用于制备食欲肽-2受体拮抗剂的方法和化合物、以及杂质少的莱博雷生与流程
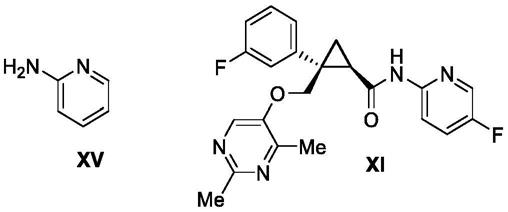
用于制备食欲肽
‑
2受体拮抗剂的方法和化合物、以及杂质少的莱博雷生
技术领域
1.本发明涉及用于制备作为食欲肽
‑
2受体拮抗剂有用的莱博雷生(lemborexant)的化合物和方法。进而,本发明涉及包含少量杂质的莱博雷生。
背景技术:
2.食欲肽受体是主要在脑中被观察到的g蛋白偶联受体。食欲肽受体的内源性配体、食欲肽
‑
a及食欲肽
‑
b是通过定位于下丘脑的神经元而表达。食欲肽
‑
a是33个氨基酸的肽,食欲肽
‑
b包含28个氨基酸(sakurai t.等人,cell[细胞],1998年,92卷,573
‑
585页)。食欲肽受体有2个亚型即ox1及ox2;ox1优先与食欲肽
‑
a结合,但ox2与食欲肽
‑
a及食欲肽
‑
b两者结合。提示了食欲肽在大鼠中刺激食物消耗,食欲肽信号传递可以在用以调节摄食行为的中心反馈机制中发挥作用(sakurai等人,上述)。还观察到食欲肽控制觉醒
‑
睡眠状态(chemelli r.m.等人,cell[细胞],1999年,98卷,437
‑
451页)。食欲肽还可以在与阿片类及烟碱依赖症(s.l.borgland等人,neuron[神经元],2006年,49卷,598
‑
601页;c.j.winrow等人,neuropharmacology[神经药理学],2010年,58卷,185
‑
194页)以及酒精依赖症(j.r.shoblock等人,psychopharmacology[心理药理学],2011年,215卷,191
‑
203页)并发的脑改变中发挥作用。进而,提示了食欲肽在一些压力反应中发挥作用(t.ida等人,biochem.biophys.res.commun.[生物化学和生物物理学研究通讯],2000年,270卷,318
‑
323页)。
[0003]
已知(1r,2s)
‑2‑
(((2,4
‑
二甲基嘧啶
‑5‑
基)氧基)甲基)
‑2‑
(3
‑
氟苯基)
‑
n
‑
(5
‑
氟吡啶
‑2‑
基)环丙基甲酰胺(莱博雷生)等化合物是强力的食欲肽受体拮抗剂,这些化合物在失眠症等睡眠障碍的治疗以及其他治疗应用中有用。引证文献清单专利文献
[0004]
[专利文献1]wo 2012/039371[专利文献2]wo 2013/123240非专利文献
[0005]
[非专利文献1]sakurai t.等人,cell[细胞],1998年,92卷,573
‑
585页[非专利文献2]chemelli r.m.等人,cell[细胞],1999年,98卷,437
‑
451页[非专利文献3]s.l.borgland等人,neuron[神经元],2006年,49卷,598
‑
601页
[非专利文献4]c.j.winrow等人,neuropharmacology[神经药理学],2010年,58卷,185
‑
194页[非专利文献5]j.r.shoblock等人,psychopharmacology[心理药理学],2011年,215卷,191
‑
203页[非专利文献6]t.ida等人,biochem.biophys.res.commun.[生物化学和生物物理学研究通讯],2000年,270卷,318
‑
323页
技术实现要素:
技术问题
[0006]
作为制备莱博雷生的方法,已知有专利文献1中所描述的方法,但是在专利文献1中,莱博雷生是由制备实例14的化合物prep 14
‑
2通过化合物prep 14
‑
3而合成。该制备方法由于产率低,因此于将上述制备方法用于工业生产时遗留有问题。
[0007]
作为制备莱博雷生的方法,已知有专利文献2中所描述的方法,但是在专利文献2中,通过使用酵素的乙酰化,由化合物5通过化合物6而合成。在该制备方法中,由于在设备洗涤中必须考虑酵素的残留,因此在将上述制备方法用于工业生产时仍有问题。
[0008]
此外,本发明人发现,在莱博雷生化合物的制备中含有作为杂质的式vi、式ix、式x、式xii、式xiii、式xiv和式xv的化合物。
[0009]
因此,需要用于制备作为药物有用的莱博雷生的合成方法和中间体。因此,本技术的目的在于提供此种合成方法和中间体。此外,需要作为药物有用且包含少量杂质的莱博
雷生。因此,本技术的目的在于提供包含少量杂质的莱博雷生。问题的解决方案
[0010]
在本说明书中,提供一种用于制备作为食欲肽
‑
2受体拮抗剂有用的莱博雷生的化合物和方法。进而,提供包含少量杂质的莱博雷生。
[0011]
本发明提供了制备式iii的化合物的方法,其包括:a)向包含式ii的化合物甲苯和四氢呋喃的混合物中添加格氏试剂;以及b)向包含乙酰氯与乙酸乙酯的混合物中添加上述混合物。
[0012]
在一个实施例中,格氏试剂为环己基氯化镁。
[0013]
本发明提供了制备式xi的化合物的方法,其包括:制备如上所述的式iii的化合物的制造步骤。
[0014]
本发明提供了制备如上所述的式xi的化合物的方法,其包括将将i)式ix的化合物ii)式x的化合物
iii)乙酸乙酯、iv)n,n
‑
二异丙基乙胺、以及v)1
‑
丙烷膦酸的混合物在60℃
‑
80℃的温度进行搅拌。
[0015]
本发明提供了制备式ix的化合物的方法,其包括:制备如上所述的式iii的化合物。
[0016]
本发明提供了制备式ix的化合物的方法,其包括:使式vii的化合物与氧化剂反应。
[0017]
在一个实施例中,使式vii的化合物与氧化剂反应的步骤是通过依序添加次氯酸钠和亚氯酸钠作为氧化剂来进行的。
[0018]
本发明提供了制备式ix的化合物的方法,
其包括:a)将式vii的化合物用次氯酸钠进行氧化而制备式viii的醛,以及b)将该醛用亚氯酸钠进行氧化。
[0019]
在一个实施例中,步骤a)中的氧化被有效量的2,2,6,6
‑
四甲基哌啶
‑1‑
烃氧基(tempo)催化。
[0020]
本发明提供了制备式vii的化合物的方法,其包括:制备如上所述的式iii的化合物的制造步骤,以及向式v的化合物中添加氢氧化钠水溶液并将两者在30℃
‑
50℃的温度进行搅拌。
[0021]
本发明提供了制备式v的化合物的方法,其包括:a)向i)式vi的化合物ii)n
‑
甲基吡咯烷酮、和iii)乙腈的混合物中添加叔丁醇钾并且在40℃
‑
70℃的温度进行搅拌;以及b)将通过向上述混合物中添加i)式iv的化合物所得的混合物在40℃
‑
70℃的温度进行搅拌。
[0022]
本发明提供了制备式iv的化合物的方法,其包括:向包含i)通过如上所述的方法制备的式iii的化合物、甲苯、三乙胺和1
‑
甲基咪唑的组合物中添加ii)对甲苯磺酰氯、甲苯和乙腈的混合
物。
[0023]
本发明提供了制备式ii的化合物的方法,其包括:向包含式i的化合物甲苯、四氢呋喃、和硼氢化钠的组合物中添加甲醇。
[0024]
本发明提供了式xi的化合物,其中式xii的化合物的含量为0.10质量%或以下。
[0025]
本发明提供了如上所述的式xi的化合物,其中式xii的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0026]
本发明提供了式xi的化合物,其中包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量为1.0质量%或以下。
[0027]
本发明提供了如上所述的式xi的化合物,其中包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量是通过高效液相色谱法(hplc)确定的。
[0028]
本发明提供了式xi的化合物,其中式xii的化合物的含量为0.10质量%或以下,包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量为1.0质量%或以
下。
[0029]
本发明提供了如上所述的式xi的化合物,其中式xii的化合物,以及包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量是通过高效液相色谱法(hplc)确定的。
[0030]
本发明提供了式xi的化合物,其中式vi的化合物的含量为0.05质量%或以下。
[0031]
本发明提供了如上所述的式xi的化合物,其中式vi的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0032]
本发明提供了式xi的化合物,其中式ix的化合物的含量为0.05质量%或以下。
[0033]
本发明提供了如上所述的式xi的化合物,其中式ix的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0034]
