间质干细胞的非病毒修饰
1.本发明一般地涉及间质干细胞的非病毒修饰。更具体地,本发明涉及用于治疗性使用,如癌症治疗的间质干细胞(msc)的非病毒修饰。
背景技术:
2.目前,在国立卫生研究院临床试验数据库登记了>700项使用间质干细胞(msc)的临床试验。尽管认为msc
‑
基治疗是安全的[2],但是临床前和临床数据已显示出最多中等效果并且经常是无效的[3
‑
5]。为了克服这种困境,新涌现的趋势将基因修饰msc。仅在英国,37%的登记的试验使用基因修饰的细胞,其中90%使用病毒载体用于向msc的基因递送[6]。使用修饰的msc产生胞嘧啶脱氨酶(cd)用于基因
‑
导向的酶前体药物疗法(gdept)的临床试验正在进行中[7]。参考到使用病毒的固有安全性和生产问题[7,8],使用非病毒方法的msc的高效修饰是所期望的,但是存在显著困难。
[0003]
多种临床前研究和临床试验[28
‑
31]已利用病毒载体作为msc修饰中的有效基因递送载体。尽管病毒基因递送是高效的,但是存在缺陷,其可以包括病毒载体向宿主基因组中的随机整合,这可能会中断重要的基因表达和细胞过程。即使使用非整合病毒载体,由于病毒抗原在转导细胞上可能的递呈可以在移植后潜在激活体内免疫应答,因此可能出现病毒转导的安全性风险。病毒载体的生产是劳动密集且技术上具有挑战性的,因此随着转基因数的增加,存在放大的困难。此外,应注意病毒载体感染的细胞通常具有低拷贝数(<10个拷贝/细胞)。尽管病毒能够保持转基因的表达[32],但是病毒感染的细胞通常具有低拷贝数(<10个拷贝/细胞)[33,34]。另一方面,研究显示可以通过非病毒方法将提高的dna拷贝数递送至各个细胞[35,36],因此提高治疗剂递送中的有效负荷,然而,这些通常具有低转染效率(通常约0
‑
35%)。临床级病毒的生产是费力的并且通常包括稳定生产系的原始细胞库的产生和认证,因此在基因
‑
细胞治疗中造成了高成本[37
‑
39]。
[0004]
非病毒方法通常具有缺陷,从而防止临床使用。非病毒方法,例如,阳离子聚合物、脂质体、电穿孔等,通常在临床治疗相关尺度,在修饰msc中具有低效率。另外,非病毒方法,如电穿孔可以具有低细胞存活力,从而防碍大规模使用。
[0005]
瞬时转染是快速获得单位细胞的高有效负荷的方法,其避免了可能引起细胞衰老的抗生素选择和数周的处理工作[17]并且降低了肿瘤的趋向性[18]以及与病毒诱导的msc转化有关的安全性问题[19]。尽管某些非病毒方法在易于生产、低成本和安全性谱方面具有优于病毒载体的优势[20],但是缺乏对msc修饰的广泛使用主要是由于转染的低效率(0
‑
35%)所造成的[21,22]。尽管可以将高拷贝的dna递送至细胞中,但是转基因表达通常保持较低。使用某些非病毒方法的转基因的低表达可以是由于质粒dna在非生产性胞内区室中的积累,从而使得用于基因转录的质粒的可用性较低。
[0006]
需要用于msc转染的其它、替代性和/或改善的方法。
技术实现要素:
[0007]
对于一些不同的治疗和非治疗应用,修饰以表达治疗基因或其它所关心的基因的干细胞是所期望的。通常,在前体药物基因疗法领域中,由于非病毒方法通常提供不良的转染效率,因此基于病毒的基因修饰方法已在临床前和临床研究中促进了修饰干细胞,如msc的方法。然而,在这些应用中基于病毒的基因修饰具有固有的安全性风险,因此临床级病毒的生产可以是费力的,并且可以通过病毒方法引入每个细胞的基因拷贝数通常较低(通常<10个拷贝/细胞)。此外,通过病毒或非病毒实现干细胞,如msc的基因修饰,同时不对所产生的细胞的表型(即多潜能性、免疫表型、趋性等)造成不期望的改变是该领域所面对的另一种困难。
[0008]
如本文中详细描述的,本发明人现已开发了用核酸构建体转染间质干细胞的方法,从核酸构建体表达一种或多种功能基因,核酸构建体是非病毒的并且在某些实施方式中,它可以提供高转染效率、高拷贝数/细胞、高细胞存活力、长持续时间的瞬时表达和/或基本无变化的多能性表型。在某些实施方式中,这些方法可以是可缩放的和/或适合于修饰的间质干细胞的大规模临床生产。本文还详细描述了转染的间质干细胞和间质干细胞群体,它们的使用、使用这些转染的干细胞治疗疾病或病症,如癌症的方法,和与之有关的试剂盒和组合物。
[0009]
在一个实施方式中,在本文中提供了用从中表达一种或多种功能基因的核酸构建体转染的间质干细胞(msc),msc具有通过核酸构建体的转染基本无变化的多能性表型并且msc不含基于病毒的转染载体材料。
[0010]
在另一实施方式中,本文提供了多种间质干细胞(msc),其中,至少约60%的多种msc用从中表达一种或多种功能基因的核酸构建体转染,多种转染的msc具有通过核酸构建体的转染基本无变化的多能性表型,并且多种msc不含基于病毒的转染载体材料。
[0011]
在多种msc的另一实施方式中,至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或至少约95%的多种msc可以用核酸构建体转染,并且多种msc表达一种或多种功能基因。在其它实施方式中,多种msc的细胞存活力可以为至少约70%、至少约75%、至少约80%或至少约85%。
[0012]
在本文中的任何一种或多种转染的msc的另一实施方式中,一种或多种msc各自可以用平均至少约1000、至少约2000、至少约3000、至少约4000、至少约5000、至少约6000、至少约7000、至少约8000、至少约9000或者至少约10000拷贝的核酸构建体转染。在另一实施方式中,一种或多种功能基因可以在转染的msc细胞中瞬时表达。在另一实施方式中,一种或多种msc可以来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。在另一实施方式中,一种或多种msc可以是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。在另一实施方式中,多种msc可以来源于人、狗、猫、马或其它物种。在另一实施方式中,核酸构建体可以包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0013]
在本文中的任何一种或多种转染的msc的另一实施方式中,一种或多种msc可以在转染后瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或至少约17天。在另一实施方式中,一种
或多种功能基因可以包括自杀基因。在另一实施方式中,一种或多种功能基因可以包含胞嘧啶脱氨酶(cdy)。在另一实施方式中,一种或多种功能基因可以包含尿嘧啶转磷酸核糖基酶(uprt)。在另一实施方式中,一种或多种功能基因可以包含cdy和uprt两者。在另一实施方式中,cdy和uprt可以作为融合构建体表达。在另一实施方式中,一种或多种功能基因可以包含荧光蛋白。在另一实施方式中,荧光蛋白可以包括绿色荧光蛋白(gfp)。在另一实施方式中,一种或多种功能基因可以包含cdy、uprt和gfp。在另一实施方式中,cdy、uprt和gfp可以作为融合构建体表达。在另一实施方式中,一种或多种功能基因可以包含单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)或者另一种胸苷激酶。在另一实施方式中,一种或多种功能基因可以包含一种或多种癌症疗法基因,或者与癌症疗法无关的一种或多种功能基因。
[0014]
在本文中的任何一种或多种转染的msc的另一实施方式中,一种或多种转染的msc可以使用阳离子聚合物、能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定一种或多种msc的微管网络的第二试剂,用核酸构建体转染。在另一实施方式中,阳离子聚合物可以包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。在另一实施方式中,阳离子聚合物可以包括直链聚亚乙基亚胺(lpei)。在另一实施方式中,第一试剂可以包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。在另一实施方式中,第二试剂可以包括组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)。在另一实施方式中,第二试剂可以包括saha(伏立诺他)。
[0015]
在本文中的任何一种或多种msc的另一实施方式中,表型可以包括一种或多种msc的肿瘤和/或癌症趋向性。在另一实施方式中,本文所描述的任何实施方式的一种或多种基因工程msc可以对使用5
‑
氟胞嘧啶(5fc)或更昔洛韦(gcv)的治疗敏感。一种或多种msc的一个或多个实施方式可以:a)将5fc转化为5
‑
氟尿嘧啶(5fu)、5
‑
氟脲嘧啶单磷酸盐(fump)或两者;b)将更昔洛韦转化为更昔洛韦单磷酸盐;或者c)上述a)和b)的组合。在另一实施方式中,表型可以包括其中在转染后cd表面标志物的表达可以基本无变化的免疫表型。
[0016]
在本文所描述的任何一种或多种msc的另一实施方式中,一种或多种转染的msc可以是塑料
‑
附着的,可以表达cd105、cd73和cd90(>95%),可以缺乏cd45、cd34、cd14和hla
‑
dr表面分子的表达(<2%)并且可以能够体外分化为成骨细胞、脂肪细胞和成软骨细胞,从而满足国际细胞疗法协会(isct)所定义的免疫表型标准。在另一实施方式中,一种或多种转染的msc可以是未分化的。
[0017]
在本文所描述的任何一种或多种msc的另一实施方式中,一种或多种msc可以处于冷冻保存状态。
[0018]
在另一实施方式中,一种或多种msc可以用于治疗癌症。在某些实施方式中,癌症可以包括淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌或它们的任意组合。在另一实施方式中,一种或多种msc可以与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0019]
在另一实施方式中,本文提供了用从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法,该方法包括:将多种msc暴露于包含与阳离子聚合物复合的核酸构建体的转染混合物;将msc暴露于能够重定向来自胞内酸性区室的内吞核酸的第一试
剂和能够稳定msc的微管网络的第二试剂;并温育msc;从而提供用核酸构建体转染的msc。
[0020]
在本文所描述的方法的另一实施方式中,在暴露于转染混合物,暴露于第一试剂和第二试剂期间,在温育期间或它们的任意组合,可以不对msc离心。在另一实施方式中,温育msc的步骤可以包括温和混合,但不离心。在另一实施方式中,温育msc的步骤可以包括将msc温育至少约2小时。在另一实施方式中,温育msc的步骤可以包括将msc温育约2小时至约48小时。在另一实施方式中,温育msc的步骤可以包括将msc温育约3小时至约24小时或者约4小时至约18小时。
[0021]
在本文所描述的任何方法的另一实施方式中,阳离子聚合物可以包括已鉴别为对msc具有低细胞毒性的阳离子聚合物。在另一实施方式中,阳离子聚合物可以具有约5kda至约200kda的大小。在另一实施方式中,阳离子聚合物可以包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。在另一实施方式中,阳离子聚合物可以包括直链聚亚乙基亚胺(lpei)。
[0022]
在本文所描述的任何方法的另一实施方式中,第一试剂可以包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。在另一实施方式中,第二试剂可以包括组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)。在另一实施方式中,第二试剂可以包括saha(伏立诺他)。
[0023]
在本文所描述的任何方法的另一实施方式中,将msc暴露于转染混合物的步骤可以包括将核酸构建体与阳离子聚合物复合以提供包含复合的核酸构建体的转染混合物,并将转染混合物添加至msc。在另一实施方式中,将msc暴露于转染混合物的步骤可以包括将转染混合物添加至msc并将msc与转染混合物温育。在另一实施方式中,将msc暴露于第一试剂和第二试剂的步骤可以包括用补充有第一试剂和第二试剂的细胞培养基替换转染混合物。在另一实施方式中,将msc暴露于转染混合物的步骤可以包括从msc除去培养基并用转染混合物替换培养基。在另一实施方式中,将msc暴露于转染混合物的步骤可以包括在轻微离心下将msc与转染混合物温育。在另一实施方式中,轻微离心可以包括约200g离心约5分钟。
[0024]
在本文所描述的任何方法的另一实施方式中,细胞培养基可以包括完全培养基。在另一实施方式中,msc可以处于约60%汇合,并且可以在暴露于转染混合物之前约24小时接种msc。在另一实施方式中,转染混合物可以包括处于无血清dmem中或新鲜培养基中的复合的核酸构建体。
[0025]
在本文所描述的任何方法的另一实施方式中,msc所暴露的转染混合物中的核酸构建体的量可以在约200至约500ng每1.9cm2表面积之间。在另一实施方式中,msc所暴露的转染混合物中的核酸构建体的量可以在约250至约400ng每1.9cm2表面积之间。在另一实施方式中,msc所暴露的转染混合物中的核酸构建体的量可以在约300至约350ng每1.9cm2表面积之间。在另一实施方式中,在转染混合物中阳离子聚合物与核酸构建体的比值可以为约1μg至约30μg阳离子聚合物每1μg核酸构建体。在另一实施方式中,可以用平均至少约1000、至少约2000、至少约3000、至少约4000、至少约5000、至少约6000、至少约7000、至少约8000、至少约9000或者至少约10000拷贝的核酸构建体各自转染转染的msc。在另一实施方式中,核酸构建体可以包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接
区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0026]
在本文所描述的任何方法的另一实施方式中,一种或多种功能基因可以包括自杀基因。在另一实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy)和/或胸苷激酶(tk)。在另一实施方式中,一种或多种功能基因可以包含尿嘧啶转磷酸核糖基酶(uprt)。在另一实施方式中,一种或多种功能基因可以包含cdy和uprt两者。在另一实施方式中,cdy和uprt可以作为融合构建体表达。在另一实施方式中,一种或多种功能基因可以包含荧光蛋白。在另一实施方式中,荧光蛋白可以包含绿色荧光蛋白(gfp)。在另一实施方式中,一种或多种功能基因可以包含cdy、uprt和gfp。在另一实施方式中,cdy、uprt和gfp可以作为融合构建体表达。在另一实施方式中,一种或多种功能基因可以包括单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)或者另一种胸苷激酶。在另一实施方式中,一种或多种功能基因可以包含一种或多种癌症疗法基因,或者与癌症疗法无关的一种或多种功能基因。在另一实施方式中,可以在转染的msc中瞬时表达一种或多种功能基因。在另一实施方式中,msc可以在转染后瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。在另一实施方式中,一种或多种功能基因可以包括荧光蛋白,并且方法还可以包括使用细胞分选或facs分离、选择或纯化转染的msc的步骤。
[0027]
在本文所描述的任何方法的另一实施方式中,通过转染转染的msc的多能性表型可以基本无变化。例如,不希望是限制性的,多能性表型可以包括分化潜能,从而修饰的细胞能够分化为与天然msc相当的成骨、生脂和/或软骨形成系。在另一实施方式中,多能性表型可以包括msc的肿瘤和/或癌症趋向性。在另一实施方式中,多能性表型可以包括其中在转染后cd表面标志物的表达基本无变化的免疫表型。在另一实施方式中,转染的msc可以是未分化的。
[0028]
在本文所描述的任何方法的另一实施方式中,转染的msc可以是塑料
‑
附着的,可以表达cd105、cd73和cd90(>95%),可以缺乏cd45、cd34、cd14和hla
‑
