1以及以下生物标志物的至少一个或由pttg1以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括pygb以及以下生物标志物的至少一个或由pygb以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括rpp25以及以下生物标志物的至少一个或由rpp25以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、scpep1、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括scpep1以及以下生物标志物的至少一个或由scpep1以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、slc46a3、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括slc46a3以及以下生物标志物的至少一个或由slc46a3以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、snx7、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括snx7以及以下生物标志物的至少一个或由snx7以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、tpbg、xbp1。还应当理解的是,所述一组生物标志物可以包括tpbg以及以下生物标志物的至少一个或由tpbg以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、xbp1。还应当理解的是,所述一组生物标志物可以包括xbp1以及以下生物标志物的至少一个或由xbp1以及以下生物标志物的至少一个组成:aspm、anln、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg。
[0068]
本领域技术人员将理解的是,anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1中的两个或更多个生物标志物的任何组合足以提供肺癌受试者的预后或确定治疗方案。
[0069]
尽管已经示出和描述了本发明的一些优选实施方案,但是本领域技术人员将理解的是,可以进行各种改变和修改,却并不背离所附权利要求定义的本发明的范围。
附图说明
[0070]
为了更好地理解本发明,并且说明如何实施本发明的实施例,现在将仅以示例的方式参考附图,其中:
[0071]
图1a是肺肿瘤的示意图,示出了说明肿瘤采样偏差问题的采样点;
[0072]
图1b是预测方法的步骤和图1a肿瘤采样偏差问题的临床意义的示意图;
[0073]
图1c是采用特定的已知特征绘制的风险分数
‑
患者图;
[0074]
图1d和1e是条形图,显示了采用两个已知特征的低、高和不一致风险患者的比例;
[0075]
图1f是9个已知特征各肿瘤区域属于同一聚类的患者百分数相对聚类数量的图形。
[0076]
图2a是开发和验证预后特征的方法的步骤流程图;
[0077]
图2b是预后方法步骤的流程图;
[0078]
图2c是实施图2b所述方法的系统组件的示意性框图;
[0079]
图2d和2e分别是对两个基因ckmt2和hoxc11来说,所有样本都属于同一聚类的患者比例相对聚类数量的图形;
[0080]
图2f是层次聚类一致性相对每个基因的图形;
[0081]
图3a说明了计算多个基因的rna肿瘤内异质性的步骤;
[0082]
图3b是每个基因的中值绝对偏差(mad)相对标准偏差分数的图形;
[0083]
图3c是每个基因的变异系数(cv)相对标准偏差分数的图形;
[0084]
图3d说明了计算rna肿瘤间异质性度量的随机抽样过程;
[0085]
图3e是多个基因的rna肿瘤内异质性数值(y
‑
轴)相对rna肿瘤间异质性数值(x
‑
轴)的图形;
[0086]
图4a示出了验证队列三个特征中每个特征的预后值;
[0087]
图4b示出了预测值相对已知风险因素的森林图;
[0088]
图4c、4d和4e示出了亚分期标准的预后值、当前的化疗临床指南和i期患者的输出特征,其中改进的风险预测可能影响临床决策;
[0089]
图4f示出了多个患者输出特征的风险分数;
[0090]
图4g示出了采用rna
‑
seq数据集和四个微阵列数据集的预后值评估图;