本发明提供了式xi的化合物,其中式x的化合物的含量为0.05质量%或以下。
[0035]
本发明提供了如上所述的式xi的化合物,其中式x的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0036]
本发明提供了式xi的化合物,其中式xiii的化合物的含量为0.05质量%或以下。
[0037]
本发明提供了如上所述的式xi的化合物,其中式xiii的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0038]
本发明提供了式xi的化合物,其中式xiv的化合物的含量为0.05质量%或以下。
[0039]
本发明提供了如上所述的式xi的化合物,其中式xiv的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0040]
本发明提供了式xi的化合物,其中式xv的化合物的含量为0.05质量%或以下。
[0041]
本发明提供了如上所述的式xi的化合物,其中式xv的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0042]
本发明提供了如上所述的式xi的化合物,其中式xi的化合物的含量为97.0质量%或以上。
[0043]
本发明提供了如上所述的式xi的化合物,其中式xi的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0044]
本发明提供了包含式xi的化合物和式xii的化合物的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式xii的化合物的含量为0.10质量%或以下。
[0045]
本发明提供了如上所述的化合物,其中式xi的化合物和式xii的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0046]
本发明提供了化合物,其含有式xi的化合物,式xii的化合物以及包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质,其中该式xi的化合物的含量为97.0质量%或以上,该式xii的化合物的含量为0.10质量%或以下,并且该包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量为1.0质量%或以下。
[0047]
本发明提供了如上所述的化合物,其中式xi的化合物,式xii的化合物,以及包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量是通过高效液相色谱法(hplc)确定的。
[0048]
本发明提供了包含式xi的化合物和式vi的化合物的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式vi的化合物的含量为0.05质量%或以下。
[0049]
本发明提供了如上所述的化合物,其中式xi的化合物和式vi的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0050]
本发明提供了具有包含式xi的化合物和式ix的化合物的式的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式ix的化合物的含量为0.05质量%或以下。
[0051]
本发明提供了如上所述的化合物,其中式xi的化合物和式ix的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0052]
本发明提供了包含式xi的化合物和式x的化合物的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式x的化合物的含量为0.05质量%或以下。
[0053]
本发明提供了如上所述的化合物,其中式xi的化合物和式x的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0054]
本发明提供了包含式xi的化合物和式xiii的化合物的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式xiii的化合物的含量为0.05质量%或以下。
[0055]
本发明提供了如上所述的化合物,其中式xi的化合物和式xiii的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0056]
本发明提供了包含式xi的化合物和式xiv的化合物的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式xiv的化合物的含量为0.05质量%或以下。
[0057]
本发明提供了如上所述的化合物,其中式xi的化合物和式xiv的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0058]
本发明提供了包含式xi的化合物和式xv的化合物的化合物,其中式xi的化合物的含量为97.0质量%或以上,并且式xv的化合物的含量为0.05质量%或以下。
[0059]
本发明提供了如上所述的化合物,其中式xi的化合物和式xv的化合物的含量是通过高效液相色谱法(hplc)确定的。发明的有利效果
[0060]
本发明能够提供用于制备食欲肽
‑
2受体拮抗剂的合成方法和中间物。进而,本发明可提供包含少量杂质的莱博雷生。
具体实施方式
[0061]
以下,对本说明书中所使用的符号、术语等的含义进行说明,从而详细地说明本说明书。
[0062]
在本说明书中,化合物的结构式不限于方便表达的式,并且该化合物还可形成盐。进而,还有存在多晶型的情况,但同样地不受限定,可以是任一单晶型或其混合物,除无水物以外,还可以是水合物或溶剂化物,均包含在本发明中。
[0063]
在本说明书中,除非另外特别说明,盐的具体事例包括氢卤酸盐(例如氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐等)、无机酸盐(例如硫酸盐、硝酸盐、高氯酸盐、磷酸盐、碳酸盐、碳酸氢盐等)等。
[0064]
在一个实施例中,本说明书中所描述的化合物可以以盐,例如药学上可接受的盐的形式提供。“药学上可接受的盐”是保留母体化合物的所需的生物活性但不会赋予不希望的毒物学作用的盐。药学上可接受的盐的具体事例包含无机酸盐(硫酸盐、硝酸盐、高氯酸盐、磷酸盐、碳酸盐、碳酸氢盐、氢氟酸盐、盐酸盐、氢溴酸盐、氢碘酸盐等)、有机羧酸盐(乙酸盐、草酸盐、马来酸盐、酒石酸盐、富马酸盐、柠檬酸盐等)、有机磺酸盐(甲磺酸盐、三氟甲磺酸盐、乙磺酸盐、苯磺酸盐、甲苯磺酸盐、樟脑磺酸盐等)、氨基酸盐(天冬氨酸盐、谷氨酸
盐等)、季胺盐、碱金属盐(钠盐、钾盐等)及碱土类金属盐(镁盐、钙盐等)。
[0065]
本发明还包含说明书中所描述的化合物的经同位素标记的化合物及使用其的制备方法。经同位素标记的化合物除了一个或多个原子经具有与自然界中通常可见的原子质量或质量数不同的原子质量或质量数的原子置换以外,其余与说明书中所描述的化合物相同。可并入本发明的化合物中的同位素例如为氢、碳、氮、氧、氟、氯、磷、硫和碘的同位素,包含2h、3h、
11
c、
14
c、
13
n、
15
o、
18
f、
32
p、
35
s、
123
i、和
125
i等。包含这些同位素和/或其他同位素的本发明的化合物及其药学上可接受的衍生物(例如盐)还包含于本技术专利范围内。
[0066]
本发明的同位素标记的化合物,例如并入有3h和/或
14
c等放射性同位素的化合物对药物和/或基质的组织分布测定有用。3h和
14
c因其易于制备和检测而被认为可用。认为同位素
11
c及
18
f在pet(正电子发射断层摄影)方面有用,认为同位素
125
i在spect(单光子发射计算机断层扫描摄影)方面有用,这些同位素在脑成像方面全部有用。利用2h等更重的同位素进行的置换会产生某种类型的治疗优势,如由更高的代谢稳定性所获得的生物体内半衰期增加或所需剂量的减少等,因此认为在某种状况下有用。
[0067]
本说明书中所表示的结构意指还包含该结构的所有对映异构体形态、非对映异构体形态及几何学(或构象)形态;例如针对各不对称中心的r型及s型构型、(z)型及(e)型双键异构物以及(z)型及(e)型构形异构物。因此,本发明的化合物的单一立体化学异构物、对映异构体、非对映异构体和几何(或构象)混合物在本发明的范围内。除非另有说明,否则本发明的化合物的互变异构形态全部在本发明的范围内。
[0068]