dr表面分子的表达(<2%)并且可以能够体外分化为成骨细胞、脂肪细胞和成软骨细胞,从而满足国际细胞疗法协会(isct)所定义的免疫表型标准。
[0029]
在本文所描述的任何方法的另一实施方式中,可以用核酸构建体转染至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或者至少约95%的msc并且msc表达一种或多种功能基因。在另一实施方式中,转染的msc的细胞存活力可以为至少约70%、至少约75%、至少约80%或者至少约85%。
[0030]
在本文所描述的任何方法的另一实施方式中,方法可以不含基于病毒的转染载体材料。
[0031]
在本文所描述的任何方法的另一实施方式中,msc可以来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。在另一实施方式中,msc可以是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。在另一实施方式中,一种或多种msc可以来源于人、狗、猫、马或其它物种。
[0032]
在本文所描述的任何方法的另一实施方式中,所产生的msc可以对使用5
‑
氟胞嘧啶(5fc)或更昔洛韦(gcv)或两者的治疗敏感。在另一实施方式中,所产生的msc可以:a)将5fc转化为5
‑
氟尿嘧啶(5fu)、5
‑
氟尿嘧啶单磷酸盐(fump)或两者;b)将更昔洛韦转化为更
昔洛韦单磷酸盐;或者c)上述a)和b)的组合。
[0033]
在本文所描述的任何方法的另一实施方式中,方法可以包括在将msc暴露于转染混合物的步骤之前,在生长培养基,如新鲜的生长培养基中培养msc的步骤。在另一实施方式中,将msc暴露于转染混合物的步骤可以包括将转染混合物添加至msc但不从msc中除去生长培养基,并且在暴露和温育步骤期间不进行离心。在另一实施方式中,将msc暴露于第一试剂和第二试剂的步骤可以包括将第一试剂和第二试剂同时、顺序或与转染混合物组合添加至msc。在另一实施方式中,可以与转染混合物向msc的添加同时将第一试剂和第二试剂添加至msc,或者第一试剂和第二试剂可以与转染混合物混合并添加至msc。在另一实施方式中,可以在将转染混合物添加至msc之后不久,将第一试剂和第二试剂添加至msc。在另一实施方式中,可以在第一试剂和第二试剂添加至msc之前不除去转染混合物。在另一实施方式中,msc向转染混合物的暴露持续时间可以与msc向第一和第二试剂暴露持续时间重叠。在另一实施方式中,可以在第一试剂和第二试剂添加至msc之前不除去转染混合物。
[0034]
在本文所描述的任何方法的另一实施方式中,方法还可以包括冷冻保存转染的间质干细胞(msc)的步骤以用于储存。在另一实施方式中,方法还可以包括融化冷冻保存的转染间质干细胞的步骤,从而为其使用作准备。
[0035]
在本文所描述的任何方法的另一实施方式中,转染的msc是可以如本文所描述的任何一种或多种msc实施方式所定义的msc。在另一实施方式中,可以通过如本文所描述的任何方法产生msc或多种msc的一个或多个实施方式。
[0036]
在另一实施方式中,本文提供了通过如本文所描述的任何方法所产生的msc或多种msc。
[0037]
在另一实施方式中,本文提供了如本文所定义的任何一种或多种msc用于治疗对其有需要的受试者中的癌症的使用。在某些实施方式中,作为非限制性实例,癌症可以包括淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌。在另一实施方式中,一种或多种msc可以与5fc、5fu、gcv或它们的任意组合进行组合使用。在另一实施方式中,一种或多种msc可以在用于治疗癌症的药剂的生产中使用。在另一实施方式中,一种或多种msc可以与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0038]
在另一实施方式中,本文提供了治疗对其有需要的受试者中的癌症的方法,其中方法可以包括:将如本文所定义的任何一种或多种msc施用至受试者的癌细胞附近的区域,其中一种或多种msc中的一种或多种功能基因可以有助于对癌细胞的抗癌作用。
[0039]
在本文所描述的任何癌症治疗方法的某些实施方式中,癌症可以(例如)包括淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌。
[0040]
在本文所描述的任何癌症治疗方法的另一实施方式中,可以与5fc、5fu、gcv或它们的任意组合同时、顺序或组合施用一种或多种msc。在另一实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy)、胸苷激酶(tk)或两者。在另一实施方式中,一种或多种功能基因可以包括尿嘧啶转磷酸核糖基酶(uprt)。在另一实施方式中,一种或多种功能基因可以包括cdy和uprt两者。在另一实施方式中,可以在一种或多种msc中作为融合构建体表达cdy和uprt。在另一实施方式中,一种或多种msc可以在转染后瞬时表达一种或多种功
能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0041]
在另一实施方式中,本文所描述的任何癌症治疗方法还可以包括向受试者施用5fc、5fu、更昔洛韦或它们的任意组合的步骤,从而将一种或多种msc暴露于5fc、5fu、更昔洛韦或其组合。
[0042]
在另一实施方式中,本文所描述的任何癌症治疗方法还可以在施用一种或多种msc的步骤之前,包括根据如本文所描述的任何实施方式中所定义的任何方法生产一种或多种msc的步骤。
[0043]
在另一实施方式中,本文提供了包含本文所描述的任何实施方式的一种或多种工程化msc和药物可用的载体、稀释剂、赋形剂、细胞培养基或缓冲剂中的至少一种的组合物。
[0044]
在另一实施方式中,本文提供了包含本文所描述的任何实施方式的任何一种或多种msc的治疗诊断试剂。
[0045]
在另一实施方式中,本文提供了用从中瞬时表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的试剂盒。在一个实施方式中,试剂盒可以包括以下中的一种或多种:msc;设计用于瞬时表达一种或多种功能基因的核酸构建体;细胞培养基;阳离子聚合物;能够重定向来自胞内酸性区室的内吞核酸的第一试剂;能够稳定msc的微管网络的第二试剂;用于实施如在本文中的任何实施方式中所描述的方法的说明书;5fc;gcv;和/或5fu。在某些实施方式中,试剂盒可以包括冷冻保存缓冲液或试剂、融化缓冲液或试剂或两者。在某些非限制性实施方式中,可以使用冷冻保存缓冲液或溶液,如cryostor10(biolife solutions usa)。在其它示例性实施方式中,融化的工程化msc可以保存在低温溶液,如hypothermosol(biolife solutions usa)中。如将对本领域技术人员显而易见的,还可以使用其它实例。
[0046]
在本文中的任何试剂盒的另一实施方式中,msc可以来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。在另一实施方式中,msc可以是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。在另一实施方式中,msc可以来源于人、狗、猫、马或其它物种。在另一实施方式中,核酸构建体可以包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。在另一实施方式中,阳离子聚合物可以包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。在另一实施方式中,阳离子聚合物可以包括直链聚亚乙基亚胺(lpei)。在另一实施方式中,第一试剂可以包括dopc、dppc或另一种促融脂质(fusogenic lipid)中的一种或多种。在另一实施方式中,第一试剂可以包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。在另一实施方式中,第二试剂可以包括组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)。在另一实施方式中,第二试剂可以包括saha(伏立诺他)。在另一实施方式中,一种或多种功能基因可以包括自杀基因。在另一实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy)或者胸苷激酶(tk)。在另一实施方式中,一种或多种功能基因可以包括尿嘧啶转磷酸核糖基酶(uprt)。在另一实施方式中,一种或多种功能基因可以包括cdy和uprt两者。在另一实施方
式中,cdy和uprt可以表达为融合构建体。在另一实施方式中,一种或多种功能基因可以包括荧光蛋白。在另一实施方式中,荧光蛋白可以包括绿色荧光蛋白(gfp)。在另一实施方式中,一种或多种功能基因可以包括cdy、uprt和gfp。在另一实施方式中,cdy、uprt和gfp可以表达为融合构建体。在另一实施方式中,一种或多种功能基因可以包括单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)。在另一实施方式中,一种或多种功能基因可以包含一种或多种癌症疗法基因,或者与癌症疗法无关的一种或多种功能基因。在另一实施方式中,阳离子聚合物可以包括已鉴别为对msc具有低细胞毒性的阳离子聚合物。在另一实施方式中,阳离子聚合物可以具有约5kda至约200kda的大小。在另一实施方式中,在试剂盒的一个或多个实施方式中阳离子聚合物与核酸构建体的比值可以为约1μg至约30μg阳离子聚合物每1μg核酸构建体。
[0047]
在本文中的任何试剂盒的另一实施方式中,试剂盒可以用于制备msc
‑
基抗癌剂。在本文中的任何试剂盒的另一实施方式中,试剂盒可以包括用于实施如本文所描述的实施方式中的任一个中所定义的任何方法的说明书和/或设备。
[0048]
在另一实施方式中,本文提供了用于用从中瞬时表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的试剂盒,其中试剂盒可以包括以下中的任何一个或多个:msc;设计用于瞬时表达一种或多种功能基因的核酸构建体;细胞培养基;阳离子聚合物;能够重定向来自胞内酸性区室的内吞核酸的第一试剂;能够稳定msc的微管网络的第二试剂;用于实施如本文所描述的任何方法的说明书;5fc;gcv;和/或5fu。在某些实施方式中,试剂盒可以包括如以上所描述的冷冻保存缓冲液或试剂、融化缓冲液或试剂或两者。
[0049]
在另一实施方式中,本文提供了用从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法,方法包括:在生长培养基中培养msc;将包含与阳离子聚合物复合的核酸构建体的转染混合物添加至msc,同时不从msc除去生长培养基;将能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂添加至msc;和温育msc,同时与转染混合物、第一试剂和第二试剂全部接触培养期;其中与转染混合物的添加同时,与转染混合物的添加顺序或者与转染混合物组合,将第一试剂和第二试剂添加至msc;并且其中在转染混合物的添加和温育期结束之间不对msc离心;从而提供用核酸构建体转染的msc。
[0050]
在用于用如本文所描述的从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法的另一实施方式中,温育期可以为至少约2小时。在另一实施方式中,温育期可以是约2小时、约3小时、约4小时、约5小时、约6小时、约7小时、约8小时、约9小时、约10小时、约11小时、约12小时、约13小时、约14小时、约15小时、约16小时、约17小时、约18小时、约19小时、约20小时、约21小时、约22小时、约23小时、约24小时、约25小时、约26小时、约27小时、约28小时、约29小时、约30小时、约31小时、约32小时、约33小时、约34小时、约35小时或约36小时或更长。
[0051]
在另一实施方式中,本文提供了通过用于用如本文所描述的从中表达一种或多种功能基因的核酸构建体转染多种间质干细胞(msc)的任何方法产生的msc细胞或多种msc细胞。在另一实施方式中,本文提供了本文所描述的任何msc细胞或多种msc细胞用于治疗对其有需要的受试者中的癌症或者用于生产癌症治疗药剂的使用。
[0052]
本文描述了治疗对其有需要的受试者中的癌症的方法。在某些实施方式中,这些方法可以包括将如本文所描述的一个或多个实施方式中所定义的任何一种或多种msc施用
至受试者的癌细胞附近的区域,其中一种或多种msc中的一种或多种功能基因有助于对癌细胞的抗癌作用。
[0053]
在某些实施方式中,癌症可以(例如)包括淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌。
[0054]
在另一实施方式中,本文提供了包含如本文所描述的任何一种或多种msc和药物可用的载体、稀释剂、赋形剂、细胞培养基或缓冲剂中的至少一种的组合物。在另一实施方式中,本文提供了包含本文所描述的任何实施方式的任何一种或多种msc的治疗诊断试剂和/或试剂盒。
附图说明
[0055]
根据其中参考附图的以下描述,这些及其它特征将变得更加显而易见,其中:
[0056]
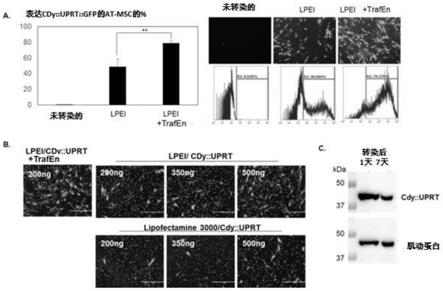
图1a
‑
c显示了通过lpei基转染方法的实施方式的cdy::uprt_at
‑
msc的产生。增强子使得cdy::uprt能够在at
‑
msc中高表达。图1a显示用1μg与lpei复合的cdy::uprt::gfp pdna(1μgdna至5μl lpei)转染6孔培养容器中的at
‑
msc。在离心后,用新鲜培养基(具有或不具有trafen)替换转染混合物。一天后,采集代表性图象并将细胞胰蛋白酶化以用于facs分析。结果表示为平均值
±
sd(n=4)。将未转染的at
‑
msc用作阴性对照。使用双尾学生t检验计算转染条件之间的显著性差异。**p<0.01。图1b显示分别使用离心或生产商的规程,使用lpei或lipofectamine3000,以不同量的cdy::uprt表达质粒转染在24
‑
孔容器中培养的at
‑
msc(lot00088)。在24h温育后,用4%多聚甲醛固定细胞并对cdy(绿色)和核(hoechst染色剂,蓝色)染色。显示了代表性图象。比例尺表示400μm。图1c:在存在trafen的情况下,用lpei/cdy::uprt多聚复合物转染的at
‑
msc。在修饰后1或7天,将细胞裂解以用于使用靶向cdy的抗体的免疫印迹法分析。将肌动蛋白用作样品加载的内源对照;
[0057]
图2a
‑
b是显示使得修饰的at
‑
msc对5fc和5fu敏感的cdy::uprt表达的柱状图。图2a显示用150μg/ml 5fc处理cdy::uprt_at
‑
msc指定的时间。通过标准mts测定测量每个时间点的细胞存活力。在各个时间点,将无5fc处理的样品用作对照。图2b显示在存在100μg/ml 5fu的情况下培养5天后,比较了未修饰和修饰的at
‑
msc之间对5fu的灵敏度。使用mts测定测量处理后的细胞存活力。将无5fu处理的条件作为100%。结果表示为平均值
±
sd(n=4)。使用双尾学生t检验计算at
‑
msc和cdy::uprt_at
‑
msc之间的细胞存活力的显著性差异。**,p<0.005;
[0058]
图3a
‑
c显示cdy::uprt表达不影响标准免疫表型谱和分化潜能。图3a显示用荧光团
‑
缀合的抗体标记at
‑
msc和cdy::uprt_at
‑
msc并根据生产商的说明书,通过流式细胞术分析。将同种型抗体用作各自对照。柱状图显示了同种型(红色),未修饰的at
‑
msc(绿色)和cdy::uprt_at
‑
msc(蓝色)的合并的轮廓图。图3b显示了按照生产商的建议,将两种细胞类型在补充有成骨分化剂的培养基中培养14天。在温育结束时,用茜素红s对细胞染色。用茜素红s染色的钙沉积是指明分化的at
‑
msc的表型之一。图3c显示将未修饰的和表达cdy::uprt的at
‑
msc在含有用于生脂分化的组分的培养基中培养。14天后,用油红
‑
o对细胞染色。该染料对细胞中可见的油滴染色,其指示了生脂分化。以20
×
放大倍数采集图像;
[0059]
图4a
‑
b显示cdy::uprt表达不影响at