[0091]
图4h示出的图表明oracle特征的任何子集都可能具有预后值;
[0092]
图5a示出了不同癌症类型的rna异质性象限的预后关联性;
[0093]
图5b比较了多种癌症类型每个象限内基因的表现,以确定这些象限是否富集或缺乏预后基因;
[0094]
图6a是基因表达ith相对拷贝数ith的图形;
[0095]
图6b示出了亚克隆染色体拷贝数变化(分别为减少和增加)的表达差异;
[0096]
图6c示出了rna异质性象限的克隆拷贝数增加;及
[0097]
图6d示出了rna异质性象限q4中富集的反应组通路(reactome pathways)。
具体实施方式
[0098]
如上所述,图1a至1f表明采样偏差会使分子生物标志物在几种癌症类型中的使用变得混乱。这是因为作为肿瘤进化的底物,肿瘤内异质性(即单个肿瘤内遗传和转录组特征的空间变异性)会影响分子生物标志物应用的结果,因为结果可能取决于被检测肿瘤样本的位置。针对肿瘤采样偏差问题,人们已经提出了多种解决方案,包括利用多区域测序来(i)汇集多个活检,以获得单个肿瘤的整体分子风险估计(如blackhall等人在neoplasia(2004)发表的“stability and heterogeneity of expression profiles in lung cancer specimens harvested following surgical resection”中所述;或(ii)鉴定具有
最大免疫逃避性的“致命”亚克隆(例如,如mlecnik等人在jncl j natl cancer inst 110,97
‑
108(2018)上发表的“comprehensive intrametastatic immune quantification and major impact of immunoscore on survival”中所述)或转移潜能(例如,如yachida等人在nature 467,1114
‑
1117(2010)上发表的“distant metastasis occurs late during the genetic evolution of pancreatic cancer”中所述)。然而,在临床上,多区域测序目前是不切实际的。
[0099]
图2a是开发一组生物标志物(或基因表达特征)的步骤流程图,所述生物标志物产生可靠的预后结果,适用于临床实践中常规采集的单区域肿瘤样本。正如下文更详细解释的那样,所述一组生物标志物包括具有低肿瘤内异质性但高肿瘤间异质性的基因,最大限度地降低采样偏差的混乱影响,但最大限度地提高了患者之间的区分能力。
[0100]
第一步s100是收集训练数据,例如来自癌症基因组图谱(tcga)的959名i至iii期nsclc患者(469名luad患者和490名lusc患者)的基因表达和生存数据。该数据形成一个训练数据集,用于导出如下所述的特征。因此可以按照rna
‑
seq预处理管道中的标准方法处理下载的数据以形成训练数据。例如,可以与人类基因组进行比对,例如采用67中描述的mapsplice包。然后,可以对基因表达进行定量,例如,采用bioconductor的genomicfeatures和genomic ranges包。然后,可以应用表达过滤器,保留至少2个肿瘤样本中至少0.5cpm的基因,如步骤s101所示。然后,采用love等人在genome biol 15,550(2014)上发表的“moderated estimation of fold change and dispersion for rna
‑
seq data with deseq2”中所述来自deseq2包的方差稳定转换,获得过滤基因的归一化计数值。应当理解的是,当开发不同疾病的预后特征时,还可以收集不同患者的数据。
[0101]
下一步s102是计算每个基因的预后度量,识别显著的预后基因。然后应用第一过滤步骤s104根据基因的预后效应去除基因(即选择预后度量高于阈值的基因)。这些基因中的每一个基因都对每位患者的总体生存率具有未知的影响。预后度量可采用任何合适的方法计算。
[0102]
例如,可以采用cox单变量回归分析。cox模型由h(t)表示的风险函数表示。风险函数可以解释为在时间t死亡的风险。其可以估计如下:
[0103]
h(t)=h0(t)
×
exp(b1x1+b2x2+
…
+b
p
x
n
)
[0104]
其中t表示生存时间,h(t)是由一组n个协变量(x1,x2,
……
,x
n
)确定的风险函数
‑
在这种情况下是基因,组(b1,b2,