在本说明书中,“化合物”意指含有90质量%或以上的该化合物,并且可能含有其制备步骤中的起始材料或可能产生的副产物作为相关物质者。例如,“式xi的化合物”是含有90质量%或以上的式xi的化合物,并且可能含有式vi的化合物、式ix的化合物、式x的化合物、式xii的化合物、式xiii的化合物、式xiv的化合物、式xv的化合物等各制备步骤中的起始材料或可能产生的副产物等。因此,本说明书中的“化合物”由于可能含有副产物等作为杂质,因此还具有称为“组合物”的情况。此处,在表示式vi的化合物、式ix的化合物、式x的化合物、式xii的化合物、式xiii的化合物、式xiv的化合物、式xv的化合物等杂质的含量的情形时,含量以式xi的化合物的总质量计。
[0069]
在本说明书中,“总相关物质的含量”是指包含式vi、式ix、式x、式xiii、式xiv或式xv的相关物质的总量(质量%)。
[0070]
在本说明书中,提供了用于制备(1r,2s)
‑2‑
(((2,4
‑
二甲基嘧啶
‑5‑
基)氧基)甲基)
‑2‑
(3
‑
氟苯基)
‑
n
‑
(5
‑
氟吡啶
‑2‑
基)环丙基甲酰胺(莱博雷生)的化合物(例如中间体化合物)及其制备方法,该莱博雷生已知为强力的食欲肽受体拮抗剂,并且在失眠症等睡眠障碍的治疗以及其他治疗应用中有用。进而,在本说明书中,提供包含少量杂质的莱博雷生。
[0071]
本发明提供了制备式iii的化合物的方法,其包括:a)向包含式ii的化合物甲苯和四氢呋喃的混合物中添加格氏试剂;以及b)向包含乙酰氯与乙酸乙酯的混合物中添加上述混合物。
[0072]
在上述方法中,步骤a)可以在60℃或以下进行,步骤b)可以在20℃或以下进行。
[0073]
在一个实施例中,格氏试剂为环己基氯化镁。
[0074]
本发明提供了制备式xi的化合物的方法,其包括:制备如上所述的式iii的化合物。
[0075]
本发明提供了制备如上所述的式xi的化合物的方法,将将i)式ix的化合物
ii)式x的化合物iii)乙酸乙酯、iv)n,n
‑
二异丙基乙胺、以及v)1
‑
丙烷膦酸的混合物在60℃
‑
80℃的温度进行搅拌。
[0076]
在上述方法中,反应溶剂可以是乙酸乙酯,搅拌上述混合物的步骤的温度可以是70℃。
[0077]
本发明提供了制备式ix的化合物的方法,其包括:制备如上所述的式iii的化合物。
[0078]
本发明提供了制备式ix的化合物的方法,其包括:使式vii的化合物
与氧化剂反应。
[0079]
在上述方法中,反应溶剂可以是甲苯和水的混合液,反应温度可以是35℃或以下。
[0080]
在一个实施例中,使式vii的化合物与氧化剂反应的步骤是通过依序添加次氯酸钠和亚氯酸钠作为氧化剂来进行的。
[0081]
本发明提供了制备式ix的化合物的方法,其包括:a)将式vii的化合物用次氯酸钠进行氧化而制备式viii的醛,以及b)将该醛用亚氯酸钠进行氧化。
[0082]
在上述方法中,a)步骤和b)步骤中的反应溶剂可以是甲苯和水的混合液,反应温度可以是25℃或以下。
[0083]
在一个实施例中,步骤a)的氧化是被有效量的2,2,6,6
‑
四甲基哌啶
‑1‑
烃氧基(tempo)催化。
[0084]
本发明提供了制备式vii的化合物的方法,
其包括:制备如上所述的式iii的化合物的制造步骤,以及向式v的化合物中添加氢氧化钠水溶液并将两者在30℃
‑
50℃的温度进行搅拌。
[0085]
在上述方法中,反应溶剂可以是甲苯、n
‑
甲基吡咯烷酮、乙腈和水的混合液,反应温度可以是40℃。
[0086]
本发明提供了制备式v的化合物的方法,其包括:a)向i)式vi的化合物ii)n
‑
甲基吡咯烷酮、和iii)乙腈的混合物中添加叔丁醇钾并且将两者在40℃
‑
70℃的温度进行搅拌;以及b)将通过向上述混合物中添加i)式iv的化合物
所得的混合物在40℃
‑
70℃的温度进行搅拌。
[0087]
在上述方法中,a)步骤中的反应溶剂可以是n
‑
甲基吡咯烷酮和乙腈的混合液,反应温度可以是50℃,b)步骤中的反应溶剂可以是甲苯、n
‑
甲基吡咯烷酮和乙腈的混合液,反应温度可以是50℃。
[0088]
本发明提供了制备式iv的化合物的方法,其包括:向包含i)通过如上所述的方法制备的式iii的化合物、甲苯、三乙胺和1
‑
甲基咪唑的组合物中添加ii)对甲苯磺酰氯、甲苯和乙腈的混合物。
[0089]
在上述方法中,反应溶剂可以是甲苯和乙腈的混合液,反应温度可以是8℃。
[0090]
本发明提供了制备式ii的化合物的方法,其包括:向包含式i的化合物甲苯、四氢呋喃、和硼氢化钠的组合物中添加甲醇。
[0091]
在上述方法中,反应溶剂可以是甲苯、四氢呋喃和甲醇的混合液,反应温度可以是
50℃或以下。
[0092]
本发明提供了式xi的化合物,其中式xii的化合物的含量为0.10质量%或以下。
[0093]
本发明提供了如上所述的式xi的化合物,其中式xii的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0094]
本发明提供了式xi的化合物,其中包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量为1.0质量%或以下。
[0095]
本发明提供了如上所述的式xi的化合物,其中包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量是通过高效液相色谱法(hplc)确定的。
[0096]
本发明提供了式xi的化合物,其中式xii的化合物的含量为0.10质量%或以下,包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量为1.0质量%或以下。
[0097]
本发明提供了如上所述的式xi的化合物,其中式xii的化合物,以及包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量是通过高效液相色谱法(hplc)确定的。
[0098]
本发明提供了式xi的化合物,其中式vi的化合物的含量为0.05质量%或以下。
[0099]
本发明提供了如上所述的式xi的化合物,其中式vi的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0100]
本发明提供了式xi的化合物,其中式ix的化合物的含量为0.05质量%或以下。
[0101]
本发明提供了如上所述的式xi的化合物,其中式ix的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0102]
本发明提供了式xi的化合物,其中式x的化合物的含量为0.05质量%或以下。
[0103]
本发明提供了如上所述的式xi的化合物,其中式x的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0104]
本发明提供了式xi的化合物,其中式xiii的化合物的含量为0.05质量%或以下。
[0105]
本发明提供了如上所述的式xi的化合物,其中式xiii的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0106]
本发明提供了式xi的化合物,其中式xiv的化合物的含量为0.05质量%或以下。
[0107]
本发明提供了如上所述的式xi的化合物,其中式xiv所表示的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0108]
本发明提供了式xi的化合物,其中式xv的化合物的含量为0.05质量%或以下。
[0109]
本发明提供了如上所述的式xi的化合物,其中式xv的化合物的含量是通过高效液相色谱法(hplc)确定的。
[0110]
上述所列举并且在本说明书中披露的方法中使用的化合物均可以立体化学纯的形态提供,应理解这些包含于本发明中。在一个实施例中,立体化学纯的化合物具有约超过
75重量%的化合物的一个立体异构体及约小于25重量%的该化合物的其他立体异构体;约超过80重量%的化合物的一个立体异构体及约小于20重量%的该化合物的其他立体异构体;或约超过85重量%的化合物的一个立体异构体及约小于15重量%的该化合物的其他立体异构体;或约超过90重量%的化合物的一个立体异构体及约小于10重量%的该化合物的其他立体异构体;或约超过95重量%的化合物的一个立体异构体及约小于5重量%的该化合物的其他立体异构体;或约超过97重量%的化合物的一个立体异构体及约小于3重量%的该化合物的其他立体异构体;或约超过98重量%的化合物的一个立体异构体及约小于2重量%的该化合物的其他立体异构体。在一个实施例中,立体化学纯的化合物具有超过99重量%的化合物的一个立体异构体及小于1重量%的该化合物的其他立体异构体。