‑
msc的迁移能力。图4a显示使用细胞侵袭测
定评价msc的迁移性。首先,将200k或400k的靶细胞在24孔容器中的补充有10%fbs的dmem中铺板。6小时后,用1
×
pbs清洗细胞培养物一次并用无血清dmem替换。将cdy::uprt_at
‑
msc(实验前一天修饰)和未修饰的at
‑
msc加载到matrigel
‑
涂覆的细胞培养小室(cell inserts)中。将该小室分别转移至靶细胞培养。24小时后,通过采集用hoechst 33342染色的细胞的荧光图像,在显微镜下评价细胞侵袭。图片提供了每帧迁移细胞的平均值(n=3)。将hek293t用作阴性对照。使用双尾学生t检验计算200,000和400,000个靶细胞之间的显著性差异。**,p<0.01。图4b显示以10
×
放大倍数采集用hoechst 33342染色的迁移的cdy::uprt_at
‑
msc的图象。比例尺代表400μm;
[0060]
图5a
‑
c显示了通过cdy::uprt_at
‑
msc/5fc介导的对癌细胞的体外选择性细胞毒性抗癌作用。图5a显示将cdy::uprt_at
‑
msc与u251
‑
mg、mb
‑
mda231或mkn1在存在或不存在150μg/ml 5fc的情况下,在补充有2%fbs的dmem中共培养。将治疗性细胞和癌细胞系以1个cdy::uprt_at
‑
msc比5、10、50、100个癌细胞的比例混合。5天后,通过标准mts测定,通过分光光度法评价增殖抑制。增殖抑制效率定义为100%
‑
(样品/对照
×
100%)。将无5fc处理的条件用作对照。图片误差线代表平均值(n=4)+sd。图5b显示了在实验结束时采集的混合培养(1个msc比10个癌细胞)的明视野。比例尺代表400μm。图5c显示通过间接共培养评价cdy::uprt_at
‑
msc或at
‑
msc对mb
‑
mda
‑
231的细胞毒性抗癌作用。将相等数目的治疗性细胞和mb
‑
mda
‑
231分别接种到转板和24孔板中。将细胞在补充有2%fbs和100μg/ml 5fc的dmem中共培养4天。然后,除去转板并用hoechst 3222对培养平板上保留的细胞染色。用酶标仪采集荧光读数。将增殖抑制效率(%)定义为100%
‑
(使用5fc的条件/无5fc的各自条件
×
100%)。将从三个生物重复的9个区域采集的相对荧光单位显示为平均值
±
sem。图片代表从每个孔中的9个区域所采集的结果,平均值+sem。显示了在24孔板上保持的癌细胞的各自图象。比例尺代表400μm;
[0061]
图6a
‑
c显示了通过使用不同转染方法所产生的cdy::uprt_at
‑
msc/5fc介导的不同的细胞毒性抗癌作用。用1μg通过lpei(具有或不具有trafen)和lipofectamine 3000介导的无cpg cdy::uprt表达质粒转染at
‑
msc(250,000个细胞)。转染后一天,将cdy::uprt_at
‑
msc与u251
‑
mg、mb
‑
mda231或mkn1在存在或不存在150μg/ml 5fc的情况下,在补充有2%fbs的dmem中共培养。将治疗性细胞和癌细胞系以1个cdy::uprt_at
‑
msc比1个(图6a)、5个(图6b)、10个(图6c)癌细胞的比例混合。5天后,通过标准mts测定,通过分光光度法评价增殖抑制。将无5fc处理的条件用作对照。增殖抑制效率定义为100%
‑
(样品/对照
×
100%)。图片误差线代表平均值(n=4)
±
sd。使用双尾学生t检验计算使用lpei+trafen和其它方法的条件之间的显著性差异。**,p<0.01;
[0062]
图7a
‑
c显示长期表达使得cdy::uprt_at
‑
msc能够具有持续抗癌效率。在存在trafen的情况下,用1μg通过lpei介导的无cpg cdy::uprt表达质粒转染at
‑
msc(250,000个细胞)。1天(图7a)和7天(图7b),收集修饰的at
‑
msc并在存在或不存在150μg/ml 5fc的情况下,以1个msc比5或10个癌细胞的比值与mkn1和mkn28细胞系共培养。温育5天后,通过标准mts测定,通过分光光度法评价增殖抑制。将无5fc处理的条件用作对照。增殖抑制效率定义为100%
‑
(样品/对照
×
100%)。图片误差线代表平均值(n=4)
±
sd。图7c显示在修饰后1或7天,将细胞裂解以用于使用靶向cdy的抗体的免疫印迹法分析。将肌动蛋白用作样品加载的内源对照。转染后1和7天收集at
‑
msc的细胞裂解液。使用免疫印迹分析评价cdy::uprt的
表达。在平行实验中,在转染后第1(a)或7(b)天采集修饰的at
‑
msc。
[0063]
图8a
‑
b显示trafen使得能够在at
‑
msc中进行有效的lpei基转染。图8a显示以不同量的pdna制备了lpei/pcmv
‑
gfp多聚复合物或者lipofectamine 3000/pcmv
‑
gfp脂质体复合物。分别按照离心规程或生产商的说明书,通过lpei(1μg pdna比10μl lpei)或者lipofectamine 3000转染at
‑
msc。以每个生物重复(n=3)9个区域,通过分光光度法(ex475/em509)测量gfp表达的荧光强度(rfu)。图片表示rfu的平均值+sem。观察到随着dna量的增加,细胞数减少。图8b显示在存在或不存在trafen的情况下,用与200ng pcmv
‑
gfp复合的lpei转染at
‑
msc。24小时后,将细胞胰蛋白酶化,成粒并在1
×
pbs中再混悬以用于流式细胞术分析。将转染效率计算为对如通过facs定量的细胞总数归一化的gfp阳性细胞的百分比。柱状图表示平均值
±
sd,n=3。采集明视野和荧光图像。显示代表性图象;
[0064]
图9显示了在分离自不同供体的at
‑
msc中的高转染效率。at
‑
msc分离自年龄31
‑
45岁的女性供体(lot00061,roosterbio)。以1μg pdna比5μl lpei的比值,以不同量的pdna制备lpei/pcmv
‑
gfp多聚复合物。1天(24小时)后,采集代表性图象,然后将细胞胰蛋白酶化,成粒并在1
×
pbs中再混悬以用于流式细胞术分析。将转染效率计算为对如通过facs定量的细胞总数归一化的gfp阳性细胞的百分比。柱状图表示平均值
±
sd,n=3。采集荧光图像。显示代表性图象;
[0065]
图10显示了cdy::uprt::gfp在at
‑
msc中的延长表达。在存在trafen的情况下,用1.25μg表达pdna的融合cdy::uprt::gfp的pei多聚复合物转染at
‑
msc。在转染后1、2、3、5、8天,采集荧光和明视野图象。以细胞培养物的9个区域,通过分光光度法(ex475/em509)测量gfp表达的荧光强度(rfu)。图片表示两个生物复制的rfu的平均值+sd。使用双尾学生t检验计算在转染后不同天的gfp表达之间的显著性差异。**p<0.01;
[0066]
图11显示了表达cdy::uprt::gfp的at
‑
msc的生脂分化。在存在trafen的情况下,用1.25μg表达pdna的融合cdy::uprt::gfp的pei多聚复合物转染at
‑
msc。转染后24小时、用生脂分化培养基替换培养基。14天后,用油红
‑
o对细胞染色。如通过gfp表达所指示的修饰的at
‑
msc显示出可见的油滴,表明转染后at
‑
msc的多潜能性保持无变化;
[0067]
图12a
‑
c显示出cdy::uprt_at
‑
msc/5fc和5fu的相当的抗癌效率。在u251
‑
mg(图12a)、mda
‑
mb
‑
231(图12b)和mkn1(图12c)中评价抗癌作用。通过将相等数目的cdy::uprt_at
‑
msc和癌细胞系(2000个u251
‑
mg,5000个mda
‑
mb
‑
231和mkn1)共培养,分析了cdy::uprt_at
‑
msc结合5fc的治疗效力(抗癌作用)。1天后,用补充有2%fbs和不同浓度的5fc(5、10、50、100μg/ml)的dmem替换培养基。另一方面,在5fu处理前24h,接种4000个u251
‑
mg,10000个mda
‑
mb
‑
231和mkn1。在补充有2%fbs的dmem中,通过5、10、50、100μg/ml 5fu处理细胞系。温育5天后,通过标准mts测定定性评价细胞毒性作用。将无5fc和5fu处理的条件用作设置为100%的阴性对照。图片表示平均值
±
sd,n=4;
[0068]
图13a
‑
b显示了cdy::uprt_at
‑
msc/5fc对癌细胞系的选择性增殖抑制。将cdy::uprt_at
‑
msc与hs738t(atcc,crl
‑
7869)、ags、mkn28、hs746t、nugc3和mkn45(由新加坡国立大学癌症研究所(national university cancer institute,singapore)的yong wei peng博士友好提供)共培养。图13a显示在存在或不存在150μg/ml 5fc的情况下,在补充有2%fbs的dmem中温育混合培养物。将治疗性细胞和癌细胞系以1个cdy::uprt_at
‑
msc比10个癌细胞的比例混合。5天后,通过标准mts测定,通过分光光度法评价增殖抑制。将无5fc处理的
条件用作对照。增殖抑制效率定义为100%
‑
(样品/对照
×
100%)。图片误差线代表平均值(n=4)
±
sd。图13b显示了在实验结束时采集的混合培养物的明视野图象。比例尺代表400μm;
[0069]
图14a
‑
b显示了在来自不同来源的干细胞中的相当的转染效率和抗癌效率。在存在trafen的情况下,用离心规程转染脂肪组织(at,roosterbio)、骨髓(bm,roosterbio)和uc(脐带,atcc)来源的msc。转染后24小时、将细胞胰蛋白酶化并采集用于免疫印迹分析(图14a)。将细胞裂解以用于使用靶向cdy和肌动蛋白的抗体的免疫印迹法分析。图14b显示在相同实验中,收获细胞以用于与多种癌细胞系以1个msc比50个癌细胞的比值的共培养研究。将细胞在含有100μg/ml 5fc的培养基中共培养5天。在温育结束时,通过以波长ex340/em488测量用hoechst 33342染色的细胞的rfu,通过分光光度法评价剩余的细胞数。将使用未修饰的msc的条件用作对照。因此计算增殖抑制百分比。图片表示从四个重复收集的数据,平均值
±
sem;
[0070]
图15显示在修饰以表达hsv
‑
tk的多种干细胞中相当的转染效率和抗癌效率。在存在trafen的情况下,用离心规程转染at
‑
、bm
‑
和uc
‑
msc。将1微克pselect
‑
zeo
‑
hsv1tk(invivogen)用于转染250,000个msc。转染后24小时、收获msc以用于与多种癌细胞系以1个msc比50个癌细胞的比值的共培养研究。将细胞在含有100μg/ml前体药物更昔洛韦(invivogen)的培养基中共培养5天。在温育结束时,通过以波长ex340/em488测量用hoechst 33342染色的细胞的rfu,通过分光光度法评价剩余的细胞数。将使用未修饰的msc的条件用作对照。因此计算增殖抑制百分比。图片表示从四个重复收集的数据,平均值
±
sem;
[0071]
图16a
‑
b显示了使用含有cpg岛的表达载体,cdy::uprt表达随时间减少。在存在trafen的情况下,根据离心规程,用1μgpselect
‑
zeo
‑
fcyfur(invivogen)转染at
‑
msc(250,000个细胞)。转染后1、3、7天,收获细胞以用于免疫印迹分析(图16a)和共培养实验(图16b)。对于共培养实验,将cdy::uprt修饰的at
‑
msc与u
‑
251mg和mda
‑
mb
‑
231细胞以1个msc比1、5或10个癌细胞的比值在补充有2%fbs和100μg/ml 5fc的dmem中培养。5天后,通过标准mts测定,通过分光光度法评价增殖抑制。将无5fc处理的条件用作对照。因此计算增殖抑制百分比。图片表示从四个重复收集的数据,平均值+sem;
[0072]
图17显示了用于msc转染的规程的示例性实施方式的图示;
[0073]
图18显示了以多种dna的量的细胞存活力和转染效率。不使用离心转染at
‑
msc。通过流式细胞术分析确定基因修饰效率和细胞存活力;
[0074]
图19显示了使用非离心规程转染的at
‑
msc中的cdy::uprt的长期表达。通过流式细胞术分析确定基因修饰效率;
[0075]
图20a
‑
b显示了聚合物与不同类型的msc:uc
‑
msc(图20a)和bm
‑
msc(图20b)的相容性。在不使用离心的情况下,将msc与转染混合物温育24h;
[0076]
图21显示随着dna和聚合物的量的升高,细胞存活力降低。在不使用离心的情况下,通过多种聚合物转染at
‑
msc。直链pei(<200kda)的浓度为1μg/μl;
[0077]
图22显示随着dna和聚合物的量的升高,细胞存活力降低。在不使用离心的情况下,通过多种聚合物转染uc
‑
msc。直链pei(<5kda)的浓度为10μg/μl;
[0078]
图23a
‑
b显示使用trafen方法,单位细胞的高表达水平。用慢病毒感染u2os细胞
(图23a)或者at
‑
msc细胞(图23b)并温育5天(图23b的左图)。在相同实验组中,在第4天,在存在trafen的情况下,用pei转染来自单独培养的细胞(图23b的右图)。在第5天,采集感染和转染的细胞的荧光图像。对于at
‑
msc,通过流式细胞术分析进一步确定了慢病毒和trafen方法的基因修饰效率。在通过trafen方法转染的at
‑
msc中,更多数目的细胞表达高水平的gfp(图23b的右图);
[0079]
图24a
‑
b显示了不同细胞容器尺寸中获得的转染的msc的个数(图24a)和msc个数&容器尺寸的良好相关性的图(图24b);
[0080]
图25是显示使用非病毒转染方法生产大量转染的msc的整体方法的开发的示意图。提供了考虑和/或优化以实现目标的因素。由于细胞的差异度,可以筛选一组trafen相容性聚合物(例如,pei)以获得高转染效率、低细胞毒性、长期表达和生产可缩放的优化制剂;
[0081]
图26显示了以多种dna的量的细胞存活力和转染效率。通过流式细胞术分析确定基因修饰效率和细胞存活力;
[0082]
图27a
‑
b显示cdy::uprt表达不影响标准免疫表型谱和分化潜能。图27a显示用荧光团
‑
缀合的抗体标记cdy::uprt_at
‑
msc并通过流式细胞术分析。将同种型用作阴性对照。图27b显示将两种细胞类型在补充有生脂分化剂和成骨分化剂的培养基中分别培养14和21天。在温育结束时,用油红
‑
o(生脂)或茜素红s(成骨)对细胞染色。油红
‑
o对细胞中可见的油滴染色,其指示了生脂分化。用茜素红s染色的钙沉积是指明分化的at
‑
msc的表型之一;
[0083]
图28与以上图8a有关。然后,将贴壁细胞胰蛋白酶化并用碘化丙啶(pi)和hoechst 33342(h33342)染色。根据生产商的规程,用nc
‑
3000细胞计数器确定细胞存活力和总贴壁细胞。将未转染的群体用作对照。细胞存活力(%)表示pi阴性细胞的百分比。相对于设置为100%的对照计算总贴壁细胞的百分比。将数据表示为以三个生物重复进行的实验的平均值+sd。使用双尾学生t检验计算对照和转染样品之间的显著性差异。**,p<0.05;
[0084]
图29与以上图8b中所示的结果有关。在相同实验中,使用nc
‑
3000细胞计数器确定了每种条件下的贴壁细胞总数(左图)和细胞存活力(右图)。将未转染的群体用作对照。结果表示为平均值
±
sd,n=3;
[0085]
图30与以上图1有关。在相同实验中,使用nc
‑
3000细胞计数器确定了每种条件下的细胞总数和细胞存活力。计算了对照(未转染)处转染群体中的总贴壁细胞百分比。数据表示为平均
±
sd,n=3;
[0086]
图31显示了用cd::uprt和cd::uprt::gfp修饰的msc的相当的抗癌效率。在存在的情况下增强子的情况下,用1μg cdy::uprt或者cd::uprt::gfp转染msc(200,000个细胞)。转染后1天,将u
‑
251mg细胞与cd::uprt_msc以1:1、5或10(msc:癌细胞)的比值在补充有2%fbs,具有或不具有100μg/ml 5fc的dmem中共培养。5天后,通过标准mts测定,通过分光光度法评价处理条件中的增殖抑制。将无5fc处理的条件用作设置为0%的对照。相对于对照计算增殖抑制(%)。将从四个重复收集的数据表示为平均值+sd;