……
,b
n
)是每个协变量的权重(或系数),术语h0称为基线风险,与所有x
i
都等于0(数量exp(0)等于1)的风险值相对应。h(t)中的“t”提醒我们风险可能会随时间变化。但是,可以消除时间方差,从而可以通过对患者i与参考组的风险比取对数而以线性形式重写该模型,该模型可以写为:
[0105][0106]
这个线性方程被称为cox比例风险模型,其中包含每个患者i的一组n个协变量(即基因)(x
1i
,x
2i
,
……
,x
ni
),和针对所有患者优化模型的一组权重(b1,b2,
……
,b
n
)。单变量分析指的是逐个考虑每个变量。通常,对于每个变量,计算系数及系数附近95%置信区间的下限和上限(分别为ci95l和ci95u)。p值是变量统计显著性的度量,采用wald检验或时序检验(log
‑
rank)计算。q值是采用benjamini&hochberg方法调整后的p值。
[0107]
如步骤s104所示,可以应用一个以上预后过滤器。例如,第一过滤器可以包括根据预后显著性阈值过滤所有基因,例如,在此实施例中p<0.05可以将基因数量从19026个减少到4240个。可以采用第二过滤器,根据中值阈值过滤基因,例如,可以去除预后度量低于预后阈值的所有基因。在这个实施例中,这可以将基因数量从19026个减少到9512个。这两个阈值可以合在一起作为一个预后阈值考虑,因此总的来说,第一个过滤步骤可以将基因的数量从19026个减少到2023个。
[0108]
然后可以采用第二过滤步骤s106。该过滤器可称为克隆表达过滤器或异质性过滤器。正如下文更详细地解释的那样,克隆表达过滤器可以去除既不具有低肿瘤内异质性又不具有高肿瘤间异质性的基因(即选择既具有低肿瘤内异质性又具有高肿瘤间异质性的基因)。在这个实施例中,这可能将基因数量从2023个减少到176个。
[0109]
然后可以采用第三过滤步骤s108。这个可以被称为一致性过滤器的过滤器可以根据基因聚类一致性分数筛选出剩余的基因。聚类一致性分数可采用任何合适的方法计算。例如,一致性可以通过对癌症表达数据的层次聚类分析来确定,其中从每个肿瘤获得多个样本,例如,采用曼哈顿度量ward法,如gyanchandani等在clin.cancer res.22,5362
–
5369(2016)上发表的“intratumor heterogeneity affects gene expression profile test prognostic risk stratification in early breast cancer”中所述。一致性在每个基因水平上确定为所有样本归为同一聚类的肿瘤百分数。聚类分析可以从2到患者总数(例如,此tracerx luad队列中的28个)迭代分析。对于每个基因,可以将所有区域都属于同一聚类的患者数量对照聚类数作图得到一条曲线。例如,如图2d和2e所示,对两个基因ckmt2和hoxc11来说,所有样本都属于同一聚类的患者比例对照聚类数作图。然后将每个基因的聚类一致性分数总结为曲线下的面积。每个基因的一致性分数可以如图2f所示作图,图中示出了每个基因的层次聚类一致性。一旦计算出每个基因的一致性分数,就可以去除一致性分数低于一致性阈值的所有基因。一致性阈值(即截止值)可以采用十倍交叉验证来确定。在这个实施例中,这可能将基因数量从176个减少到90个。
[0110]
对于实用的预后试剂盒来说,此基因数量可能仍然太多,因此可以采用标准方法,如套索回归(lasso regression)(s110)任选进一步减少基因的数量。可以采用glmnet包在rsoftware环境中应用套索回归,该glmnet包在friedman等人发表在j stat softw 33,1
‑
22(2010)上的“regularised paths for generalized linear models via coordinate descent”中进行了描述,用于采用套索惩罚(α=1)的cox比例风险模型(例如,如simon等人在j stat softw 39,1
‑
13(2011)上发表的“regularisation paths for cox’s proportional hazards model via coordinate descent”中所述)。在这个实施例中,这可能将基因数量从90个减少到23个。然后,输出得到的23个基因组(即特征)(s112)。得到的特征可称为oracle特征(结果风险相关的克隆肺表达)。输出特征的预后准确性可以采用验证数据评估(s114)。