[0111]
如上所述,在一个实施例中,本说明书所说明的化合物可以以盐,例如药学上可接受的盐的形式提供。
[0112]
某一实施例使制备式xi的化合物成为可能,其中式xii的化合物的含量为0.10质量%或以下,式vi、式ix、式x、式xiii、式xiv或式xv的化合物的各含量为0.05%或以下,并且包含式vi、式ix、式x、式xiii、式xiv和式xv的化合物的总相关物质的含量为1.0质量%或以下。
[0113]
某一实施例使制备式xi的化合物成为可能,其中式xi的化合物的含量为97.0质量%或以上、98.0质量%或以上或98.5质量%或以上,并且为102.0质量%或以下、100.0质量%或以下、99.9质量%或以下、99.0质量%或以下或98.9质量%或以下。此处,式xi的化合物的含量是通过高效液相色谱法(hplc)确定的。更具体而言,在下述实例的“纯度测试”部分的中所述的条件下进行hplc,用样品的式xi的化合物的峰面积除以色谱图上的式xi的化合物的标准品的峰面积,所得的数值表示式xi的化合物的含量。
[0114]
本发明的另一实施例是含有式xi的化合物或其结晶、及药学上可接受的添加物的药物组合物。药物组合物可通过将药学上可接受的添加物与式xi的化合物或其结晶进行混合而制备。根据本发明的药物组合物例如可以根据日本药典第17修订版的制剂总则中所描述的方法等已知的方法进行制备。实例
[0115]
以下,根据实例来详细地说明本发明。然而,本发明不限于这些实例。此外,以下实例中所使用的简称为从业者众所周知的惯用的简称,一些简称如下所示。
[0116]
质子核磁共振(1h
‑
nmr)图谱的化学位移是以相对于四甲基硅烷的δ单位(ppm)进行记录,偶合常数是以赫兹(hz)进行记录。图案中,s、d、br和m分别表示单峰、双峰、宽峰和多重峰。
[0117]1h
‑
nmr图谱和
13
c
‑
nmr图谱是使用varian 400或500mhz分光计、或者由日本电子股份有限公司(jeol ltd.)制造的jnm
‑
al400型核磁共振装置(400mhz)测定的。
[0118]
在以下实例中,“室温”通常表示约10℃至约35℃。除非特别说明,“%”表示重量百分比。
[0119]
(a)(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮(式i的化合物)的制备
[0120]
(a
‑
1)(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲腈(式ib的化合物)的合成在
‑
10℃
‑
0℃,将由(3
‑
氟苯基)乙腈(120kg,888mol,1.0equiv.)与四氢呋喃(9.0kg)混合而成的溶液滴加至由叔丁醇钠(188kg,1954mol,2.2equiv.)与四氢呋喃(890kg)混合而成的溶液中。搅拌1小时以上后,在
‑
12℃
‑
18℃向其中滴加(2r)
‑2‑
(氯甲基)环氧乙烷(94.5kg,1021mol,1.15equiv.)。在通过高效液相色谱法(以下称为“hplc”)分析确认反应结束后,添加水(50kg)并进行搅拌。在减压下蒸馏去除溶剂而获得(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲腈溶液,直接用于下一步骤。
[0121]
用以确认上述反应的hplc条件:试剂和流动相:
·
纯水
·
乙腈(关东化学公司(kanto chemical co.,inc.)目录编号01031
‑
2b高速液相色谱或同等产品)
·
高氯酸(70%)(和光纯药工业公司(wako pure chemical industries,ltd.)目录编号162
‑
00695特级化学品或同等产品)
·
流动相a液:水、乙腈和高氯酸(70%)混合液(950:50:1,v/v/v)
·
流动相b液:水、乙腈和高氯酸(70%)混合液(100:900:1,v/v/v)
·
溶液:水和乙腈混合液(50:50,v/v)
·
hplc装置的针头冲洗剂:水和乙腈混合液(50:50,v/v)
·
(3
‑
氟苯基)乙腈标准样品hplc条件:检测器:shimadzu spd
‑
20a紫外线吸收计(测定波长:262nm)或同等产品柱:内径4.6mm、长度15cm、填充2.7μm的液相色谱用十八烷基硅烷化硅胶的不锈钢管实例:meteoric core c18(ymc),(相当于usp包装类型l1)柱温:接近30℃的恒温流动相:a液:水、乙腈和高氯酸(70%)混合液(950:50:1,v/v/v)
b液:水、乙腈和高氯酸(70%)混合液(100:900:1,v/v/v)流速:0.9ml/min梯度条件:注入量:10μl样品架温度:接近5℃的恒温面积测定范围:30分钟
[0122]
(a
‑
2)(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮(式i的化合物)的合成(a
‑2‑
1)晶种的制备在0
‑
7℃,向由(3
‑
氟苯基)乙腈(20g,0.15mol,1.00equiv.)与四氢呋喃(150ml)混合而成的溶液中滴加双(三甲基甲硅烷基)酰胺钠(四氢呋喃溶液)(150ml,0.30mol,2.00equiv.)。搅拌0.5小时后,在17℃向其中滴加(2r)
‑2‑
(氯甲基)环氧乙烷(11.8ml,0.15mol,1.00equiv.)。在5℃搅拌2小时,在室温下搅拌3天后,向其中添加水(10ml)并进行搅拌。在减压下蒸馏去除溶剂而获得(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲腈,直接用于下一步骤。向所获得的(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲腈中添加乙醇(150ml)及2m氢氧化钾水溶液(148ml,0.30mol,2.00equiv.),将混合物在80℃搅拌4小时后,向其中添加水(66ml)和浓盐酸(47.5ml,0.57mol,3.80equiv.)。在60℃搅拌1小时,进而在室温下搅拌45小时。过滤出所沉淀的固体,用水进行洗涤直至确认ph为5,在氮气气流下进行干燥,以产率81%(由(3
‑
氟苯基)乙腈获得的总产率)获得(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮(23g)。
[0123]
(a
‑2‑
2)(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮(式i的化合物)的合成(a
‑2‑2‑
步骤1)向(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲腈溶液中添加乙醇
(237kg)、水(237kg)和25%氢氧化钠水溶液(220kg,1376mol,1.55equiv.),在内温77℃
‑
87℃进行搅拌。通过hplc分析确认反应结束后,在减压下蒸馏去除溶剂,向其中添加甲苯(624kg)和水(60kg)并进行搅拌,然后静置。将有机层排出后,向水层中添加甲苯(624kg)并进行搅拌,然后静置。将有机层排出,直接用于下一步骤。
[0124]
用以确认上述反应的hplc条件:试剂和流动相:纯水、乙腈、高氯酸(70%)、流动相a液、流动相b液、溶液、和hplc装置的针冲洗液与(a
‑
1)相同。(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲腈标准样品hplc条件:与(a
‑
1)相同。
[0125]
(a
‑2‑2‑
步骤2)向(a
‑2‑2‑
步骤1)中所获得的溶液中添加甲苯(409kg)及35%盐酸(287kg,2753mol,3.10equiv.)。将混合物在内温55℃
‑
65℃进行搅拌,通过hplc分析确认反应结束。在确认内温到35℃以下后,将混合物静置并排出水层,其后,向有机层中添加甲苯(172kg)及7%碳酸氢钠水溶液(482kg)并进行搅拌,然后静置。将水层排出后,向有机层中添加水(480kg)并进行搅拌,然后静置。将水层排出,算出有机层中的(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮含量。在减压下蒸馏去除溶剂后,向其中添加乙醇(614kg)。在减压下蒸馏去除溶剂后,向其中添加乙醇使得总体积为(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮含量的2倍量。将(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮加热并在内温60℃
‑