[0087]
图32显示了在存在5
‑
氟尿嘧啶(5
‑
fu)的情况下,cd::uprt_at
‑
msc的体内抗
‑
肿瘤作用。为了建立s.c肿瘤,将5
×
106个替莫唑胺耐受性u
‑
251mg细胞皮下注射至背侧区。当肿瘤达到目标尺寸时,将1
×
106个cd::uprt_at
‑
msc或者msc直接注射至s.c肿瘤。1天后,连续4天每天施用500mg/kg/天的5fc。在msc施用后第7、11、15天用数字卡尺测量s.c肿瘤的尺
寸。将仅前体药物组用作对照组。根据标准公式v=(w(2)
×
l)/2计算肿瘤体积(mm3)。(a)箱须柱状图(box and whisker bar graph)显示了从来自每组的n=5个样品测量的肿瘤体积分布。在第7、11、15天,处理组(cd::uprt_at
‑
msc/5
‑
fu)中的肿瘤体积显示出统计学显著差异(p<0.05)。(b)在实验结束时,将小鼠安乐死。提取肿瘤并用4%pfa固定。图像显示了从每个组提取的肿瘤(n=5);
[0088]
图33显示了表达的持续时间和基于转染效率的杀伤效率的比较。使用添加或不添加trafen的pei衍生物(聚合物),用不同的dna量(200ng
‑
400ng)转染24孔培养容器中的ad
‑
msc。转染后2天,将细胞胰蛋白酶化以用于facs分析。提供了(a)cduprtgfp+%和(b)%细胞存活力,%pi
‑
两者。结果表示为平均值
±
sd(n=3)。将未转染的at
‑
msc用作阴性对照。(c)在多种转染条件下,收获后cd:uprt:gfp阳性细胞%,使用facs进行分析。(d)u251
‑
mg与用不同转染条件转染的msc的共培养的细胞存活力%。结果表示为平均值
±
sd(n=6);
[0089]
图34显示了转染后的msc的表型。(a)对于未处理过的msc(左图)和cd:uprt:gfp msc(右图)的cd标志物(cd90、cd74、cd105、cd14、cd20、cd34和cd45)的表达,将同种型对照用作facs分析的阴性对照。(b)对于cd:uprt:gfp msc,对于成骨分化的茜素红s染色(顶部)和对于生脂分化的油红o染色(中间)的代表性图象,还对于生脂分化显示了gfp图象和油红o染色的重叠(底部)。白色箭头指向分化后14天被油红o染色的表达cd:uprt:gfp的细胞(c)相对于成纤维细胞,朝u251
‑
mg迁移的未治疗过和修饰的ad
‑
msc的数目的变化倍数。使用未配对,双尾学生t检验计算两组之间的显著性差异。n.s.表示p
‑
值>0.05,并因此是不显著的;
[0090]
图35显示了cduprtgfp_msc/5
‑
fc对于tmzr神经胶质瘤的细胞毒性。将转染的ad
‑
msc与神经胶质瘤细胞系(a)u251
‑
mg和u251
‑
mgtmzr40,(b)u87
‑
mg和u87
‑
mgtmzr40,(c)hgcc细胞系和(d)成纤维细胞共培养。通过以不同的msc:癌症或成纤维细胞比值,与100μg/ml 5
‑
fc温育7天,确定共培养的细胞存活力。结果表示为平均值
±
sd(n=6);
[0091]
图36显示了cduprtgfp_msc/5
‑
fc对于u251
‑
mg
tmzr40
的体内细胞毒性。(a)在治疗前和治疗后最长15天测量肿瘤体积,(b)一旦在治疗后15天收获,测量肿瘤重量,(c)在治疗前和治疗后最长15天测量小鼠体重的变化%。结果表示为平均值
±
sem(小鼠个数为至少6)。使用未配对,双尾学生t检验计算来自未治疗过的msc和不同数目的cd:uprt:gfp_msc治疗的肿瘤体积和重量之间的显著性差异。通过*表示p
‑
值<0.05,同时通过**表示p
‑
值<0.01,并且通过***表示p
‑
值<0.005。n.s.表示p
‑
值>0.05,并因此是不显著的;
[0092]
图37显示了cd::uprt::gfp_msc结合5fc作为tmz耐受性成胶质细胞瘤(u251
‑
mg
tmzr40
)的治疗方式的应用。对于治疗方案,瘤内注射1
×
106个治疗性细胞或天然细胞(第0天)。1天后,小鼠接受4天,每天1次的500mg 5fc/kg/天的腹膜内注射。在5fc的最后一次剂量后第3天,对小鼠再次注射工程化干细胞,并在实验持续期间内重复该循环。在3个循环后(肿瘤诱导后50天或者第一次msc注射后36天),终止实验。(a)在msc注射后所指明的天数,测量肿瘤体积,(b)图象显示在第一次msc注射后36天时的肿瘤尺寸。(c)实验过程期间的小鼠体重;
[0093]
图38显示了肛周癌治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2020年1月):存活,未报道复发;
[0094]
图39显示了口腔黑素瘤治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。
最近一次修订(2020年1月):存活;
[0095]
图40显示了甲状腺癌治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2019年6月):存活;
[0096]
图41显示了软组织肉瘤(癌症溃疡)治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2018年11月):存活,未报道复发。14
‑
11
‑
2018的超声报告:在左侧肛围区存在轮廓分明的低回声圆形块,测量为4
×3×
2cm。与周围或更深的器官无粘连。不存在转移,特别是在腰下淋巴结中。膀胱中存在少量1.5mm尿石,少数存在于前列腺部。其它器官正常。目前已完全缓解;
[0097]
图42显示了鼻肿瘤治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2020年1月):存活;
[0098]
图43显示了胃肠癌治疗数据。施用途径为狗cd::uprt::gfp_msc的静脉内输注。最近一次修订(2019年7月):存活。根据超声报告,尽管存在二次生长,但是原始生长显著减小;
[0099]
图44显示用含有gfp转基因的载体修饰来自不同的商品化来源/协会的msc类型。图片中的柱显示了如通过流式细胞术所测量的gfp+群体的%;
[0100]
图45显示修饰来自不同来源的msc以表达cd::uprt::gfp;
[0101]
图46显示了ad
‑
msc和uc
‑
msc在平底表面上的放大的线性度。(a)将转染的活细胞个数对容器表面积作图。(b)来自ad和uc
‑
msc的facs分析的gfp+%的代表性图象。(c)不同培养容器中的转染百分比;
[0102]
图47显示了在ad msc中研究不同微载体的结果。(a)所使用的微载体的描述,(b)将不同天,在不同微载体上生长的活细胞数作图;
[0103]
图48显示了微载体上转染的增强。将msc在微载体上的1.9cm^2上接种,并用不同的dna量和添加增强子转染。(a)将转染效率,%gfp+和%pi
‑
作图,并且(b)以4
×
放大倍数采集代表性图象;
[0104]
图49显示了影响微载体放大的不同速度。(a)提供了%转染效率,%gfp+和细胞存活力,%pi
‑
。结果表示为平均值
±
sd(n=3)。将未转染的ad
‑
msc用作阴性对照。(b)以4
×
放大倍数采集转染细胞的代表性图象;
[0105]
图50显示了通过慢病毒或者trafen介导的转染方法修饰的at
‑
msc的cd::uprt::gfp表达和抗癌效率的比较结果。(a)感染后3天,对msc进行1μg/ml嘌罗霉素选择2
‑
周。在建立稳定表达cd::uprt::gfp的msc后,设立另一组实验以通过trafen介导的转染产生cd::uprt::gfp_msc。转染后2天,采集修饰的msc的荧光图像。(b)此后,收获两个培养物并进行(b)facs分析和(c,d)共培养研究。图片中的柱表示通过mts测定获得的在1个msc比1、5、50、100个癌细胞的多种比值下的癌症杀伤效率。通过双尾学生t检验评价通过慢病毒相对于trafen方法产生的cd::uprt::gfp_msc的癌症杀伤效力的显著性差异;**,p
‑
值<0.005;*,p
‑
值<0.05。在共培养实验结束时,采集明视野图象;
[0106]
图51显示了对患有复发性明细胞癌的46岁患者进行的同情性使用治疗的结果。通过如本文所描述的表达cd::uprt::gfp的msc的瘤内注射治疗受试者;
[0107]
图52显示了与非离心/旋转离心转染方法的实例(底部)相比,典型的离心/旋转离心
‑
基转染方法(顶部)的示意图。还提供了对根据这些方法治疗的细胞收集的数据(参见实
施例11);
[0108]
图53显示了冷冻保存修饰的msc(使用trafen制备)以允许其长期储存的工作流程的示意图。如所示的,可以将修饰的msc置于冷冻保存储存。当需要时,可以从储存中移除细胞并通过在低温溶液中融化来准备用于使用;和
[0109]
图54显示了如图53所示的冷冻保存,然后融化的修饰的msc的细胞存活力(a)、表达水平(b)和功能活性(c)的结果。如所示的,在冷冻保存并在低温溶液中维持最多72h之后,修饰的msc保留了高细胞存活力和表达水平。
具体实施方式
[0110]
本文描述了用从中表达一种或多种功能基因的核酸构建体转染间质干细胞的方法。还描述了转染的间质干细胞和间质干细胞群体,它们的使用、使用这些转染的干细胞治疗疾病或病症,如癌症的方法,和与之有关的试剂盒和组合物。将理解出于旨在对于本领域技术人员说明性的目的提供实施方式和实施例,并且这些实施方式和实施例不旨在以任何方式进行限制。
[0111]
对于一些不同的治疗和非治疗应用,修饰以表达治疗基因或其它所关心的基因的干细胞是所期望的。一个实例是前体药物基因疗法领域,其旨在提供修饰的干细胞,修饰的干细胞表达能够在修饰的干细胞引入受试者或患者的位点处将非活性前体药物转化为活性治疗形式的外源酶。通常,在前体药物基因疗法领域中,由于非病毒方法通常提供不良的转染效率,因此基于病毒的基因修饰方法已在临床前和临床研究中促进了修饰干细胞,如msc的方法。的确,多种临床前研究和临床试验已利用病毒载体作为用于干细胞修饰的基因递送载体。尽管病毒可以能够维持转基因的表达,但是被病毒感染的细胞通常具有单位细胞的低转基因有效负荷(<10个拷贝/细胞)。高拷贝的抄录单元通常是所期望的,因为这可以导致更高的转基因表达,其可以改善细胞载体在递送治疗剂中的有效负荷。临床级病毒的生产可以是费力的并且经常包括稳定生产系的原始细胞库的产生和认证,因此在基因
‑
细胞治疗中造成了高成本。病毒载体生产中的瓶颈已影响了细胞和基因疗法的开发和商业化。
[0112]
尽管就单位细胞的高有效负荷,避免可能引起细胞衰老的抗生素选择(和潜在地数周的处理工作)[40]和/或可能降低肿瘤趋向性[41]以及与病毒诱导的msc转化有关的安全性问题[42]来说,瞬时转染可以具有优势,但是本领域中非病毒转染效率通常较低。的确,尽管非病毒方法在易于生产和/或低成本和安全性谱方面具有优于病毒载体的优势[43],但是在本领域中缺乏对非病毒msc修饰的广泛使用可能是由于通常在本领域中所观察到的低转染效率(0
‑
35%)所造成的[44,45]。例如,由于某些化学品基转染方法的较差表现(<5%的效率)[46],已通过反转录病毒转导工程化人脂肪组织来源的msc(at
‑
msc)来表达cd::uprt[47,48]。
[0113]
在这些应用中基于病毒的基因修饰具有固有的安全性风险,因此临床级病毒的生产可以是费力的,并且可以通过病毒方法引入每个细胞的基因拷贝数通常较低(通常<10个拷贝/细胞)。此外,通过病毒或非病毒实现干细胞,如msc的基因修饰,同时不对所产生的细胞的表型(即多潜能性、免疫表型、趋向性等
……
)造成不期望的改变,并且同时获得高转染效率是本领域所面对的另一种困难。
[0114]
如本文中详细描述的,本发明人现已开发了用核酸构建体转染间质干细胞的方法,从核酸构建体表达一种或多种功能基因,核酸构建体是非病毒的并且在某些实施方式中,它可以提供高转染效率、高拷贝数/细胞、高细胞存活力、长持续时间的瞬时表达和/或基本无变化的多能性表型。在某些实施方式中,这些方法可以是可缩放的和/或适合于修饰的间质干细胞的大规模临床生产。本文还详细描述了转染的间质干细胞和间质干细胞群体,它们的使用、使用这些转染的间质干细胞治疗疾病或病症,如癌症的方法,和与之有关的试剂盒和组合物。
[0115]
本发明的方面可以包括多个实施方式,其包括(但不限于)以下示例性实施方式:
[0116]
实施方式1.一种用从中表达一种或多种功能基因的核酸构建体转染的一种间质干细胞(msc),msc具有其中通过核酸构建体的转染,多潜能性(例如,分化潜能)、免疫表型和/或癌症趋向性表型特征中的任何一种或多种基本无变化的表型,并且msc不含基于病毒的转染载体材料。
[0117]
实施方式2.多种间质干细胞(msc),其中至少约60%的多种msc用从中表达一种或多种功能基因的核酸构建体转染,多种转染的msc具有其中通过核酸构建体的转染,多潜能性(例如,分化潜能)、免疫表型和/或癌症趋向性表型特征中的任何一种或多种基本无变化的表型,并且多种msc不含基于病毒的转染载体材料。
[0118]
实施方式3.根据实施方式2所述的多种msc,其中用核酸构建体转染了至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或者至少约95%的多种msc并且多种msc表达一种或多种功能基因。
[0119]
实施方式4.根据实施方式2或3所述的多种msc,其中多种msc的细胞存活力为至少约70%、至少约75%、至少约80%或者至少约85%。
[0120]
实施方式5.根据实施方式1
‑
4中任一项所述的一种或多种msc,其中用平均至少约1000、至少约2000、至少约3000、至少约4000、至少约5000、至少约6000、至少约7000、至少约8000、至少约9000或至少约10000拷贝的核酸构建体各自转染一种或多种转染的msc。
[0121]
实施方式6.根据实施方式1
‑
5中任一项所述的一种或多种msc,其中在转染的msc细胞中瞬时表达一种或多种功能基因。
[0122]
实施方式7.根据实施方式1
‑
6中任一项所述的一种或多种msc,其中一种或多种msc来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。
[0123]
实施方式8.根据实施方式1
‑
7中任一项所述的一种或多种msc,其中一种或多种msc是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。
[0124]
实施方式9.根据实施方式1
‑
8中任一项所述的一种或多种msc,其中核酸构建体包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0125]
实施方式10.根据实施方式1
‑
9中任一项所述的一种或多种msc,其中一种或多种转染的msc在转染后瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0126]
实施方式11.根据实施方式1
‑
10中任一项所述的一种或多种msc,其中使用阳离子聚合物、能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定一种或多种msc
的微管网络的第二试剂,用核酸构建体转染一种或多种转染的msc。
[0127]
实施方式12.根据实施方式11所述的一种或多种msc,其中阳离子聚合物包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。
[0128]
实施方式13.根据实施方式12所述的一种或多种msc,其中阳离子聚合物包括直链聚亚乙基亚胺(lpei)。
[0129]
实施方式14.根据实施方式11
‑
13中任一项所述的一种或多种msc,其中第一试剂包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。
[0130]
实施方式15.根据实施方式11
‑
14中任一项所述的一种或多种msc,其中第二试剂包括组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)。