[0111]
应当理解的是,图2a中的每个过滤步骤可以以任何顺序应用。所示顺序仅仅是示例性的而非限制性的。
[0112]
预后生物标志物特征包括以下基因:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1。有五个基因与细胞增殖有关:anln、aspm、cdca4、plk1、prkca以
及6个基因(errfi、furin、itga6、jag1、ppp1r13l、pttg1)与致癌信号通路有关。这些基因中只有7个基因以前被用于luad预后特征,即aspm、furin、plk1、pnp、prkca、pttg1和ttbg。预后生物标志物预测独立于治疗的生存风险。
[0113]
为肺癌受试者提供预后或预测风险水平的方法,所述方法包括:
[0114]
a)将来自受试者的生物样本与和一组生物标志物中的每个成员特异性结合的试剂接触,所述一组生物标志物包括以下生物标志物或由以下生物标志物组成:anln、aspm、cdca4、errfi1、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pttg1、pygb、rpp25、scpep1、slc46a3、snx7、tpbg、xbp1;
[0115]
(b)根据样本中生物标志物的核酸表达水平,确定受试者的风险分数;及
[0116]
(c)根据受试者的风险分数提供肺癌的预后。
[0117]
所述方法还可包括从患者采集样本。样本可以是肿瘤样本。此处提供的方法、试剂盒和组合物中使用的试剂可以是核酸,例如寡核苷酸或引物。
[0118]
此处所述预后涉及临床结果,例如总体生存率、中期或长期死亡率(例如1、2、3、4或5年)或无病生存率。
[0119]
应当理解的是,图2a示出了一种用于生成预后特征的方法,但是,所述方法可以通过用预测性度量代替预后度量而容易地适用于预测。
[0120]
图2b提供了预后的方法实施例。图中示出了使用输出特征的预后方法中可以执行的步骤。第一步是接触生物样本(步骤s200)。生物样本可以是采用任何合适的方法获得的肿瘤样本,例如活检获得的供体样本。然后,采用标准技术确定肿瘤样本中23个基因中每个基因的数值(步骤s202)。
[0121]
下一步(s204)是根据23个基因中每个基因数值的加权总和来确定风险分数。因此,风险分数可以通过以下方式计算:
[0122]
风险分数=b1x
1i
+b2x
2i
+
…
+b
n
x
ni
[0123]
式中x
1i
、x
2i
、
……
、x
ni
是每个患者i的23个选择基因的数值,b1、b2、
……
、b
n
是每个基因的一组相关权重。权重可以采用如上所述的套索回归确定。
[0124]
例如,下面示出了特征中每个基因的合适权重。具有正β系数的基因与风险比>1相关(即是“不利基因”,预测较差的生存率),具有负系数的基因(“有利基因”)则与之相反。应当理解的是,这些权重指示合适的数值而不是限制性的。
[0125]
[0126][0127]
回到图2b,一旦确定了风险分数,就将其与风险分数阈值进行比较(步骤s206)。如果风险分数等于或高于阈值,则认为患者无法存活的风险较高,因此将患者归类为高风险患者(步骤s210)。相反,如果风险分数低于阈值,则认为患者无法存活的风险较低,因此将患者归类为低风险患者(步骤s212)。阈值可以例如是用于导出特征的数据的中值风险分数和/或最显著分割的中值风险分数(时序检验p<0.01)。换句话说,阈值可以是最显著地将训练队列分成复发和非复发(即治愈)患者的风险分数。
[0128]
作为步骤s208的替代,可以将风险分数与上阈值和下阈值进行比较。如果风险分数等于或高于上阈值,则患者归类为高风险患者。如果风险分数低于下阈值,则患者归类为低风险患者。如果风险分数位于这两个阈值之间,则患者归类为中风险患者。正如下文所解释的那样,上阈值和下阈值可被确定为训练队列确定的风险分数的三分位数。
[0129]
一旦确定了风险分数,这可以任选用于决定最合适的治疗。例如,对于高风险患者,建议采用辅助化疗以补充手术。与单纯化疗相比,这种治疗可提高总体生存率。这对于i期患者尤其重要,因为在i期患者中,缺乏识别高风险患者的临床指标。目前,i期患者倾向于不接受化疗,导致约25%的i期患者在5年内复发。相比之下,对于低风险患者,治疗可以从单独手术或以上指定的联合手术方法中选择。在这种情况下,两种选择都同样有效。
[0130]