65℃的搅拌下进行溶解,向其中添加(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮的含量的0.8倍的水。冷却后,在内温40℃
‑
50℃向其中添加晶种60g,确认结晶析出。将混合物在内温40℃
‑
50℃搅拌1小时以上后,在
‑
10℃/h
‑‑
20℃/h冷却至
‑
5℃
‑
5℃或以下,过滤出所沉淀的固体,利用乙醇进行洗涤。将所获得的固体在外温45℃或以下进行减压干燥,以产率60%(由(3
‑
氟苯基)乙腈获得的总产率)获得(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮(103kg)。
[0126]
用以确认上述反应的hplc条件:试剂和流动相:纯水、乙腈、高氯酸(70%)、流动相a液、流动相b液、溶液、和hplc装置的针冲洗液与(a
‑
1)相同。(1s,2r)
‑1‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙烷
‑1‑
甲酸溶液(浓度约0.5mg/ml)hplc条件:检测器、柱、柱温、流动相、流速、注入量、样品架温度与(a
‑
1)相同。梯度条件:
面积测定范围:40分钟
[0127]
nmr数据(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮:1h nmr(500mhz,dmso
‑
d6)δ7.43
‑
7.36(m,1h),7.35
‑
7.31(m,1h),7.31
‑
7.28(m,1h),7.14
‑
7.08(m,1h),4.46(dd,j=9.1,4.6hz,1h),4.25(d,j=9.1hz,1h),2.80(dt,j=8.0,4.6hz,1h),1.72(dd,j=7.9,4.8hz,1h),1.37(t,j=4.8hz,1h);
13
c nmr(126mhz,dmso
‑
d6)δ175.35,161.98(d,jcf=243.1hz),137.67(d,jcf=8.2hz),130.15(d,jcf=8.6hz),124.19(d,jcf=2.8hz),115.01(d,jcf=22.4hz),113.95(d,jcf=20.9hz),67.93,30.70(d,jcf=2.4hz),25.57,19.99。
[0128]
(b)2,4
‑
二甲基嘧啶
‑5‑
醇(式vi的化合物)的制备
[0129]
(b
‑
1)1
‑
(4
‑
硝基苯氧基)丙烷
‑2‑
酮(式via的化合物)的制备将4
‑
硝基苯酚(305.0kg,2192.5mmol)、碳酸钾(318.2kg,2302.1mmol)和乙腈(1678kg)的混合物在60℃搅拌30分钟。在60℃,向反应混合物中滴加氯丙酮(净重213.0kg,2302.1mmol),将所获得的混合物在60℃(外温)搅拌7小时。将反应混合物在冰浴中冷却后,以同一温度向反应混合物中添加甲苯(1847kg)和水(1830kg)。在室温下搅拌所获得的反应
混合物后,将有机层分离,利用盐水(水1525kg和盐170.8kg)进行洗涤。在减压下蒸馏去除溶剂,将所获得的残余物在55℃(内温)溶解于甲苯(1319kg)后,冷却至内温5℃,过滤出所沉淀的固体。将所获得的固体用适量的甲苯进行洗涤,获得标题化合物的湿体(等价于干燥产率343.9kg,产率80%)。1h
‑
nmr(cd3cl)δ(ppm):2.31(3h,s),4.67(2h,s),6.95(2h,d,j=9.3hz),8.22(2h,d,j=9.3hz)。
[0130]
(b
‑
2)(z)
‑4‑
(二甲氨基)
‑3‑
(4
‑
硝基苯氧基)丁
‑3‑
烯
‑2‑
酮(式vib的化合物)的制备向1
‑
(4
‑
硝基苯氧基)丙烷
‑2‑
酮(等价于干燥343.9kg,1762.1mmol)中添加甲苯(1785kg)并将一部分浓缩后,添加n,n
‑
二甲基甲酰胺二甲基乙缩醛(231.0kg,1938.3mmol),并在78℃(内温)搅拌20小时。以同一温度向反应混合物中添加甲苯(595kg),在内温55℃向其中添加甲醇(163kg)并熟化30分钟后,冷却至5℃。过滤出所沉淀的固体。将所获得的固体用适量的甲苯洗涤后,获得标题化合物的湿体(等价于干燥产率159.0kg,产率36%)。1h
‑
nmr(cd3cl)δ(ppm):2.00(3h,brs),3.01(6h,s),6.90
‑
7.16(2h,brm),7.16
‑
7.46(1h,brs),8.22(2h,d,j=8.8hz)。
[0131]
此外,式vib的化合物也可以通过如下所述的方法进行制备。(b
‑2‑
a)(z)
‑4‑
(二甲氨基)
‑3‑
(4
‑
硝基苯氧基)丁
‑3‑
烯
‑2‑
酮(式vib的化合物)的制备向1
‑
(4
‑
硝基苯氧基)丙烷
‑2‑
酮(15.0g,76.9mmol)与4
‑
硝基苯酚(802mg,5.76mmol)中添加乙腈(15ml),并向其中添加n,n
‑
二甲基甲酰胺二甲基乙缩醛(15.4ml,115mmol),在78℃(外温)搅拌19小时。在50℃(外温)向反应混合物中添加甲苯(300ml),冷却至5℃(外温),过滤出所沉淀的固体。将所获得的固体用适量的甲苯洗涤后,在减压下在50℃进行干燥而获得标题化合物(12.4g,产率64%)。
[0132]
(b
‑2‑
b)(z)
‑4‑
(二甲氨基)
‑3‑
(4
‑
硝基苯氧基)丁
‑3‑
烯
‑2‑
酮(式vib的化合物)的制备
在60℃(外温),将4
‑
硝基苯酚(50.0g,359mmol)、碳酸钾(52.2g,377mmol)和乙腈(350ml)的混合物搅拌1小时。以同一温度向反应混合物中滴加氯丙酮(31.5ml,377mmol),将所获得的混合物以同一温度搅拌25小时。在冰冷水下搅拌反应混合物,在13℃(内温)向反应混合物中添加甲苯(350ml)和水(300ml)。在室温下搅拌所获得的反应混合物后,将有机层分离,利用盐水(水250ml和盐27.8g)进行洗涤。在减压下从该有机层(168.5g)蒸馏去除溶剂,向所获得的残余物(1
‑
(4
‑
硝基苯氧基)丙烷
‑2‑
酮(式via的化合物);净重16.7g,85.6mmol)中添加乙腈(60ml),在减压下蒸馏去除溶剂。向所获得的残余物中添加乙腈(60ml),在减压下蒸馏去除溶剂后,添加乙腈(16.7ml)并进行溶解,然后向其中添加n,n
‑
二甲基甲酰胺二甲基乙缩醛(17.2ml,128mmol),在78℃(外温)搅拌19.5小时。以同一温度向反应混合物中添加甲苯(200ml),冷却至6℃(外温),过滤出所沉淀的固体。将所获得的固体用适量的甲苯洗涤后,在减压下在50℃进行干燥而获得标题化合物(12.3g,产率58%)。
[0133]
(b
‑2‑
c)(z)
‑4‑
(二甲氨基)
‑3‑
(4
‑
硝基苯氧基)丁
‑3‑
烯
‑2‑
酮(式vib的化合物)的制备在18℃(内温),向4
‑
硝基苯酚(5.57g,40.1mmol)和乙腈(17ml)的混合物中添加三乙胺(5.3ml,38.2mmol)和氯丙酮(3.0ml,36.4mmol),在55℃(外温)搅拌69小时。在30℃(外温),向反应混合物中添加n,n
‑
二甲基甲酰胺二甲基乙缩醛(7.3ml,54.6mmol),并在80℃(外温)搅拌43小时。在36℃(内温),向反应混合物中添加n,n
‑
二甲基甲酰胺二甲基乙缩醛(7.3ml,54.6mmol),在80℃(外温)搅拌18.5小时。将内温冷却至45℃或以下后,添加甲苯(40ml),在减压下蒸馏去除溶剂。向所获得的残余物中添加甲苯(111ml)和乙腈(5.6ml)并在80℃(外温)进行搅拌,向其中添加甲醇(4.4ml)后冷却至6℃(外温),过滤出所沉淀的固体。将所获得的固体用适量的甲苯洗涤后,在减压下在50℃进行干燥而获得标题化合物(3.97g,产率44%)。
[0134]
(b
‑2‑
d)(z)
‑4‑
(二甲氨基)
‑3‑
(4
‑
硝基苯氧基)丁
‑3‑
烯
‑2‑
酮(式vib的化合物)的制备
在室温下,将4
‑
硝基苯酚(50.0g,359mmol)、碳酸钾(49.6g,359mmol)和乙腈(350ml)的混合物搅拌25分钟。以同一温度向混合物中滴加氯丙酮(33.3g,359mmol),将该混合物在60℃(外温)搅拌14小时。在室温下向所获得的反应混合物(1
‑
(4
‑
硝基苯氧基)丙烷
‑2‑
酮(式via的化合物)和4
‑
硝基苯酚的混合物(97:3))中添加甲苯(350ml)和水(300ml)并进行搅拌。将有机层分离,用水(250ml)进行洗涤。在减压下蒸馏去除该有机层的溶剂,向所获得的残余物(284g)中添加甲苯(500ml),在减压下蒸馏去除溶剂。向所获得的残余物(149g)中添加甲苯(103g)与n,n