[0131]
实施方式16.根据实施方式11
‑
15中任一项所述的一种或多种msc,其中第二试剂包括saha(伏立诺他)。
[0132]
实施方式17.根据实施方式1
‑
16中任一项所述的一种或多种msc,其中一种或多种功能基因包括自杀基因。
[0133]
实施方式18.根据实施方式1
‑
17中任一项所述的一种或多种msc,其中一种或多种功能基因包括胞嘧啶脱氨酶(cdy)。
[0134]
实施方式19.根据实施方式1
‑
18中任一项所述的一种或多种msc,其中一种或多种功能基因包括尿嘧啶转磷酸核糖基酶(uprt)。
[0135]
实施方式20.根据实施方式1
‑
19中任一项所述的一种或多种msc,其中一种或多种功能基因包括cdy和uprt两者。
[0136]
实施方式21.根据实施方式20的一种或多种msc,其中cdy和uprt作为融合构建体表达。
[0137]
实施方式22.根据实施方式1
‑
21中任一项所述的一种或多种msc,其中一种或多种功能基因包括荧光蛋白。
[0138]
实施方式23.根据实施方式22所述的一种或多种msc,其中荧光蛋白包括绿色荧光蛋白(gfp)。
[0139]
实施方式24.根据实施方式20所述的一种或多种msc,其中一种或多种功能基因包括cdy、uprt和gfp。
[0140]
实施方式25.根据实施方式24所述的一种或多种msc,其中cdy、uprt和gfp作为融合构建体表达。
[0141]
实施方式26.根据实施方式1
‑
25中任一项所述的一种或多种msc,其中一种或多种功能基因包括单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)或者另一种胸苷激酶。
[0142]
实施方式27.根据实施方式1
‑
26中任一项所述的一种或多种msc,其中表型包括msc的肿瘤和/或癌症趋向性。
[0143]
实施方式28.根据实施方式1
‑
27中任一项所述的一种或多种msc,一种或多种msc对使用5
‑
氟胞嘧啶(5fc)或更昔洛韦(gcv)的治疗敏感。
[0144]
实施方式29.根据实施方式1
‑
28中任一项所述的一种或多种msc,一种或多种msc:
a)将5fc转化为5
‑
氟尿嘧啶(5fu)、5
‑
氟脲嘧啶单磷酸盐(fump)或两者;b)将更昔洛韦转化为更昔洛韦单磷酸盐;或者c)上述a)和b)的组合。
[0145]
实施方式30.根据实施方式1
‑
29中任一项所述的一种或多种msc,用于在治疗癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌中的使用。
[0146]
实施方式31.用于根据实施方式30所述的使用的一种或多种msc,其中一种或多种msc用于与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0147]
实施方式32.根据实施方式1
‑
31中任一项所述的一种或多种msc,其中表型包括其中在转染后cd表面标志物的表达基本无变化的免疫表型。
[0148]
实施方式33.根据实施方式32所述的一种或多种msc,其中一种或多种转染的msc是塑料
‑
附着的,表达cd105、cd73和cd90(>95%),缺乏cd45、cd34、cd14和hla
‑
dr表面分子的表达(<2%)并且能够体外分化为成骨细胞、脂肪细胞和成软骨细胞,从而满足国际细胞疗法协会(isct)所定义的免疫表型标准。
[0149]
实施方式34.根据实施方式1
‑
33中任一项所述的一种或多种msc,其中一种或多种转染的msc是未分化的。
[0150]
实施方式35.一种用从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法,方法包括:
[0151]
将msc暴露于包含与阳离子聚合物复合的核酸构建体的转染混合物;
[0152]
将msc暴露于能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂;和
[0153]
温育msc;
[0154]
从而提供用核酸构建体转染的msc。
[0155]
实施方式36.根据实施方式35所述的方法,其中在暴露于转染混合物,暴露于第一试剂和第二试剂期间,在温育期间或它们的任意组合,不对msc离心。
[0156]
实施方式37.根据实施方式35或36所述的方法,其中温育msc的步骤包括温和混合,但不离心。
[0157]
实施方式38.根据实施方式35
‑
37中任一项所述的方法,其中温育msc的步骤包括温育msc至少约2小时。
[0158]
实施方式39.根据实施方式38所述的方法,其中温育msc的步骤包括温育msc约2小时至约48小时、或者约3小时至约24小时。
[0159]
实施方式40.根据实施方式39所述的方法,其中温育msc的步骤包括温育msc约4小时至约18小时。
[0160]
实施方式41.根据实施方式35
‑
40中任一项所述的方法,其中阳离子聚合物包括已鉴别为对msc具有低细胞毒性的阳离子聚合物。
[0161]
实施方式42.根据实施方式35
‑
41中任一项所述的方法,其中将msc暴露于转染混合物的步骤可以包括将核酸构建体与阳离子聚合物复合以提供包含复合的核酸构建体的转染混合物,并将转染混合物添加至msc。
[0162]
实施方式43.根据实施方式35
‑
42中任一项所述的方法,其中将msc暴露于转染混合物的步骤包括将转染混合物添加至msc并将msc与转染混合物温育。
[0163]
实施方式44.根据实施方式35
‑
43中任一项所述的方法,其中将msc暴露于第一试剂和第二试剂的步骤包括用补充有第一试剂和第二试剂的细胞培养基替换转染混合物。
[0164]
实施方式45.根据实施方式44所述的方法,其中细胞培养基包括完全培养基。
[0165]
实施方式46.根据实施方式35
‑
45中任一项所述的方法,其中msc处于约60%汇合,并且在暴露于转染混合物之前约24小时接种msc。
[0166]
实施方式47.根据实施方式35
‑
46中任一项所述的方法,其中阳离子聚合物具有约5kda至约200kda的大小。
[0167]
实施方式48.根据实施方式35
‑
47中任一项所述的方法,其中阳离子聚合物包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。
[0168]
实施方式49.根据实施方式35
‑
48中任一项所述的方法,其中阳离子聚合物包括直链聚亚乙基亚胺(lpei)。
[0169]
实施方式50.根据实施方式35
‑
49中任一项所述的方法,其中第一试剂包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。
[0170]
实施方式51.根据实施方式35
‑
50中任一项所述的方法,其中第二试剂包括组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)。
[0171]
实施方式52.根据实施方式51所述的方法,其中第二试剂包括saha(伏立诺他)。
[0172]
实施方式53.根据实施方式35
‑
52中任一项所述的方法,其中转染混合物包括处于无血清dmem中或新鲜培养基中的复合的核酸构建体。
[0173]
实施方式54.根据实施方式35
‑
53中任一项所述的方法,其中将msc暴露于转染混合物的步骤包括从msc除去培养基并用转染混合物替换培养基。
[0174]
实施方式55.根据实施方式35所述的方法,其中将msc暴露于转染混合物的步骤包括在轻微离心下将msc与转染混合物温育。
[0175]
实施方式56.根据实施方式55所述的方法,其中轻微离心包括约200g离心约5分钟。
[0176]
实施方式57.根据实施方式35
‑
56中任一项所述的方法,其中msc所暴露的转染混合物中的核酸构建体的量在约200至约500ng每1.9cm2表面积之间。
[0177]
实施方式58.根据实施方式57所述的方法,其中msc所暴露的转染混合物中的核酸构建体的量在约250至约400ng每1.9cm2表面积之间。
[0178]
实施方式59.根据实施方式58所述的方法,其中msc所暴露的转染混合物中的核酸构建体的量在约300至约350ng每1.9cm2表面积之间。
[0179]
实施方式60.根据实施方式35
‑
59中任一项所述的方法,其中在转染混合物中阳离子聚合物与核酸构建体的比值为约1μg至约30μg阳离子聚合物每1μg核酸构建体。
[0180]
实施方式61.根据实施方式35
‑
60中任一项所述的方法,其中一种或多种功能基因包括自杀基因。
[0181]
实施方式62.根据实施方式35
‑
61中任一项所述的方法,其中一种或多种功能基因包括胞嘧啶脱氨酶(cdy)和/或胸苷激酶(tk)
[0182]
实施方式63.根据实施方式35
‑
63中任一项所述的方法,其中一种或多种功能基因包括尿嘧啶转磷酸核糖基酶(uprt)。
[0183]
实施方式64.根据实施方式35
‑
64中任一项所述的方法,其中一种或多种功能基因包括cdy和uprt两者。
[0184]
实施方式65.根据实施方式64所述的方法,其中cdy和uprt作为融合构建体表达。
[0185]
实施方式66.根据实施方式35
‑
66中任一项的方法,其中一种或多种功能基因包括荧光蛋白。
[0186]
实施方式67.根据实施方式66所述的方法,其中荧光蛋白包括绿色荧光蛋白(gfp)。
[0187]
实施方式68.根据实施方式64所述的方法,其中一种或多种功能基因包括cdy、uprt和gfp。
[0188]
实施方式69.根据实施方式68所述的方法,其中cdy、uprt和gfp作为融合构建体表达。
[0189]
实施方式70.根据实施方式35
‑
69中任一项所述的方法,其中一种或多种功能基因包括单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)。
[0190]
实施方式71.根据实施方式35
‑
70中任一项所述的方法,其中在转染的msc中瞬时表达一种或多种功能基因。
[0191]
实施方式72.根据实施方式35
‑
71中任一项所述的方法,其中用平均至少约1000、至少约2000、至少约3000、至少约4000、至少约5000、至少约6000、至少约7000、至少约8000、至少约9000或者至少约10000拷贝的核酸构建体各自转染转染的msc。
[0192]
实施方式73.根据实施方式35
‑
72中任一项所述的方法,其中转染的msc的表型,如包括多潜能性、免疫表型和/或癌症趋向性表型特征中的任何一种或多种的表型通过转染基本无变化。
[0193]
实施方式74.根据实施方式73所述的方法,其中表型包括msc的肿瘤和/或癌症趋向性。
[0194]
实施方式75.根据实施方式73或74所述的方法,其中表型包括其中在转染后cd表面标志物的表达基本无变化的免疫表型。
[0195]
实施方式76.根据实施方式75所述的方法,其中转染的msc是塑料
‑
附着的,表达cd105、cd73和cd90(>95%),缺乏cd45、cd34、cd14和hla
‑
dr表面分子的表达(<2%)并且能够体外分化为成骨细胞、脂肪细胞和成软骨细胞,从而满足国际细胞疗法协会(isct)所定义的免疫表型标准。
[0196]
实施方式77.根据实施方式35
‑
76中任一项所述的方法,其中用核酸构建体转染至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或者至少约95%的msc并且msc表达一种或多种功能基因。
[0197]
实施方式78.根据实施方式35
‑
77中任一项所述的方法,其中转染的msc的细胞存活力为至少约70%、至少约75%、至少约80%或者至少约85%。
[0198]
实施方式79.根据实施方式35
‑
78中任一项所述的方法,其中转染的msc是未分化的。
[0199]
实施方式80.根据实施方式35
‑
79中任一项所述的方法,其中方法不含基于病毒的
转染载体材料。
[0200]
实施方式81.根据实施方式35
‑
80中任一项所述的方法,其中msc来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。
[0201]
实施方式82.根据实施方式35
‑
81中任一项所述的方法,其中msc是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。
[0202]
实施方式83.根据实施方式35
‑
82中任一项所述的方法,其中核酸构建体包括无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0203]
实施方式84.根据实施方式35
‑
83中任一项所述的方法,其中msc在转染后瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0204]
实施方式85.根据实施方式35
‑
84中任一项所述的方法,其中所产生的msc对使用5
‑
氟胞嘧啶(5fc)或更昔洛韦(gcv)或两者的治疗敏感。
[0205]
实施方式86.根据实施方式35
‑
84中任一项所述的方法,其中所产生的msc:a)将5fc转化为5
‑
氟尿嘧啶(5fu)、5
‑
氟尿嘧啶单磷酸盐(fump)或两者;b)将更昔洛韦转化为更昔洛韦单磷酸盐;或者c)上述a)和b)的组合。
[0206]
实施方式87.根据实施方式35
‑
86中任一项所述的方法,其中一种或多种功能基因包括荧光蛋白,并且方法另外包括使用细胞分选或facs分离、选择或纯化转染的msc的步骤。
[0207]
实施方式88.根据实施方式35
‑
87中任一项所述的方法,其中转染的msc是如根据实施方式1
‑
34中任一项所定义的msc。
[0208]
实施方式89.通过根据实施方式35
‑
88中任一项所述的方法产生的msc或多种msc。
[0209]
实施方式90.如根据实施方式1
‑
34或89中任一项所定义的一种或多种msc用于治疗对其有需要的受试者中的癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌的使用。
[0210]
实施方式91.根据实施方式90所述的使用,其中一种或多种msc用于与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0211]
实施方式92.如根据实施方式1
‑
34或89中任一项所定义的一种或多种msc在用于治疗癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌的药剂的生产中的使用。
[0212]
实施方式93.根据实施方式92所述的使用,其中一种或多种msc用于与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0213]
实施方式94.治疗对其有需要的受试者中的癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌的方法,方法包括:
[0214]
将如根据实施方式1
‑
34或89中任一项所定义的一种或多种msc施用至受试者的癌细胞附近的区域,
[0215]
其中一种或多种msc中的一种或多种功能基因有助于对癌细胞的抗癌作用。
[0216]
实施方式95.根据实施方式94所述的方法,其中一种或多种msc与5fc、5fu、gcv或它们的任意组合同时、顺序或组合施用。
[0217]
实施方式96.根据实施方式94或95所述的方法,其中一种或多种功能基因包括胞嘧啶脱氨酶(cdy)、胸苷激酶(tk)或两者。
[0218]
实施方式97.根据实施方式94
‑
96中任一项所述的方法,其中一种或多种功能基因包括尿嘧啶转磷酸核糖基酶(uprt)。
[0219]
实施方式98.根据实施方式94
‑
97中任一项所述的方法,其中一种或多种功能基因包括cdy和uprt两者。
[0220]
实施方式99.根据实施方式98所述的方法,其中在一种或多种msc中作为融合构建体表达cdy和uprt。
[0221]
实施方式100.根据实施方式94
‑
99中任一项所述的方法,其中一种或多种msc在转染后瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0222]
实施方式101.根据实施方式94
‑