用于执行该方法的相关系统的示意图如图2c所示。所述系统包括计算装置210,所述计算装置210可以是手持便携式装置,临床医生可以将其携带用于不同的患者,并且可以在所述装置上安装计算风险分数的应用程序。计算装置210包括标准组件(例如处理单元或处理器220)、用于允许用户输入信息(例如确定分数)的用户界面单元222,以及用于存储执行计算的代码和/或用于比较计算风险分数的阈值的存储器224。用户界面可以显示信息或者替代地,可以有显示器224用于向用户显示信息,例如计算的风险分数和/或如上所述的治疗建议,以及用于与其他设备通信和/或访问云端240的通信模块228,例如,用于处理风险分数。还示意性地示出了组织样本230。
[0131]
所述示意系统可以部分或全部采用专用硬件构建。此处使用的例如“模块”或“单元”等术语可包括但不限于执行某些任务或提供相关功能的硬件设备,例如分立或集成组件形式的电路、现场可编程门阵列(fpga)或专用集成电路(asic)。在一些实施例中,所描述的元件可以被配置为驻留在有形的、持久的、可寻址的存储介质上,并且可以被配置为在一个或多个处理器上执行。在一些实施例中,这些功能元件可以包括,例如,组件(例如软件组件、面向对象的软件组件、类组件和任务组件)、过程、功能、属性、程序、子程序、程序代码段、驱动程序、固件、微代码、电路、数据、数据库、数据结构、表格、数组和变量。尽管已经参考此处讨论的组件描述了示例实施例,但是,这些功能元件可以组合成更少的元件或分开成其它的元件:
[0132]
图3a至3e说明了如何创建克隆表达过滤器。对于每个基因,计算rna肿瘤内异质性值和rna肿瘤间异质性值。这些每基因指标可以通过同一肿瘤内各区之间的标准偏差来量化变异性,以生成肿瘤内异质性值,并通过不同肿瘤相同肿瘤区域之间的标准偏差来量化变异性,以生成肿瘤间异质性值。它们可以采用多区域rnaseq数据(归一化计数值)计算。
[0133]
图3a至3e绘制了来自伦敦大学学院赞助的tracerx肺癌研究中100名nsclc患者数据集的肿瘤样本数据。进行多区域取样以从同一组织中依次获得dna和rna。对dna样本进行全外显子组测序。在100个肿瘤的队列中,从68个肿瘤的174个区域获得了足够质量的rna样本。其中,至少有两个样本来自48个肿瘤。
[0134]
可以根据需要进行进一步处理。例如,进行比对,例如,采用dobin等人在bioinformatics29,15to 21(2013)发表的“star;ultrafast universal rna
‑
seq aligner”中描述的star包,将读长映射到人类基因组。例如,采用li等人在bmc bioinformatics 12,323(2011)上发表的“rsem;accurate transcript quantification from rna
‑
seq data with or without a reference genome”描述的rsem包对转录表达进行量化,以生成计数和每百万表达值的转录本数(tpm)。采用表达过滤器,保留至少20%(30/156)肿瘤样本中至少1tpm的基因。最后,采用上述deseq2包将方差稳定转换应用于来自过滤基因的计数(假设计数值呈负二项式分布)。输出同方差和文库大小归一化计数值按如下所述使用。在此实施例中,可能有19206个基因需要考虑。
[0135]
如图3a所示,对于每个患者(例如cruk0003),确定了每个基因在多个位置(例如r1至r8)的基因表达。例如,图3a左侧的图示出了edc4、calm2和prom1在多个位置的基因表达。对于给定的肿瘤,可以计算特定基因在各肿瘤区域的表达值的标准偏差,产生基因特异性、患者特异性的rna肿瘤内异质性度量(σ
g,p
)。对于示例患者,这些都显示在图3a中心的表格中,三个基因分别是0.075、0.552和2.248。因此,edc4在整个肿瘤中几乎没有变化,但prom1
seed
‑
and
‑
vote”中描述的subread包。采用durinck等人在nat protoc 4,1184
‑
1191(2009)发表的“mapping identifiers for the integration of genomic datasets with the r/bioconductor package biomart”中描述的biomart包将基因id转换为hgnc id。然后选择最大值用于多映射探针。将上述训练数据集中识别出的低表达基因从验证数据集中过滤掉,并采用上述deseq2包应用方差稳定转换,以输出归一化计数值。还获得了额外的临床信息(例如治疗状态和肿瘤大小)。
[0142]