‑
二甲基甲酰胺二甲基乙缩醛(52.9ml,395mmol),并在80℃(外温)搅拌15小时。将所获得的反应混合物冷却至室温并熟化3小时后,冷却至5℃(外温)并熟化2小时。过滤出所沉淀的固体,利用适量的甲苯进行洗涤。在减压下将该固体在50℃进行干燥,而获得标题化合物(55.7g,产率62%)。
[0135]
(b
‑
3)2,4
‑
二甲基
‑5‑
(4
‑
硝基苯氧基)嘧啶(式vic的化合物)的制备添加乙醇钠20%乙醇溶液(518.8kg,1524.9mol)、乙醇(213kg)和乙脒盐酸盐(净重144.2kg,1524.9mmol),并在25℃(内温)搅拌94分钟。添加(z)
‑4‑
(二甲氨基)
‑3‑
(4
‑
硝基苯氧基)丁
‑3‑
烯
‑2‑
酮(等价于干燥159.0kg,635.4mmol),将混合物在60℃(内温)搅拌5小时。将反应混合物冷却至5℃(内温)后,添加水(2385kg),过滤出所沉淀的固体。将所获得的固体用适量的水洗涤后,获得标题化合物的湿体(等价于干燥产率146.1kg,产率94%)。1h
‑
nmr(cd3cl)δ(ppm):2.40(3h,s),2.75(3h,s),6.96(2h,d,j=9.0hz),8.24(2h,d,j=9.0hz),8.32(1h,s)。
[0136]
(b
‑
4)2,4
‑
二甲基嘧啶
‑5‑
醇(式vi的化合物)的制备在60℃(内温),将2,4
‑
二甲基
‑5‑
(4
‑
硝基苯氧基)嘧啶(等价于干燥146.1kg,595.8mmol)、甲醇(579kg)及氢氧化钠水溶液(氢氧化钠71.5kg、水77.5kg,1787.3mmol)的混合物搅拌13小时。向反应混合物中添加甲苯(1264kg)和水(584kg)并进行搅拌、静置后,将水层分离,在减压下将水层浓缩。将所获得的浓缩液冷却至8℃(内温)后,向其中添加甲苯(316kg),在内温0℃
‑
15℃添加浓盐酸(310.3kg,2978.8mmol)。向其中添加乙酸乙酯(328kg)以分离液体。将水层进行分离,利用甲苯
‑
乙酸乙酯(316kg
‑
328kg)进行洗涤。向所获得的水层中添加乙酸乙酯(1311kg)后,冷却至5℃(内温),添加5mol/l氢氧化钠水溶液直
至反应混合物的ph为6.5。将有机层进行分离,用乙酸乙酯(1311kg)再次萃取水层。将所获得的有机层合并后,用水(44kg)进行洗涤,在减压下蒸馏去除溶剂。将甲苯(379kg)添加至所获得的残余物中以悬浮,在50℃(内温)搅拌2小时。将混合物冷却至10℃(内温)以下,搅拌2小时后进行过滤。将所获得的固体用适量甲苯洗涤后,在减压下在45℃进行干燥而获得标题化合物(63.7kg,86%)。1h
‑
nmr(cd3cl)δ(ppm):2.51(3h,s),2.65(3h,s),8.02(1h,s),10.01(1h,brs)。
[0137]
(c)(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸(式ix的化合物)的制备
[0138]
(c
‑
1)[(1s,2r)
‑1‑
(3
‑
氟苯基)环丙烷
‑
1,2
‑
二基]二甲醇(式ii的化合物)的制备在50℃或以下,向(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮(70kg,364mol,1.00equiv.)、甲苯(350l)、四氢呋喃(350l)、硼氢化钠(13.8kg,364mol,1.00equiv.)的混合物中添加甲醇(58.3kg,1821mol,5.00equiv.)并进行搅拌。通过hplc分析确认反应结束,向20%柠檬酸水溶液(350kg)中添加反应液及甲苯(140l)并进行搅拌,然后静置。将水层排出,向有机层中添加5%碳酸氢钠水溶液(140kg),然后静置。将水层排出,向有机层中添加5%氯化钠水溶液(140kg)并进行搅拌,然后静置。将水层排出,在减压下蒸馏去除溶剂而获得[(1s,2r)
‑1‑
(3
‑
氟苯基)环丙烷
‑
1,2
‑
二基]二甲醇(相当于67.2kg,产率94%)甲苯溶液,直接用于下一步骤。
[0139]
用以确认上述反应的hplc条件:试剂和流动相:纯水乙腈(关东化学公司(kanto chemical co.,inc.)目录编号01031
‑
2b高速液相色谱或同等产品)三氟乙酸(和光纯药工业公司目录编号208
‑
02741特级化学品或同等产品)流动相a液:水和三氟乙酸混合液(1000/1,v/v)流动相b液:乙腈和三氟乙酸混合液(1000/1,v/v)溶液:水、乙腈和三氟乙酸混合液(500/500/1,v/v/v)
hplc装置的针头冲洗剂:水和乙腈混合液(100/900,v/v)(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮标准样品hplc条件:检测器:shimadzu spd
‑
20a紫外线吸收计(测定波长:220nm)或同等产品柱:内径4.6mm、长度15cm、填充3.5μm的液相色谱用十八烷基硅烷化硅胶的不锈钢管实例:sunfire c18(沃特斯公司(waters))(相当于usp包装l1)柱温:接近40℃的恒温流动相:a液:水和三氟乙酸混合液(1000/1,v/v)b液:乙腈和三氟乙酸混合液(1000/1,v/v)流速:约1.0ml/分钟的固定流速梯度条件:注入量:5μl样品架温度:接近10℃的恒温针冲洗液:水和乙腈混合液(10/90,v/v)面积测定对象范围:长达34分钟
[0140]
(c
‑
2)[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙基]甲基=乙酸酯(式iii的化合物)的制备在60℃或以下,向[(1s,2r)
‑1‑
(3
‑
氟苯基)环丙烷
‑
1,2
‑
二基]二甲醇的甲苯溶液(相当于[(1s,2r)
‑1‑
(3
‑
氟苯基)环丙烷
‑
1,2
‑
二基]二甲醇67.2kg,342mol,1.00equiv.)与四氢呋喃(269l)的混合物中添加环己基氯化镁(1mol/kg四氢呋喃溶液)(349kg,349mol,1.02equiv.)并进行搅拌。在20℃或以下,将所获得的溶液添加至乙酸乙酯(336l)和乙酰氯(29.6kg,377mol,1.10equiv.)的混合物中并进行搅拌。通过hplc分析确认反应结束,添加
水(202l)进行搅拌,然后静置。将水层排出后,向有机层中添加6%碳酸氢钠水溶液(215kg)并进行搅拌,然后静置。将水层排出后,向有机层中添加4%氯化钠水溶液(175kg)并进行搅拌,然后静置。将水层排出,在减压下蒸馏去除溶剂后,向其中添加甲苯(134l)。在减压下蒸馏去除溶剂而获得[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙基]甲基=乙酸酯的甲苯溶液,直接用于下一步骤。
[0141]
用以确认反应的hplc条件:试剂和流动相:纯水、乙腈、三氟乙酸、流动相a液、流动相b液及hplc装置的针冲洗液与(c
‑
1)相同。溶液:水/乙腈(500/500,v/v)[(1s,2r)
‑1‑
(3
‑
氟苯基)环丙烷
‑
1,2
‑
二基]二甲醇溶液(浓度约0.5mg/ml)hplc条件检测器、柱、柱温、流动相、流速、注入量、样品架温度与(c
‑
1)相同。梯度条件:面积测定对象范围:长达44分钟
[0142]
(c
‑
3)[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
({[(4
‑
甲基苯基)磺酰基]氧基}甲基)环丙基]甲基=乙酸酯(式iv的化合物)的制备在30℃或以下,向[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙基]甲基=乙酸酯的甲苯溶液(相当于[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙基]甲基=乙酸酯81.6kg,342mol,1.00equiv.)、甲苯(294l)(还包含甲苯溶液中的甲苯量)、三乙胺(69.3kg,685mol,2.00equiv.)和1
‑
甲基咪唑(1.41kg,17mol,0.05equiv.)的混合物中添加对甲苯磺酰氯
(71.8kg,377mol,1.10equiv.)的甲苯(34l)和乙腈(69l)溶液后,在外温8℃进行搅拌。通过hplc分析确认反应结束后,添加乙腈(34l)进行搅拌。通过过滤去除三乙胺盐酸盐,利用甲苯(51l)和乙腈(51l)的混合液对滤渣进行洗涤并混合,从而获得[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