100中任一项所述的方法,还包括向受试者施用5fc、5fu、更昔洛韦或它们的任意组合的步骤,从而将一种或多种msc暴露于5fc、5fu、更昔洛韦或其组合。
[0223]
实施方式102.根据实施方式94
‑
101中任一项所述的方法,还包括在施用一种或多种msc的步骤之前,根据如实施方式35
‑
88中任一项所定义的方法产生一种或多种msc的步骤。
[0224]
实施方式103.一种组合物,其包含根据实施方式1
‑
34或89中任一项的一种或多种msc和药物可用的载体、稀释剂、赋形剂、细胞培养基或缓冲液中的至少一种。
[0225]
实施方式104.一种治疗诊断试剂,其包含根据实施方式1
‑
34或89中任一项的一种或多种msc。
[0226]
实施方式105.一种用于用从中瞬时表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的试剂盒,试剂盒包含以下中的一种或多种:
[0227]
一种msc;
[0228]
设计用于瞬时表达一种或多种功能基因的核酸构建体;
[0229]
细胞培养基;
[0230]
阳离子聚合物;
[0231]
能够重定向来自胞内酸性区室的内吞核酸的第一试剂;
[0232]
能够稳定一种msc的微管网络的第二试剂;
[0233]
用于实施如实施方式35
‑
88中任一项所定义的方法的说明书;
[0234]
5fc;
[0235]
gcv;和/或
[0236]
5fu。
[0237]
实施方式106.根据实施方式105所述的试剂盒,其中msc来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。
[0238]
实施方式107.根据实施方式105或106所述的试剂盒,其中msc是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。
[0239]
实施方式108.根据实施方式105或106所述的试剂盒,其中核酸构建体包括无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0240]
实施方式109.根据实施方式105
‑
108中任一项所述的试剂盒,其中阳离子聚合物包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。
[0241]
实施方式110.根据实施方式105
‑
180中任一项所述的试剂盒,其中阳离子聚合物包括直链聚亚乙基亚胺(lpei)。
[0242]
实施方式111.根据实施方式105
‑
110中任一项所述的试剂盒,其中第一试剂包括dopc、dppc或另一种促融脂质(fusogenic lipid)中的一种或多种。
[0243]
实施方式112.根据实施方式105
‑
111中任一项所述的试剂盒,其中第一试剂包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。
[0244]
实施方式113.根据实施方式105
‑
112中任一项所述的试剂盒,其中第二试剂包括组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)。
[0245]
实施方式114.根据实施方式113所述的试剂盒,其中第二试剂包括saha(伏立诺他)。
[0246]
实施方式115.根据实施方式105
‑
114中任一项所述的试剂盒,其中一种或多种功能基因包括自杀基因。
[0247]
实施方式116.根据实施方式105
‑
115中任一项所述的试剂盒,其中一种或多种功能基因包括胞嘧啶脱氨酶(cdy)或者胸苷激酶(tk)。
[0248]
实施方式117.根据实施方式105
‑
116中任一项所述的试剂盒,其中一种或多种功能基因包括尿嘧啶转磷酸核糖基酶(uprt)。
[0249]
实施方式118.根据实施方式105
‑
116中任一项所述的试剂盒,其中一种或多种功能基因包括cdy和uprt两者。
[0250]
实施方式119.根据实施方式118所述的试剂盒,其中cdy和uprt作为融合构建体表达。
[0251]
实施方式120.根据实施方式105
‑
119中任一项所述的试剂盒,其中一种或多种功能基因包含荧光蛋白。
[0252]
实施方式121.根据实施方式120所述的试剂盒,其中荧光蛋白包括绿色荧光蛋白(gfp)。
[0253]
实施方式122.根据实施方式118所述的试剂盒,其中一种或多种功能基因包括cdy、uprt和gfp。
[0254]
实施方式123.根据实施方式122所述的试剂盒,其中cdy、uprt和gfp作为融合构建体表达。
[0255]
实施方式124.根据实施方式105
‑
123中任一项所述的试剂盒,其中一种或多种功能基因包括单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)。
[0256]
实施方式125.根据实施方式105
‑
124中任一项所述的试剂盒,其中阳离子聚合物
包括已鉴别为对msc具有低细胞毒性的阳离子聚合物。
[0257]
实施方式126.根据实施方式105
‑
125中任一项所述的试剂盒,其中阳离子聚合物具有约5kda至约200kda的大小。
[0258]
实施方式127.根据实施方式105
‑
126中任一项所述的试剂盒,其中试剂盒中阳离子聚合物与核酸构建体的比值为约1μg至约30μg阳离子聚合物每1μg核酸构建体。
[0259]
实施方式128.根据实施方式105
‑
124中任一项所述的试剂盒,其中试剂盒用于制备msc
‑
基抗癌剂。
[0260]
实施方式129.根据实施方式128所述的试剂盒,其中试剂盒还包括用于实施如实施方式94
‑
102中任一项所定义的方法的说明书和/或设备。
[0261]
实施方式130.根据实施方式35
‑
43、46
‑
53或者57
‑
88中任一项所述的方法,其中方法包括在将msc暴露于转染混合物的步骤之前,在生长培养基,如新鲜生长培养基中培养msc的步骤。
[0262]
实施方式131.根据实施方式130所述的方法,其中将msc暴露于转染混合物的步骤包含将转染混合物添加至msc但不从msc中除去生长培养基,并且在暴露和温育步骤期间不进行离心。
[0263]
实施方式132.根据实施方式130或131所述的方法,其中将msc暴露于第一试剂和第二试剂的步骤包括将第一试剂和第二试剂同时、顺序或与转染混合物组合添加至msc。
[0264]
实施方式133.根据实施方式132所述的方法,其中与转染混合物向msc的添加同时将第一试剂和第二试剂添加至msc,或者其中将第一试剂和第二试剂与转染混合物混合并添加至msc。
[0265]
实施方式134.根据实施方式132所述的方法,其中在将转染混合物添加至msc之后不久,将第一试剂和第二试剂添加至msc。
[0266]
实施方式135.根据实施方式132
‑
134中任一项所述的方法,其中在将第一试剂和第二试剂添加至msc之前,不除去转染混合物。
[0267]
实施方式136.根据实施方式130
‑
135中任一项所述的方法,其中msc向转染混合物的暴露持续时间与msc向第一和第二试剂暴露持续时间重叠。
[0268]
实施方式137.根据实施方式136所述的方法,其中在将第一试剂和第二试剂添加至msc之前,不除去转染混合物。
[0269]
实施方式138.一种用从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法,方法包括:
[0270]
在生长培养基中培养msc;
[0271]
将包含与阳离子聚合物复合的核酸构建体的转染混合物添加至msc,同时不从msc除去生长培养基;
[0272]
将能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂添加至msc;和
[0273]
温育msc,同时与转染混合物、第一试剂和第二试剂全部接触培养期。
[0274]
其中与转染混合物的添加同时,与转染混合物的添加顺序或者与转染混合物的添加组合,将第一试剂和第二试剂添加至msc;和
[0275]
其中在转染混合物的添加和温育期结束之间不对msc离心;
[0276]
从而提供用核酸构建体转染的msc。
[0277]
实施方式139.根据实施方式138所述的方法,其中温育期为至少约2小时。
[0278]
实施方式140.根据实施方式138所述的方法,其中温育期为约2小时、约3小时、约4小时、约5小时、约6小时、约7小时、约8小时、约9小时、约10小时、约11小时、约12小时、约13小时、约14小时、约15小时、约16小时、约17小时、约18小时、约19小时、约20小时、约21小时、约22小时、约23小时、约24小时、约25小时、约26小时、约27小时、约28小时、约29小时、约30小时、约31小时、约32小时、约33小时、约34小时、约35小时或约36小时或更长。
[0279]
实施方式141.通过根据实施方式130
‑
140中任一项所述的方法产生的msc细胞或多种msc细胞。
[0280]
实施方式142.如根据实施方式141所定义的一种或多种msc用于治疗对其有需要的受试者中的癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌的使用。
[0281]
实施方式143.如根据实施方式141所定义的一种或多种msc在用于治疗癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌的药剂的生产中的使用。
[0282]
实施方式144.一种治疗对其有需要的受试者中的癌症,例如,淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌的方法,方法包括:
[0283]
将如实施方式141中所定义的一种或多种msc施用至受试者的癌细胞附近的区域,
[0284]
其中一种或多种msc中的一种或多种功能基因有助于对癌细胞的抗癌作用。
[0285]
实施方式145.一种组合物,其包含根据实施方式141所述的一种或多种msc,和药物可用的载体、稀释剂、赋形剂、细胞培养基或缓冲剂中的至少一种。
[0286]
实施方式146.一种治疗诊断试剂,其包含根据实施方式141所述的一种或多种msc。
[0287]
实施方式147.一种用于用从中瞬时表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的试剂盒,试剂盒包含以下中的一种或多种:
[0288]
一种msc;
[0289]
设计用于瞬时表达一种或多种功能基因的核酸构建体;
[0290]
细胞培养基;
[0291]
阳离子聚合物;
[0292]
能够重定向来自胞内酸性区室的内吞核酸的第一试剂;
[0293]
能够稳定一种msc的微管网络的第二试剂;
[0294]
用于实施如实施方式130
‑
140中任一项所定义的方法的说明书;
[0295]
5fc;
[0296]
gcv;和/或
[0297]
5fu。
[0298]
转染的间质干细胞以及用于其生产的方法和试剂盒
[0299]
在一个实施方式中,本文提供了用从中表达一种或多种功能基因的核酸构建体转染的间质干细胞(msc),msc具有通过核酸构建体的转染基本无变化的多能性表型并且msc
不含基于病毒的转染载体材料。
[0300]
在另一实施方式中,本文提供了多个间质干细胞(msc),其中用从中表达一种或多种功能基因的核酸构建体转染至少约60%的多种msc,多种转染的msc具有通过核酸构建体的转染基本无变化的多能性表型,并且多种msc不含基于病毒的转染载体材料。
[0301]
如将理解的,msc可以包括任何适合的msc,如来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源的那些。msc的来源可以包括人、狗、猫、马等。在某些实施方式中,msc可以是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)中的一种或多种。
[0302]
在某些实施方式中,当将表达功能基因的修饰的(转染的)msc用于治疗受试者时,考虑转染的msc可以包括最初来源于要治疗的受试者的msc或者来源不同来源或受试者的msc。在某些实施方式中,可以出于与要治疗的受试者的相容性(以避免,例如,过敏反应)选择msc,并且msc可以或可以不最初来源于要治疗的受试者。在某些实施方式中,可以使用自体同源或同种异体的msc。
[0303]
在某些实施方式中,可以用核酸构建体转染msc。如将理解的,在某些实施方式中,可以用核酸构建体瞬时转染msc(即可以将核酸构建体引入细胞的一个位置,在此可以在细胞中表达它所编码的一个或多个功能基因,但是核酸构建体未整合到细胞基因组中),或者可以用核酸构建体稳定转染(即核酸构建体可以整合到细胞基因组中,在此可以在细胞中表达它所编码的一个或多个功能基因;其实施或不实施选择步骤(例如,抗生素抗性,在此该基因与核酸构建体一起包括)。
[0304]
在某些实施方式中,可以使用如下文中详细描述的试剂和/或方法转染msc。
[0305]
如将理解的,核酸构建体可以包括适合于特定应用并且适合于编码一个或多个所关心的功能基因的任何适合的核酸序列。在某些实施方式中,核酸构建体通常可以包括在引入细胞后,可以导致一种或多种功能性基因/核酸构建体编码的多肽的产生的任何适合的质粒、表达载体或其它可表达的核酸序列。在其中期望延长表达的实施方式中,可以使用设计用于长期表达和/或防止细胞沉默的核酸构建体。
[0306]
在某些实施方式中,可以设计核酸构建体,从而编码区(即编码一种或多种所关心的功能基因的区域)使用对于在特定所关心的生物中的表达优化的密码子(例如,当使用人msc时,密码子可以对人细胞中的表达优化)。在某些实施方式中,核酸构建体可以是可表达的核酸(即当引入或存在于给定细胞中时,核酸构建体可以设计以导致多肽的表达)。在某些实施方式中,核酸构建体可以是dna或rna。在某些实施方式中,核酸构建体可以是质粒、表达载体、mrna(在某些实施方式中,其可以包括适合于在所关心的细胞中翻译的序列,如起始密码子,多聚
‑
a尾、rbs序列等)、微环dna、单链或双链dna的片段等,其具有合适的上游和/或下游序列,从而一旦将核酸构建体引入细胞,则可以发生核酸构建体的翻译或转录和翻译以提供一种或多种功能基因的多肽。
[0307]
用于使特定功能性基因/多肽过表达或者将它们引入细胞中的适合的表达载体技术在本领域中是已知的(参见,例如,molecular cloning:alaboratory manual(第4版),2012,cold spring harbor laboratory press)。如本领域技术人员将已知的,用于表达特定多肽的核苷酸序列可以编码或包括以下文献中所描述的特征:"genes vii",lewin,b.oxford university press(2000)或者"molecular cloning:a laboratory manual",
sambrook等人,cold spring harbor laboratory,第3版(2001)。可以将编码所关心的特定功能性基因/多肽的核苷酸序列引入适合的载体,如可商购的载体。还可以单独构建或使用标准分子生物学技术修饰载体,如(例如)sambrook等人(cold spring harbor laboratory,第3版(2001))中所列。本领域技术人员将认识到载体可以包括编码可以可操作性地连接至编码功能性基因/多肽的核苷酸序列的所期望的元件的核苷酸序列。这些编码所期望的元件的核苷酸序列可以包括转录启动子(例如,组成型或诱导型启动子)、转录增强子、转录终止子和/或复制起点。适合的载体的选择可以基于一些因素,其无限制地包括引入载体的核酸的尺寸,所期望的转录和翻译控制元件的类型、所期望的表达水平、所期望的拷贝数、是否期望染色体整合、所期望的选择过程的类型(如果有)或者旨在转化的宿主细胞或宿主范围。
[0308]
如将理解的,核酸构建体可以编码一种或多种功能基因。一种或多种功能基因通常可以包括任何适合的功能基因,其编码一种或多种所关心的功能性rna、肽、多肽或蛋白。如将理解的,通常将选择一种或多种功能基因以适合修饰的间质干细胞将应用的具体应用。举例来说,当在前体药物基因疗法方法中使用修饰的msc时,一种或多种功能基因可以包括能够将非活性或低活性前体药物转化为活性形式的酶,从而一旦修饰的msc暴露于前体药物,则将形成活性药物并且活性药物能够治疗周围的细胞和/或组织。在某些实施方式中,一种或多种功能基因可以包括自杀基因,其可以将前体药物转化为(例如)损害修饰的msc和周围患病细胞两者的活性形式。在另一实施方式中,一种或多种功能基因可以包括一种或多种癌症疗法基因,或者与癌症疗法无关的一种或多种功能基因,并且(例如)可以具有其它治疗或非治疗功能。