图4a比较了输出23个基因特征的性表现与基于已知论文的类似特征的表现。特征a基于shukla等在jncl j natl cancer inst 109(2017)发表的“development of a rna
‑
seq based prognostic signature in lung adenocarcinoma”中描述的特征构建管道。所述特征源自shukla论文中确定的基因,并采用标准技术选择所述特征的几个基因。例如,采用来自tcga数据库的训练数据集,特别是luad患者,执行单变量cox回归分析,并应用一级预后过滤器(单变量cox分析p<0.00025),以将shukla论文中鉴定的基因数量减少到108个。另一个预后过滤器,这次是单变量cox分析fdr<0.02,将108个基因减少到15个。最后,应用正向条件逐步回归以产生6
‑
基因特征。因此遵循了shukla论文中概述的步骤,但不同的训练数据产生了6
‑
基因特征,而不是shukla第4页上包含4个基因的预后模型。
[0143]
特征b基于kratz等人在lancet 379,823
‑
832(2012)上发表的“a practical molecular assay to predict survival in resected non
‑
squamous,non
‑
small lung cancer;development and international validation studies”中描述的特征构建管道。在特征b的开发过程中,首先将背景部分表格所列论文中确定的所有基因整理到一个列表中。例如,采用来自tcga数据库的训练数据集,特别是luad患者,执行单变量cox回归分析,并应用一级预后过滤器(单变量cox分析p<0.00025),将鉴定基因数量减少到249个。采用二级预后过滤器,通过仅列出与癌症相关的基因,将基因数量从249个减少到56个。最后,应用套索回归,得到24个基因预后特征。与特征a一样,此特征b是采用kratz论文中描述的方法推导出来的,但由于训练队列而导致不同的基因选择。这两个特征都与上述24个基因特征相当。
[0144]
图4a示出了三个特征中每个特征的预后值。采用来自uppsala数据集的验证数据对这三个特征的预后准确性进行测试。如图所示,图2a所示过程产生的特征预测了显著的生存风险(时序检验p=0.006),并优于特征a和b。换句话说,图4a表明,与采用特征a和b的风险分数相比,采用图2a过程推导的特征计算风险分数,验证队列中的患者可能更成功地分成生存时间明显不同的亚组。
[0145]
图4b是森林图,显示了该新特征与其他已知风险因素结合时的预测值。在图4b中,执行多变量(而不是之前的单变量)分析,以证明计算的风险分数(作为连续变量的输入)即使在整合临床信息以预测生存率时也保持显著性。死亡的相对风险(风险分数)以实心块显示,是肿瘤分期(例如i至iii期)、治疗状态(不采用或采用一些辅助治疗)和使用输出(oracle)特征计算的风险分数的综合函数。95%的置信水平也由条形图指示。风险比越高,死亡风险就越大,不出所料,iii期患者的值最高。图4b显示,当采用肿瘤分期和治疗状态的多变量分析时,输出特征是显著的(cox mva p=0.0247),因为该特征提供了额外的预后信息。
[0146]
图4c至4e显示了i期患者的临床可操作信息。验证数据集中大约有60名这样的患
者。图4c显示了i期患者分为两组:根据亚分期标准(时序检验p=0.52)分为ia(n=42)和ib(n=18)。以这种方式对患者进行分类并不能有效地将患者分为总生存率高和总生存率低的患者。类似地,图4f示出了根据肿瘤大小将i期患者分为高风险患者和低风险患者。目前的临床指南认为对i期luad患者来说,ib期肿瘤大于4cm的患者是高风险患者,而其他患者(即ia期肿瘤或尺寸小于4cm的ib期肿瘤)是低风险患者。在60名患者中,只有5名患者属于高风险患者。如图4d所示,这些患者未能很好地分为总生存率高和总生存率低的患者。
[0147]
图4e显示了采用输出(oracle)特征将i期患者分为两组:高风险组(红色)和低风险组(蓝色)。如图所示,这种划分在预测患者的生存率方面有效很多。
[0148]
图4f说明了肿瘤采样偏差对输出(oracle)特征的影响。采用计算的风险分数将肿瘤区域分类为“高风险”或“低风险”。然后,对单个患者的不一致分类进行评估,由此来自同一肿瘤的不同区域可能被分类为具有不同的分子风险特征。如图中所示,只有3/28名患者(即11%)不一致,这比图1c和1d中显示的不一致率低很多。
[0149]