({[(4
‑
甲基苯基)磺酰基]氧基}甲基)环丙基]甲基=乙酸酯的溶液,直接用于下一步骤。
[0143]
用以确认上述反应的hplc条件:试剂和流动相:
·
纯水、乙腈、三氟乙酸、流动相a液、流动相b液及hplc装置的针冲洗液与(c
‑
1)相同。
·
溶液:乙腈
·
[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
(羟基甲基)环丙基]甲基=乙酸酯的甲苯溶液(上一步骤之最终产物)hplc条件与(c
‑
1)相同。
[0144]
(c
‑
4)[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲基=乙酸酯(式v的化合物)的制备向2,4
‑
二甲基嘧啶
‑5‑
醇(51.0kg,411mol,1.20equiv.)、n
‑
甲基吡咯烷酮(273l)和乙腈(85l)的混合物中添加叔丁醇钾(46.1kg,411mol,1.20equiv.),在外温50℃进行搅拌。向所获得的溶液中添加[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
({[(4
‑
甲基苯基)磺酰基]氧基}甲基)环丙基]甲基=乙酸酯溶液,在外温50℃进行搅拌。通过hplc分析确认反应结束后,在30℃或以下向其中添加水(547l)并进行搅拌,然后静置。在分离液体后存放有机层,向水层中添加甲苯(220l)并进行搅拌,然后静置。排出水层,混合有机层而获得[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲基=乙酸酯溶液,直接用于下一步骤。
[0145]
用以确认上述反应的hplc条件:试剂和流动相:
·
纯水、乙腈、三氟乙酸、流动相a液、流动相b液及hplc装置的针冲洗液与(c
‑
1)相同。
·
溶液:乙腈
·
[(1r,2s)
‑2‑
(3
‑
氟苯基)
‑2‑
({[(4
‑
甲基苯基)磺酰基]氧基}甲基)环丙基]甲基=乙酸酯的溶液(浓度约1mg/ml)hplc条件与(c
‑
1)相同。
[0146]
(c
‑
5)[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇(式vii的化合物)的制备向[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲基=乙酸酯溶液中添加水(734l)和25%氢氧化钠水溶液(107l,856mol,2.50equiv.),在外温40℃进行搅拌。通过hplc分析确认反应结束后,进行冷却并静置。排出水层后,向有机层中添加磷酸二氢钠水溶液(磷酸二氢钠41.1kg和水255l)并进行搅拌,然后静置。排出水层后,向有机层中添加水(204l)并进行搅拌,然后静置。排出水层,向其中添加甲苯(86l)而获得[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇的甲苯溶液,直接用于下一步骤。
[0147]
用以确认上述反应的hplc条件:试剂和流动相:
·
纯水、乙腈、三氟乙酸、流动相a液、流动相b液及hplc装置的针冲洗液与(c
‑
1)相同。
·
溶液:水和乙腈混合液(1/1,v/v)
·
[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲基=乙酸酯溶液(浓度约1mg/ml)hplc条件除了检测器的测定波长为283nm之外,其余均与(c
‑
1)相同。
[0148]
(c
‑
6)(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸(式ix的化合物)的制备(1)晶种的制备将[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇的甲苯溶液(相当于[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇46.48kg,154mol,1.00equiv.)、2,2,6,6
‑
四甲基哌啶
‑1‑
烃氧基游离基(0.48kg,3.1mol,0.02equiv.)、甲苯(186l)和磷酸二氢钠水溶液(磷酸二氢钠6.1kg,
51mol,0.33equiv.和水56kg)的混合物在外温7℃进行冷却,添加12%次氯酸钠水溶液(9.1kg,15.4mol,0.10equiv.)进行搅拌。在25℃,添加25%亚氯酸钠水溶液(61.2kg,169mol,1.10equiv.)。通过hplc分析确认反应结束后,向其中添加亚硫酸钠水溶液(亚硫酸钠9.7kg和水93l)进行搅拌。向其中添加25%氢氧化钠水溶液(39l),在外温70℃搅拌1小时,冷却至50℃或以下并静置。存放水层,向有机层中添加水(23l)并进行搅拌,然后静置。排出有机层,混合水层,添加甲苯(139l)和5n盐酸水溶液而制备成ph 2.5
‑
4.0,并静置。排出水层后,向有机层中添加水(93l)并进行搅拌,然后静置。排出水层,将所获得的有机层在减压下蒸馏去除溶剂。进行一次以上的添加乙腈(232l)并在减压下蒸馏去除溶剂的操作。添加乙腈(232l),获得(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸溶液后,在减压下将溶剂蒸馏去除一部分。向包含基本上47.7kg的(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸的浓缩残余物中添加乙腈而使总体积成为232l。将该混合物在外温65℃的搅拌下溶解。冷却后确认结晶析出。加热至内温40℃或以上后,以
‑
20℃/h冷却至内温
‑
15℃或以下而沉淀出固体,过滤出该固体,用乙腈进行洗涤。将所获得的固体在外温50℃进行减压干燥,而获得43.5kg的(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸。(2)(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸(式ix的化合物)的制备将[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇的甲苯溶液(相当于[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇82.8kg,274mol,1.00equiv.)、2,2,6,6
‑
四甲基哌啶
‑1‑
烃氧基游离基(0.856kg,5.48mol,0.02equiv.)、甲苯(25l)和磷酸二氢钠水溶液(磷酸二氢钠10.8kg,90mol,0.33equiv.和水82.8kg)的混合物在外温8℃进行冷却,在35℃或以下添加12%次氯酸钠水溶液(48.5kg,82mol,0.30equiv.)并进行搅拌。通过hplc分析确认反应进行后,静置并排出水层。在35℃或以下,向有机层和磷酸二氢钠水溶液(磷酸二氢钠16.4kg,137mol,0.50equiv.和水82.8kg)的混合物中添加25%亚氯酸钠水溶液(119kg,329mol,1.20equiv.)。通过hplc分析确认反应结束后,添加亚硫酸钠水溶液(亚硫酸钠17.3kg和水166l)并进行搅拌。添加25%氢氧化钠水溶液(85.9l),在外温70℃进行加热并搅拌,冷却至50℃或以下并静置。存放水层,向有机层中添加水(41l)并进行搅拌,然后静置。排出有机层,混合水层,添加甲苯(248l)和5n盐酸水溶液而调整成ph 2.5
‑
4.0并静置。排出水层后,向有机层中添加水(166l)并进行搅拌,然后静置。排出水层,将所获得的有机层在减压下蒸馏去除溶剂。进行一次以上的添加乙腈(414l)并在减压下蒸馏去除溶剂的操作。添加乙腈(414l)而获得(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸溶液后,在减压下将溶剂蒸馏去除一部分。向包含基本上84.9kg的(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸的浓缩残余物中添加乙腈而使总体积成为497l。将该混合物在外温65℃的搅拌下溶解。冷却后,向其中添加晶种83g,确认结晶析出。加热至内温40℃或以上后,以
‑
20℃/h冷却至10℃或以下而析出固体,过滤出该固体,利用乙腈进行洗涤。将所获得的固体在外温60℃或以下进行减压干燥而以产率54%(由(1s,5r)
‑1‑
(3
‑
氟苯基)
‑3‑
氧杂双环[3.1.0]己烷
‑2‑