[0309]
考虑本文教导内容,前体药物基因疗法系统,包括适合的前体药物和相应功能性基因/自杀基因两者的多个实例将对本领域技术人员是已知的。下表1中列出了可以使用的功能性基因以及它们相应前体药物(当使用时)的一些实例:
[0310]
表1
‑
功能性基因/自杀基因和前体药物系统的多种实例(从j.clin.invest.,2000,105(9):1161
‑
1167改编,该文献以其全部内容作为参考并入本文)。
[0311]
cdept系统的定量数据
[0312]
[0313][0314]
缩写:2
‑
aa,2
‑
氨基蒽;acv,阿昔洛韦;ara
‑
c,阿糖胞苷;ara
‑
m,6
‑
甲氧基嘌呤阿糖核苷;ca,羧基酯酶;cb1954,5
‑
氮丙啶基
‑
2,4
‑
二硝基苯甲酰胺;cd,胞嘧啶脱氨酶;cmba,(2
‑
氯乙基)(2
‑
甲磺酰氧基乙基)氨苯甲酸;cmda,(2
‑
氯乙基)(2
‑
甲磺酰氧基乙基)氨基苯甲酰基
‑
l
‑
谷氨酸;cp,环磷酰胺;cpg2,羧肽酶g2;cpt
‑
11,伊立替康;cyc
‑
450,细胞色素p450;dck,脱氧胞苷激酶;5
‑
fc,5
‑
氟胞嘧啶;5'
‑
dfur,5'
‑
脱氧
‑5‑
氟脲嘧啶;5
‑
fudr,5
‑
氟
脱氧尿苷;5
‑
fu,5
‑
氟尿嘧啶;gcv,更昔洛韦;gcvtp,更昔洛韦三磷酸酯;hsv
‑
tk,单纯疱疹病毒胸苷激酶;if,异环磷酰胺;6
‑
mep,6
‑
甲基嘌呤;6
‑
mepdr,6
‑
甲基嘌呤
‑
2'
‑
脱氧核糖核苷;nr,硝基还原酶;pnp,嘌呤核苷磷酸化酶;sn
‑
38,7
‑
乙基
‑
10
‑
羟基
‑
喜树碱;6
‑
tg,6
‑
硫代鸟嘌呤;tp,胸嘧啶脱氧核苷磷酸化酶;6
‑
tx,6
‑
硫代黄嘌呤;vzv
‑
tk,水痘带状疱疹病毒胸苷激酶;xgprt,黄嘌呤
‑
鸟嘌呤转磷酸核糖基酶。
a
以字母顺序。
b
前体药物的ic50/野生型细胞系中药物的ic50的比值;
c
野生型细胞系中前体药物的ic50/转染(感染)细胞系中前体药物的ic50的比值;
d
pmol/mg/min;
e
细菌来源;
f
酵母来源;
g
μm/min/μg;
h
n
‑
[4
‑
(l
‑
谷氨酰基羰基氨基)苄氧羰基]多柔比星;
i
s
‑
1;
j
在相同系统中未确定;
k
无数据可获得;
l
如果细胞色素p450与p450还原酶共表达;
m
pmol/mg/min;
n
对于v79细胞中的5
‑
(氮丙啶
‑1‑
基)
‑2‑
硝基
‑4‑
羟氨基
‑
苯甲酰胺;
o
对于肌苷、腺嘌呤和鸟嘌呤核苷;
p
μm/min/mg;
q
根据体外实验推断;
r
相对最大速度。
[0315]
可以使用的功能性基因的其它实例可以包括产生可以在治疗所关心的疾病或病症中有用的核酸或多肽产物的任何适合的功能基因。如将理解的,具有治疗活性的多种核酸、肽、多肽和蛋白将是本领域技术人员已知的并且可以包括在如本文所描述的核酸构建体中。举例来说,已引入用于本领域中的癌症疗法的msc中并且可以引入本文所描述的构建体和方法中的基因可以包括以下:
[0316]
表2:与癌症疗法中的msc的修饰有关的干细胞和自杀基因疗法以及相应参考文献
[0317]
[0318]
[0319][0320]
可以使用的功能性基因的其它实例可以包括用于癌症疗法的基因(表3)和/或用于其它治疗适应症的基因(表4)。
[0321]
表3:用于癌症疗法的msc的治疗性修饰(从cytotherapy,2016,18(11):1435
‑
1445改编,该文献以其全部内容作为参考并入本文)
[0322]
评价基因修饰的msc在癌症中的应用的临床前研究
[0323]
[0324][0325]
表4:在用于多种适应症的正在进行的临床前和临床研究中用于修饰msc的基因
[0326]
[0327][0328]
msc固有的肿瘤趋向性[9,10]表明可以将msc用作细胞载体来将抗癌剂特异性递送至肿瘤和它们的转移性位点。一些msc
‑
驱动的gdept临床试验已提供了有希望的结果,其可以保证向ii期试验的进一步发展[7,11]。这些方法可以帮助在靶细胞附近局部和/或可控地酶促转化无毒前体药物。“旁观者效应”可以提高对靶细胞的细胞毒性[7]。已在广谱实体癌[7,8],包括胃癌[12
‑
14]、乳腺癌[15,16]和成胶质细胞瘤[17
‑
19]中证实了某些产生cd的msc的抗癌潜能。临床前研究已证明胞嘧啶脱氨酶/5
‑
氟胞嘧啶(cd/5fc)是非常稳健的,其中肿瘤块中低至4%的cd阳性细胞足以消除肿瘤[20
‑
22]。使用cd/5fc系统的进展在于包括尿嘧啶转磷酸核糖基酶(uprt)、直接将5fu转化为5
‑
氟脲嘧啶单磷酸盐(fump)的嘧啶补救酶,因此绕过了限速酶二氢嘧啶脱氢酶(dpd)和乳清酸转磷酸核糖基酶(oprt)[23
‑
26]。与cd/5fc和5fu相比,cd::uprt/5fc可以将5fc向其活性代谢产物的转化提高30
‑
1500倍[24,27]。
[0329]
在某些实施方式中,一种或多种功能基因可以包括自杀基因。举例来说,在某些实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy)、尿嘧啶转磷酸核糖基酶(uprt)或两者。在某些实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy)、尿嘧啶转磷酸核糖基酶(uprt)、单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)或者另一种胸苷激酶或它们的任意组合。在某些实施方式中,一种或多种功能基因可以分别单独表达,或者可以作为融合构建体表达。在某些实施方式中,核酸构建体可以包含两个或更多个功能基因,或者核酸构建体可以作为两个或更多个单独的核酸构建体的混合物提供,例如,核酸构建体分别表达不同的所关心的功能基因。
[0330]
在某些实施方式中,一种或多种功能基因可以包括荧光蛋白或其它标志物或标签。在某些实施方式中,荧光蛋白可以用于识别和/或评价成功转染的msc。在某些实施方式中,荧光蛋白可以用于从未转染的msc或其它细胞中分离(separating)、分离(isolating)、选择或纯化转染的msc。在某些实施方式中,例如,荧光蛋白可以允许facs
‑
基细胞分选来定量、纯化或分离转染的msc。在某些实施方式中,例如,一种或多种功能基因可以包括绿色荧光蛋白(gfp)。
[0331]
在某些实施方式中,核酸构建体的一种或多种功能基因可以包括cdy和uprt,其可以作为融合构建体表达或可以不作为融合构建体表达。在某些实施方式中,一种或多种功能基因还可以包括荧光蛋白,如绿色荧光蛋白(gfp),其可以作为融合构建体表达或可以不作为融合构建体表达。
[0332]
在某些实施方式中,核酸构建体的一种或多种功能基因可以包括选择基因,如抗生素抗性基因,其可以用于选择转染的msc或者选择稳定转染的msc。
[0333]
尽管任何给定转染的msc细胞中的准确拷贝数可以稍微不同,但是考虑在某些实施方式中,可以用平均至少约1000、至少约2000、至少约3000、至少约4000、至少约5000、至
少约6000、至少约7000、至少约8000、至少约9000或者至少约10000拷贝的核酸构建体各自转染一种或多种转染的msc。
[0334]
当转染多种msc或msc群体时,考虑在某些实施方式中,可以用核酸构建体转染至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或者至少约95%的多种msc并且msc在转染后表达一种或多种功能基因。因此,在某些实施方式中,当转染多种msc或msc群体时,转染效率可以是约60%至约100%之间的任何值,包括它们之间圆整至最近的0.1的任何值,或者它们之间的任何子范围。在某些实施方式中,转染效率可以为至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或者至少约95%。在某些实施方式中,转染效率可以计算为表达所关心的一种或多种功能基因的细胞%。
[0335]
在其它实施方式中,考虑msc的细胞存活力可以为至少约70%、至少约75%、至少约80%或者至少约85%。在某些实施方式中,考虑本文中的教导内容,可以使用通常本领域技术人员已知的任何适合的技术确定细胞存活力,技术如(例如)碘化丙啶测定,如cold spring harb protoc.2016 jul 1;2016(7),doi:10.1101/pdb.prot087163中所描述的。
[0336]
在某些实施方式中,转染的msc可以具有通过使用核酸构建体的转染基本无变化的多能性表型。如将理解的,可以用核酸构建体转染转染的msc,并且可以在转染的msc中表达通过核酸构建体所编码的一种或多种功能基因。然而,在多种应用中,期望与转染之前的msc相比,转染的msc的表型另外基本无变化。举例来说,msc具有多能性表型,这对于转染的msc的某些应用可以是所期望的。因此,在某些实施方式中,转染的msc的表型是多能性的并且与msc预转染相比,不会进一步分化。
[0337]
在某些实施方式中,与msc预转染的多能性表型相比,可以基本无变化的转染的msc的多能性表型可以包括其中在转染后,通过msc的cd表面标志物的表达基本无变化的免疫表型。举例来说,在某些实施方式中,一种或多种转染的msc可以是塑料
‑
附着的,可以表达cd105、cd73和cd90(>95%),可以缺乏cd45、cd34、cd14和hla
‑
dr表面分子的表达(<2%)并且可以能够体外分化为成骨细胞、脂肪细胞和成软骨细胞,从而满足国际细胞疗法协会(isct)所定义的免疫表型标准(参见cytotherapy,2006,8(4):315
‑
7,和https://www.celltherapysociety.org/news/390154/fda
‑
grand
‑
rounds
‑
cites
‑
iscts
‑
minimal
‑
criteria
‑
for
‑
defining
‑
mscs.htm,该文献以其全部作为参考并入本文)。出于这些目的,考虑在某些实施方式中,对于阳性标志物鉴定可接受的%(即对于被认为表达的cd标志物)为转染后表达标志物的细胞群体的至少约95%,并且对于阴性标志物表达可接受的%(即对于被认为不表达的cd标志物)为转染后在至少98%的细胞群体中,群体缺乏特异性标志物的表达。例如,在某些实施方式中,在转染后细胞可以缺乏hla
‑
dr标志物的表达,就像未修饰的msc缺乏该标志物的表达一样,这表明通过转染表型基本不变,并且通过转染msc的质量和表型未消极地改变。
[0338]
一般而言,在某些实施方式中,免疫表型标志物或其它表型标志物可以被认为是通过转染无变化的,其中转染的细胞的相关标志物的表达谱相对于相同细胞预转染或者天然细胞(即未修饰/未处理的对照细胞)或者未转染的等价或相当的细胞的表达谱基本无变化(即小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或者小于约1%的变化)。在某些实施方式中,免疫表型标志物或其它
表型标志物可以被认为是通过转染无变化的,其中转染的msc细胞的相关标志物的表达谱相对于天然msc细胞(即未修饰/未处理的msc细胞)的表达谱基本无变化(即小于约10%、小于约9%、小于约8%、小于约7%、小于约6%、小于约5%、小于约4%、小于约3%、小于约2%或者小于约1%的变化)。
[0339]
如将理解的,在转染后如本文所描述的转染的msc可以表达一种或多种功能基因。因此,在这些实施方式中,转染细胞可以在转染后表达一种或多种功能基因,从而在这方面与等价的未转染的msc相区别。照此,本文中对通过转染基本无变化的多能性表型的提及可以反映在转染后,除功能基因的表达以外的一种或多种表型特征(包括(但不限于)多能性表型)可以在msc中是基本无变化的。在本文中详细描述了转染后可以基本无变化的表型特征的实例,并且它们可以包括(例如)多潜能性特征、免疫表型特征、癌症趋向性特征和/或其它表型特征中的任何一种或多种。
[0340]
在某些实施方式中,并且具体地当转染的msc将用于癌症治疗时,通过转染基本无变化的msc的多能性表型可以包括msc的肿瘤和/或癌症趋向性。在某些实施方式中,可以通过细胞侵袭测定确定msc的肿瘤和/或癌症趋向性,如以下实施例1中进一步详细描述的。在某些实施方式中,当在转染后肿瘤和/或癌症的趋向性无显著损失(即在转染后肿瘤和/或癌症的趋向性可以基本相同或提高)时,转染的msc的肿瘤和/或癌症趋向性可以被认为是无变化的。
[0341]
在某些实施方式中,转染的msc可以不含基于病毒的转染载体材料。如将理解的,在下文中详细提供了如本文所描述的用于制备转染的msc的无病毒转染方法。因此,在某些实施方式中,如本文所描述的转染的msc可以不含(即可以不含有)基于病毒的转染载体材料,基于病毒的转染载体材料可以包括(例如)通常存在于基于病毒的基因或核酸递送方法中的噬菌体蛋白和/或核酸、病毒膜组分、病毒核酸和/或病毒蛋白质。
[0342]
在某些实施方式中,考虑可以用核酸构建体转染本文所描述的转染的msc,并且转染的msc可以将一种或多种功能基因表达适合于对用转染的msc治疗的受试者实现益处的一段时间。已在本文中发现本发明所发展的方法可以提供转染的msc,包括瞬时转染的msc,其将一种或多种功能基因表达延长的一段持续时间。因此,在某些实施方式中,转染的msc可以在转染后将一种或多种功能基因瞬时表达至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0343]
在某些实施方式中,本文提供了msc细胞群体或包含多种msc细胞的组合物,其中在约90%的细胞或以上中存在表达msc标志物;如通过标准存活力测试所确定的,细胞存活力为约80%或以上;约70%或以上的多种msc对于转基因是阳性的,如通过流式细胞术所测试的;或其任意组合。优选地,对于msc细胞群体或包含多种msc细胞的组合物,在约90%的细胞或以上中存在表达msc标志物,如通过标准存活力测试所确定的,细胞存活力为约80%或以上;并且约70%或以上的多种msc对于转基因是阳性的,如通过流式细胞术所测试的。
[0344]
在某些实施方式中,考虑了本文所描述的方法可以用于提供即使在瞬时转染的情况下,将一种或多种功能基因表达延长的一段持续时间的转染的msc。在某些实施方式中,考虑了当延长的一段持续时间的表达是所期望的时,可以设计核酸构建体以提供一种或多种功能基因的延长的瞬时表达。举例来说,在某些实施方式中,在本文中发现当核酸构建体包含无cpg表达质粒时,可以实现一种或多种功能基因的延长的一段持续时间的表达。基于
这些发现,本领域技术人员考虑本文的教导内容将意识到用于提高瞬时表达的持续时间的多种选择。在某些实施方式中,考虑核酸构建体可以包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。延长表达的特征的实例进一步存在于molecular therapy,2006.14(5):613
‑
626页;jbiol chem,2000.275(39):30408
‑
16页;nucleic acids research,2014.42(7):e53
‑
e53页和doi:https://doi.org/10.1016/j.ymthe.2006.03.026中,以上文献中的每一篇以其全部内容作为参考并入本文。在某些实施方式中,可以在如本文所描述的核酸构建体的设计中实现这些特征中的一些或全部。在某些实施方式中,可以作为可以添加至如本文所描述的核酸构建体的模块实现这些特征中的一些或全部。例如,在如本文所描述和使用的某些cd::uprp:gfp构建体中,在核酸构建体中使用了无cpg和s/mar的特征/模块。
[0345]