图4g示出了采用rna
‑
seq数据集和四个微阵列数据集的预后值评估图。为了研究多个队列的一致性,将输出(oracle)特征应用于四个微阵列数据集。具体而言,在luad患者的五个验证队列(n=904名i
‑
iii期luad患者)的荟萃分析中对输出(oracle)特征的预后值进行了评估。在一个rna
‑
seq数据集和四个微阵列数据集中开展单变量cox分析。在微阵列队列中,23个基因中有19个基因可用于分析(aspm、cdca4、furin、golga8a、itga6、jag1、lrp12、maff、mrps17、plk1、pnp、ppp1r13l、prkca、pygb、scpep1、slc46a3、snx7、tpbg、xbp1)。显示了每个队列具有95%置信区间的风险比,并以自然对数标度绘制。预计输出特征的表现会更差,因为23个基因中只有19个基因与微阵列探针集匹配,并且使用了在rna
‑
seq数据上训练的特征权重。但是,在四个微阵列数据集中的三个数据集中,oracle与生存率显著相关。荟萃分析考虑了所有验证队列—菱形表示五个验证队列荟萃分析的风险比—这表明oracle与结果显著相关,总体风险比为3.57。这些数据表明,通过在生物标志物设计中控制rna
‑
ith,可以获得不因表达谱技术差异而受到影响的生存关联。有关这种分析的更多信息,请参见d.biswas等人在nature medicine 25,1540
‑
1548(2019)上发表的“a clonal expression biomarker associates with lung cancer mortality”。
[0150]
图4h绘制了从oracle特征中选择的1至23个基因组合的预后值。考虑了从完整的oracle特征中选择基因组合的两种程序,作为对23个基因的每个组合进行详尽搜索的计算高效替代方案。反向构建子集从包含所有23个基因的完整模型开始,评估所有22个基因组合,然后选择具有最高预后意义的最佳组合。该程序反复进行,一次去除一个基因,直到留下一个基因。正向构建子集从不包含任何基因的模型开始,然后加入对模型产生最高预后意义的基因,一次加入一个基因,直到包括所有23个基因。重要的是,每个基因的权重都没有重新训练,因此每个组合都作为上面定义的完整oracle特征的一个子集进行评估。这些数据表明,oracle特征的23个基因中的两个或多个基因的任何组合都可能具有预后值。这两个程序的数据见附录a。
[0151]
图5a和5b说明了上述图2a中描述的方法可能对其他癌症类型具有预后相关性。图3a至3f中描述的克隆表达过滤器是通过利用来自tracerx肺队列的完整多区域rnaseq数据集生成的,所述数据集包含来自多区域lusc肿瘤和其他nsclc组织学的数据。然后,采用上述相同的逐基因指标,将该数据用于计算每个基因的肿瘤内异质性分数和肿瘤间异质性分
数。如上所述,这些基因被分为四个象限。
[0152]
然后,对于每个象限中给出泛癌显著性预后值的每个基因的比例进行评估,并显示在图5a中。例如,可以从gentles等人在nat med 21,938
‑
945(2015)发表的“the prognostic landscape of genes and infiltrating immune cells across human cancers”中描述的precog资源下载泛癌逐基因预后值。precog资源是一个荟萃数据集,汇总了166个微阵列数据集,涵盖39种不同的恶性组织学。该数据集包括之前采用cox单变量回归分析计算的z分数。选择|z|分数>1.96(相当于双侧p<0.05)的基因。与图3a至3f中的分析一致,与所有其他象限相比,q4象限中的基因(即高肿瘤间异质性值和低肿瘤内异质性值的基因)表现出明显更高的泛癌z分数(反映显著的预后能力)。
[0153]
图5b还比较了每个象限内基因的表现,以确定这些基因是否富集或缺乏预后基因。图5b中的每个点对应于来自precog数据库的33种癌症类型中的一种。每种癌症类型每个nsclc rn异质性象限中预后显著的基因数量(|z|分数>1.96)以不显著(灰色)、显著富集(红色)或显著缺乏(蓝色)表示。如图5b所示,q4中的基因在49%(19/39)的癌症类型中显著富集预后基因,仅在3/%(1/39)的头颈癌中显著缺乏。相反的是,q1中的基因(低肿瘤间异质性值和高肿瘤内异质性值的基因)在任何癌症类型中都没有显著富集,且在56%(22/39)的癌症中缺乏。q2中的基因(低肿瘤间异质性值和低肿瘤内异质性值的基因和q3中的基因(高肿瘤间异质性值和高肿瘤内异质性值的基因)显示出相似数量的缺乏和富集癌症类型。