酮获得的总产率)获得62kg的(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸。
[0149]
用以确认上述反应的hplc条件:试剂和流动相:
·
纯水、乙腈、三氟乙酸、流动相a液、流动相b液及hplc装置的针冲洗液与(c
‑
1)相同。
·
溶液:水和乙腈混合液(1/1,v/v)
·
[(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙基]甲醇的溶液(浓度约1mg/ml)hplc条件除了检测器的测定波长为283nm之外,其余均与(c
‑
1)相同。
[0150]
nmr数据(1r,2s)
‑2‑
(((2,4
‑
二甲基嘧啶
‑5‑
基)氧基)甲基)
‑2‑
(3
‑
氟苯基)
‑
环丙烷甲酸:1h nmr(500mhz,dmso
‑
d6)δ12.47(s,1h),8.17(s,1h),7.39(td,j=8.0,6.4hz,1h),7.29(d,j=7.9hz,1h),7.27
‑
7.22(m,1h),7.10(td,j=8.3,2.1hz,1h),4.63(d,j=10.2hz,1h),4.30(d,j=10.2hz,1h),2.46(s,3h),2.26(s,3h),2.13(dd,j=7.7,6.6hz,1h),1.63
‑
1.54(m,2h);
13
c nmr(126mhz,dmso
‑
d6)δ172.65,162.48(d,jcf=243.6hz),159.08,156.24,149.45,145.15(d,jcf=7.5hz),139.60,130.71(d,jcf=8.5hz),124.79(d,jcf=2.6hz),115.60(d,jcf=21.8hz),114.32(d,jcf=20.8hz),71.15,33.92(d,jcf=2.0hz),26.46,24.96,19.72,18.70。
[0151]
(d)(1r,2s)
‑2‑
(((2,4
‑
二甲基嘧啶
‑5‑
基)氧基)甲基)
‑2‑
(3
‑
氟苯基)
‑
n
‑
(5
‑
氟吡啶
‑2‑
基)环丙基甲酰胺(莱博雷生:式xi的化合物)的制备将(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸(62.4kg,197mol,1.00equiv.)、5
‑
氟吡啶
‑2‑
胺(24.3kg,217mol,1.10equiv.)、乙酸乙酯(499l)、n,n
‑
二异丙基乙胺(53.5kg,414mol,2.10equiv.)和1
‑
丙烷膦酸酐(50%乙酸乙酯溶液)(188kg,296mol,1.50equiv.)的混合物在外温70℃进行加热,通过hplc分析确认反应结束。冷却反应液后,向其中添加纯水312l并进行搅拌,然后静置。排出水层后,向有机层中添加碳酸钠水溶液(碳酸钠68.9kg和水312l)并进行搅拌,然后静置。排出水层后,向有机层中添加纯水(187l)并进行搅拌,然后静置。排出水层,向有机层中添加纯水(187l)并进行搅拌,然后静置。排出水层,过滤有机层。利用乙酸乙酯对澄清过滤管线进行冲洗后,在减压下将溶剂蒸馏去除一部分。将浓缩残余物(包含基本上莱博雷生75.3kg)中添加乙酸乙酯使得总体积达到256l的方式制备混合物,将该混合物在外温60℃的搅拌下加热溶解后,添加正庚烷(12.8kg),冷却至45℃或以下。向其中添加乙酸乙酯(31l),冷却至35℃或以下后添加
正庚烷(670kg)。然后,将悬浮液冷却至10℃或以下,过滤出混合物中的固体,用乙酸乙酯和正庚烷的混合液进行洗涤。将所获得的固体在外温60℃以下进行减压干燥而以产率87%获得莱博雷生(70kg)。
[0152]
用以确认上述反应的hplc条件:试剂和流动相:
·
纯水、乙腈、三氟乙酸、流动相a液、流动相b液及hplc装置的针冲洗液与(c
‑
1)相同。
·
溶液:水和乙腈混合液(1/1,v/v)
·
(1r,2s)
‑2‑
{[(2,4
‑
二甲基嘧啶
‑5‑
基)氧基]甲基}
‑2‑
(3
‑
氟苯基)环丙烷甲酸标准样品hplc条件除了检测器的测定波长为283nm之外,其余均与(c
‑
1)相同。
[0153]
nmr数据(1r,2s)
‑2‑
(((2,4
‑
二甲基嘧啶
‑5‑
基)氧基)甲基)
‑2‑
(3
‑
氟苯基)
‑
n
‑
(5
‑
氟吡啶
‑2‑
基)环丙基甲酰胺:1h nmr(500mhz,dmso
‑
d6)δ11.19(s,1h),8.31(d,j=3.0hz,1h),8.12(s,1h),7.94
‑
7.85(m,1h),7.62(tt,j=8.7,3.1hz,1h),7.44(dd,j=10.6,1.5hz,1h),7.41
‑
7.40(m,1h),7.39(s,1h),7.14
‑
7.06(m,1h),4.67(d,j=10.2hz,1h),4.29(t,j=9.9hz,1h),2.63(t,j=7.0hz,1h),2.38(s,3h),2.03(s,3h),1.76
‑
1.64(m,1h),1.49(dd,j=8.0,4.8hz,1h);
13
c nmr(125mhz,dmso
‑
d6)δ168.68,161.98(d,jcf=242.3hz),158.46,155.15,155.38(d,jcf=247.9hz),148.90,148.51,145.00(d,jcf=7.7hz),139.37,135.15(d,jcf=24.9hz),130.06(d,jcf=8.4hz),125.05(d,jcf=19.5hz),124.70(d,jcf=2.6hz),115.71(d,jcf=21.7hz),114.20(d,jcf=4.1hz),113.70(d,jcf=20.9hz),70.80,34.09(d,jcf=1.9hz),26.90,24.38,18.37,17.78。
[0154]
纯度试验将通过国际公开wo.2013/123240的方法制备的式xi的化合物的标准品用作外部对照,将标准品与试样中的杂质的峰面积进行比较,有次算出试样中的各化合物的质量%。将被检测出超过0.05质量%的杂质的合计作为总相关物质的含量。
[0155]
试验方法:液相色谱法、紫外线吸收计、峰面积式xi的化合物的定量方法式xi的化合物(%)=a
t
/c
t
×
c
s
/a
s
×
100a
t
:样品溶液中的式xi的化合物的峰面积a
s
:标准溶液中的式xi的化合物的峰面积c
t
:样品溶液中的式xi的化合物的浓度(mg/ml)c
s
:标准溶液中的式xi的化合物的浓度(mg/ml)各相关物质(%)=a
t
/c
t
×
c
s
/a
s
总相关物质(%)=样品溶液的相关物质合计a
t
:样品溶液中的各相关物质的峰面积
a
s
:1%标准溶液中的式xi的化合物的峰面积c
t
:样品溶液中的式xi的化合物的浓度(mg/ml)c
s
:标准溶液中的式xi的化合物的浓度(mg/ml)
[0156]
分析方法样品溶液:用稀释液将本品溶解(约0.5mg/ml)式xi的化合物的标准溶液:用稀释液溶解式xi的化合物的标准物质(约0.5mg/ml)1%标准溶液:用稀释液将式xi的化合物的标准溶液(约0.5mg/ml)稀释至100倍稀释液:甲醇和水混合液(7:3)注入量:10μl面积测定范围:注入样品溶液后70分钟
[0157]
试验条件hplc条件:测定波长:281nm柱:内径4.6mm、长度15cm、填充2.7μm的液相色谱用十八烷基硅烷化硅胶的不锈钢管实例:meteoric core c18(ymc)(或同等产品)柱温:接近40℃的恒温流动相:a液:水、乙腈和甲酸铵混合液(950:50:1(v/v/w)b液:乙腈、水和甲酸铵混合液(900:100:1(v/v/w)流速:约1.0ml/分钟的固定流速
[0158]
[表1]流动相的给液注入后的时间(分钟)流動相a(vol%)流動相b(vol%)0
‑
1095
‑
675
‑
3310
‑
40673340
‑
6567
‑
033
‑
10065
‑
700100
[0159]
对于上述实例中所获得的式xi所表示的化合物、和进行了与上述实例相同的实验的不同实施例1
‑
5中所获得的式xi的化合物,使用液相色谱法对其纯度进行分析,将所得的结果示于表2。
[0160]
[表2]
[0161]
式xi所表示的化合物的保留时间为28
±
3分钟。相对于式xi的化合物的保留时间,各杂质的相对保留时间分别如下所述。式xv的化合物:0.08式vi的化合物:0.11式x的化合物:0.16式ix的化合物:0.39式xiv的化合物:0.41式xii的化合物:0.76式xiii的化合物:1.36
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1