[0154]
图6a至6c探索了支持rna
‑
ith的基因组机制。首先考虑了如上所述采用多区域rnaseq数据计算的rna
‑
ith分数与采用多区域wes数据量化的拷贝数异质性之间的关系,这些数据在jamal
‑
hanjani等人在n engl j med 376,2109
‑
2121(2017)上发表的“tracking the evolution of non
‑
small cell lung cancer”中进行了描述。图6a是基因表达ith与拷贝数ith的关系图。从tracerx luad队列中,将逐个患者的rna
‑
ith分数对照逐个患者的scna
‑
ith分数作图。图6a显示每个患者的中值rna
‑
ith分数与每个患者的亚克隆scna事件百分数之间存在显著相关性(rs=0.48,p=0.0162)。这表明scna
‑
ith可能导致转录组异质性。
[0155]
图6b显示亚克隆拷贝数增加与表达增加之间以及亚克隆拷贝数缺失与表达减少之间存在高度显著相关性(p<0.001)。该数据表明亚克隆水平的染色体拷贝数增加和缺失与基因转录之间存在关联,并且rna
‑
ith反映了正在进行的cin和异质dna拷贝数事件的可能选择。
[0156]
图6c显示了每个象限的克隆拷贝数增加优势比。图6c对tracerx队列中最常显示克隆拷贝数增加(上四分位数)与很少显示克隆拷贝数增加(下四分位数)的每个象限内基因的相对富集进行了评估。图6c显示,在tracerx中出现克隆拷贝数增加事件的q4基因高度显著富集(p=1.18e
‑
05,费希尔(fisher)精确检验),而q3基因富集程度更低(p=0.000109,费希尔精确检验)。相比之下,q2基因缺乏(p=6.86e
‑
08,费希尔精确检验)。该数据表明,肿瘤中的均质表达可能源于肿瘤进化早期选择的克隆dna拷贝数的改变。
[0157]
图6d显示了q4中富集的反应组通路,这些通路与细胞增殖有关,包括有丝分裂、核小体组装和表观遗传调控。相比之下,对其他象限中基因通路的相同分析表明,q1基因没有显著富集,q2基因显示参与rna剪接过程,q3基因显示参与gpcr配体结合和细胞外基质组织。这种分析表明q4基因可能与肿瘤侵袭性的特定生物学特征有关,这可能解释了它们的
预后差别通路。
[0158]
此处已经描述了任选特征的各种组合,并且应当理解的是,所描述的这些特征可以以任何合适的组合进行组合。特别是,任何一个示例实施例的特征可以视情况与任何其他实施例的特征组合,除非这种组合是相互排斥的。在本说明书中,术语“包括”是指包括指定的一个或多个组件,但不排除其他组件的存在。
[0159]
应注意与此说明书同时提交、或在本说明书之前提交的与本技术有关的、可供公众查阅的所有论文和文件,通过引用,所有这些论文和文件的内容都纳入本发明中。
[0160]
本说明书(包括任何所附权利要求、摘要和附图)中公开的所有特征和/或如此公开的任何方法或过程的所有步骤可以在任何组合中组合,除非组合中至少一些这样的特征和/或步骤是相互排斥的。
[0161]
除非另外明确说明,本说明书(包括任何所附权利要求书、摘要和附图)中公开的每个特征可以被具有相同、相当或类似用途的替代特征所代替。因此,除非另外明确说明,公开的每个特征仅仅是相当或类似特征通用系列的一个实施例。
[0162]
本发明不限于前述实施例的细节。本发明扩展到本说明书(包括任何所附权利要求书、摘要和附图)中公开特征的任何新颖的一个或任何新颖的组合,或此处公开的任何方法或过程的步骤的任何新颖的一个或任何新颖的组合。
[0163]
附录a
–
具有预后值的生物标志物的特定组合数据,如采用图4h的正向和反向构建子集程序获得的数据。
[0164]
正向分析
[0165]
[0166]
[0167]
[0168]
[0169]
[0170]
[0171]
[0172]
[0173]
[0174]
[0175]
[0176]
[0177]
[0178]
[0179][0180][0181]
反向分析
[0182]
[0183]
[0184]
[0185]
[0186]
[0187]
[0188]
[0189]
[0190]
[0191]
[0192]
[0193]
[0194]
[0195]
[0196]
[0197]
[0198]
[0199]
[0200]
[0201]
[0202]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1