在某些实施方式中,可以通过如本文所描述的任何方法产生转染的msc。举例来说,在某些实施方式中,可以使用阳离子聚合物、能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂,用核酸构建体转染转染的msc。在下文中提供了这些方法和组分的进一步描述。举例来说,在某些实施方式中,阳离子聚合物可以包括直链或支链聚亚乙基亚胺(pei);第一试剂可以包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems);和/或第二试剂可以包括组蛋白脱乙酰酶抑制剂(hdaci),如saha(伏立诺他)。
[0346]
在某些实施方式中,如在功能基因包括胞嘧啶脱氨酶(cdy)、尿嘧啶转磷酸核糖基酶(uprt)或者单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)中的一种或多种的情况下,msc可以对使用5
‑
氟胞嘧啶(5fc)或更昔洛韦(gcv)的治疗敏感。在某些实施方式中,转染的msc可以能够:a)将5fc转化为5
‑
氟尿嘧啶(5fu)、5
‑
氟脲嘧啶单磷酸盐(fump)或两者;b)将更昔洛韦转化为更昔洛韦单磷酸盐;或者c)上述a)和b)的组合。在某些实施方式中,msc可以用于治疗癌症。在某些实施方式中,转染的msc可以与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0347]
在某些实施方式中,转染的msc可以是基本未分化的。
[0348]
在另一实施方式中,本文提供了用从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法,方法包括:
[0349]
将msc暴露于包含与阳离子聚合物复合的核酸构建体的转染混合物;
[0350]
将msc暴露于能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂;和
[0351]
温育msc;
[0352]
从而提供用核酸构建体转染的msc。
[0353]
已在本文中详细描述了适合的msc、核酸构建体和功能基因的实例。举例来说,在某些实施方式中,一种或多种功能基因可以包括自杀基因;胞嘧啶脱氨酶(cdy)和/或胸苷激酶(tk);尿嘧啶转磷酸核糖基酶(uprt);可以或可以不作为融合构建体提供的cdy和uprt两者;荧光蛋白,如绿色荧光蛋白(gfp);cdy、uprt和gfp,其可以或可以不作为融合构建体提供;单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk);或其任意组合。
[0354]
在某些实施方式中,msc可以来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。在某些实施方式中,msc可以是脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。在另一实施方式中,msc可
以来源于人、狗、猫、马或其它物种。
[0355]
在某些实施方式中,核酸构建体可以包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0356]
在某些实施方式中,阳离子聚合物可以包括与核酸构建体复合并且一旦对其暴露,能够将核酸构建体递送至msc中的任何适合的阳离子或多聚阳离子或部分阳离子聚合物。在某些实施方式中,阳离子聚合物可以选自聚乙烯亚胺、多聚阳离子两亲分子、deae
‑
葡聚糖、阳离子聚合物、它们的衍生物或它们的任意组合。在某些实施方式中,阳离子聚合物可以包括阳离子聚合物,如树枝状聚合物、支链聚乙烯亚胺(bpei)、直链
‑
聚亚乙基亚胺(lpei)、聚(酰胺胺)(pamam)、xtremegene或它们的任意组合。在某些实施方式中,阳离子聚合物可以包括lpei。在某些实施方式中,阳离子聚合物可以(例如)是均聚物、共聚物或者嵌段
‑
共
‑
聚物。在某些实施方式中,阳离子聚合物可以具有约5kda至约200kda的大小。在某些实施方式中,阳离子聚合物可以具有等于或小于约5kda的大小。在某些实施方式中,阳离子聚合物可以具有等于或大于约200kda的大小。在某些实施方式中,阳离子聚合物可以包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物或它们的任意组合。在某些实施方式中,阳离子聚合物可以包括直链聚亚乙基亚胺(lpei)。
[0357]
在某些实施方式中,msc所暴露的转染混合物中的核酸构建体的量可以在约200至约500ng每1.9cm2表面积之间。在某些实施方式中,msc所暴露的转染混合物中的核酸构建体的量可以在约250至约400ng每1.9cm2表面积之间。在某些实施方式中,msc所暴露的转染混合物中的核酸构建体的量可以在约300至约350ng每1.9cm2表面积之间。在某些实施方式中,msc所暴露的核酸构建体的量可以是约200至约500ng每1.9cm2之间圆整至最近的0.1的任何值或者它们之间的任何子范围。
[0358]
在任何上述方法的某些实施方式中,在转染混合物中阳离子聚合物与核酸构建体的比值可以为约1μg至约30μg阳离子聚合物每1μg核酸构建体,或者它们之间圆整至最近的0.1的任何值,或者它们之间的任何子范围。
[0359]
在某些实施方式中,阳离子聚合物和核酸n/p可以在约5至约100的范围内,例如,约5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98或99、或100,或者它们之间圆整至最近的0.1的任何值,或者它们之间的任何子范围。
[0360]
在某些实施方式中,转染混合物可以包括复合缓冲液、细胞培养基或细胞缓冲液或它们的任意组合。
[0361]
在某些实施方式中,能够重定向来自胞内酸性区室的内吞核酸的第一试剂可以包括能够将基因材料导向远离细胞的非生产性酸性区室(non
‑
productive acidic compartment)的任何适合的试剂。在另一实施方式中,第一试剂可以包括脂质、肽促融试剂或它们的组合。在某些实施方式中,第一试剂可以包括dope、chems、dppc或dopc或它们的任意组合。在某些实施方式中,第一试剂可以包括血细胞凝集素(ha2
‑
肽)、流感
‑
来源的融合肽diinf
‑
7、白喉毒素的t域或者多聚阳离子肽,如聚赖氨酸和/或聚精氨酸或它们的任意组合。在某些实施方式中,第一试剂可以包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺
(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)。可以使用dope:chem的多种比值,例如,在某些实施方式中,可以使用约9:1至1:9之间的比值,如约9:1、8:2、7:3、6:4、5:5、4:6的比值。在某些实施方式中,可以使用促融脂质(fusogenic lipid)和辅助脂质的混合物,如dope和chems的混合物。在某些实施方式中,可以使用3种脂质。例如,在某些实施方式中,可以使用处于多种比例的dppc:dope:chems的混合物。
[0362]
在某些实施方式中,能够稳定msc的微管网络的第二试剂可以包括稳定其微管或网络的任何适合的试剂。在某些实施方式中,第二试剂可以能够提高微管蛋白的乙酰化。在某些实施方式中,第二试剂可以选自组蛋白脱乙酰酶抑制剂(hdaci),如组蛋白脱乙酰酶6抑制剂(hdac6i)、微管蛋白结合剂(tba)和能够直接或间接影响微管网络稳定性的sirna。在某些实施方式中,hdaci可以包括tubastatin a、贝利司他、丁苯羟酸、帕比司他、pci
‑
24781、saha(伏立诺他)、scriptaid、曲古抑菌素a、丙戊酸、b2、salermide、sirtinol或它们的任意组合。在某些实施方式中,第二试剂可以包括组蛋白脱乙酰酶抑制剂(hdaci),如saha(伏立诺他)。
[0363]
在某些实施方式中,第一试剂和第二试剂可以一起形成trafen
tm
,其代表用于将基因材料或含有基因材料的复合物导向至用于有效转染的生产途径的运输增强剂(trafficking enhancer)。
[0364]
适合的能够重定向来自胞内酸性区室的内吞核酸的第一试剂和适合的能够稳定msc的微管网络的第二试剂和trafen
tm
的实例详细描述于标题为“a novel reagent for gene
‑
drug therapeutics”的wo2014/070111中和ho y.k.等人,enhanced non
‑
viral gene delivery by coordinated endosomal release and inhibition ofβ
‑
tubulin deactylase,nucleic acids research,2017,45(6):e38中,以上两篇文献以其全部内容作为参考并入本文。
[0365]
在上述方法的某些实施方式中,在暴露于转染混合物,暴露于第一试剂和第二试剂期间,在温育期间或它们的任意组合,不对msc离心。在上述方法的某些实施方式中,温育msc的步骤可以任选地包括温和混合,但不离心。
[0366]
在某些实施方式中,离心可以用于帮助将聚合物
‑
复合的dna快速沉积在细胞上。然后,在离心后立即或不久除去转染混合物,从而最大程度降低游离的阳离子聚合物(其通过离心基本不旋转沉淀)对细胞的毒性。因此,在某些实施方式中,具体地当使用易于进行离心的小规模操作时,离心可以用于细胞的转染。举例来说,离心方法可以包括以下步骤:将转染混合物添加至细胞;离心以将核酸复合物沉积在细胞上;除去转染混合物以除去游离的聚合物,从而使其不接触细胞;和替换(例如)可以包括trafen或者可以对其添加trafen的新鲜载体。
[0367]
在某些实施方式中,可以期望大规模操作,如在兽医和/或人疗法适应症中。在这种大规模下,离心可以是不期望的。在其它情况下,离心也可以是不期望的,如(例如)其中离心设备不可用、不方便和/或昂贵的应用。如本文详细描述的,现已发现在某些实施方式中,可以省略离心。当省略离心时,在某些实施方式中,温育时间可以延长至(例如)约2至约24小时、以使聚合物
‑
复合的dna充分接触细胞。在温育时间期间,基于正在使用的细胞和聚合物,游离聚合物的存在可能是有毒的。因此,在某些实施方式中,对于特定细胞类型和温育时间,可以适当调整阳离子聚合物的选择以降低或避免对细胞的毒性,如本文详细描述
的。
[0368]
如本文详细描述的,可以或可以不使用离心的本文所描述的方法可以提供高转染效率(例如,>约70%)。
[0369]
在省略离心的实施方式中,方法可以更易于可缩放以适应(例如)用于临床前和/或临床试验的大生产规模。然而,本发明人已发现当在转染期间省略离心时,可以延长转染期间的温育时间以实现高转染效率。因此,在如省略离心的那些的某些实施方式中,温育msc的步骤可以包括温育msc至少约2小时。在某些实施方式中,温育msc的步骤可以包括温育msc约2小时至约24小时、或者约4小时至约18小时、或者2至24小时之间圆整至最近的0.1的任何值,或者它们之间的任何子范围。
[0370]
在实施方式中,本发明人已进一步识别,当省略离心和/或当延长温育时间时,可以适当调整阳离子聚合物的选择,因为某些阳离子聚合物可以引起毒性,这特别是在延长温育时间的情况下可以是不期望的。此外,不同类型的msc(即类型、来源、细胞系和生长条件的改变)可以对阳离子聚合物和/或延长的温育时间段显示出不同的耐受性。因此,在某些实施方式中,可以对正在使用的特定msc调整转染方法。以下实施例2描述了其中实施dna的量/条件和阳离子聚合物的选择以在某些msc的转染期间避免毒性并获得高转染效率的一些实施例。因此,在本文所描述的方法的某些实施方式中,阳离子聚合物可以包括已鉴别为对特定应用的msc具有低细胞毒性的阳离子聚合物。在某些实施方式中,例如,可以按尺寸和/或电荷数筛选阳离子聚合物。在某些实施方式中,例如,某些较大的阳离子聚合物可以对于一些细胞是优先的,但是可以对其它细胞是稍微有毒的。在一些情况下,较小的阳离子聚合物和/或荷电较少的阳离子聚合物通常可以是低毒性的,但是可以在某些细胞系中显示出低转染效率和/或转染率。在某些实施方式中,例如,trafen可以用于加强转染效率。
[0371]
在某些实施方式中,当选择阳离子聚合物和/或聚合物
‑
dna复合物时,可以考虑可以在细胞类型之间改变的载体的浮力密度,因为这可以对聚合物
‑
dna复合物在细胞上的沉积速率有影响。在某些实施方式中,可以选择复合物和/或载体以有利于在细胞上的良好沉积,和/或可以选择阳离子聚合物,从而游离的阳离子聚合物对特定细胞无毒或低毒。
[0372]
在某些实施方式中,考虑可以筛选阳离子聚合物以鉴别为所关心的特定msc提供适合的转染效率和/或细胞存活力的那些阳离子聚合物,因为对于在不离心的情况下提供有效转染,这些是本文中所识别的确定阳离子聚合物与特定msc类型/供体的相容性水平的两个显著特征。在某些实施方式中,可以选择阳离子聚合物,从而在(例如)至少约2小时或约4小时的温育期期间,它不引起明显或不利的细胞毒性水平。如果阳离子聚合物对细胞无毒,则可以使温育期进行更长时间。在某些实施方式中,可以通过任何适合的方法,如碘化丙啶测定评价毒性。在某些实施方式中,转染后的细胞存活力(或者细胞存活力靶标)可以等于或大于约70%。
[0373]
在任何上述方法的某些实施方式中,将msc暴露于转染混合物的步骤可以包括将核酸构建体与阳离子聚合物复合以提供包含复合的核酸构建体的转染混合物,并将转染混合物添加至msc。换言之,将msc暴露于转染混合物的步骤将优选地包括在向msc添加前,使核酸构建体和阳离子聚合物预复合或合并。
[0374]
在任何上述方法的另一实施方式中,将msc暴露于转染混合物的步骤可以包括将转染混合物添加至msc并将msc与转染混合物温育。
[0375]
在任何上述方法的另一实施方式中,将msc暴露于第一试剂和第二试剂的步骤可以包括在将msc添加至或暴露于转染混合物的同时或之后立即添加第一试剂和第二试剂。在某些实施方式中,这可以在省略离心时进行。
[0376]
在任何上述方法的另一实施方式中,将msc暴露于第一试剂和第二试剂的步骤可以包括与将msc暴露于转染混合物的步骤中的转染混合物一起添加第一试剂和第二试剂,或者可以包括将第一试剂和第二试剂添加至已与转染混合物接触的msc(即在添加第一试剂和第二试剂之前可以不除去转染混合物)。在某些实施方式中,这可以在省略离心时进行。
[0377]
在任何上述方法的另一实施方式中,将msc暴露于第一试剂和第二试剂的步骤可以包括用补充有第一试剂和第二试剂的细胞培养基替换转染混合物。在某些实施方式中,这可以在使用离心来帮助聚合物
‑
复合的dna在细胞上的快速沉积的情况下进行,以减少对细胞的游离聚合物毒性。在某些实施方式中,细胞培养基可以包括完全培养基。
[0378]
在任何上述方法的某些实施方式中,msc可以处于约60%汇合,并且可以在暴露于转染混合物之前约24小时接种msc。
[0379]
在任何上述方法的某些实施方式中,转染混合物可以包括处于无血清dmem中或新鲜培养基中的复合的核酸构建体。
[0380]
在任何上述方法的其它实施方式中,将msc暴露于转染混合物的步骤可以包括将转染混合物(其可以或可以不另外包括新鲜培养基)添加至细胞,而在添加转染混合物之前未从细胞除去培养/生长载体。在某些实施方式中,这可以在省略离心时进行。
[0381]
在上述方法的某些实施方式中,将msc暴露于转染混合物的步骤可以包括:
[0382]
任选地,用新鲜培养/生长载体替换其中正在培养细胞的培养/生长载体;和
[0383]
将转染混合物(其可以或可以不另外包括新鲜培养基或者其可以与新鲜培养基同时或顺序添加)添加至细胞,而在添加转染混合物(如果存在)之前未从细胞除去培养/生长载体。
[0384]
因此,在某些实施方式中,考虑可以在向细胞添加复合的核酸构建体之前替换或更新细胞培养/生长载体,从而在进行转染之前提供新鲜培养基。在某些实施方式中,这可以在省略离心时进行。
[0385]
在任何上述方法的其它实施方式中,将msc暴露于转染混合物的步骤可以包括从msc除去培养基和用转染混合物替换培养基。在某些实施方式中,这可以在使用离心来帮助将聚合物
‑
复合的dna快速沉积在细胞上的情况下进行。
[0386]
在某些实施方式中,考虑将msc暴露于转染混合物的步骤可以包括在轻微离心下将msc与转染混合物温育。例如,在某些实施方式中,当小规模实施方法和/或当适合的离心设备可用时,考虑可以实施离心。可以在添加聚合物
‑
核酸构建体后,实施温和离心以避免游离聚合物的毒性。在某些实施方式中,方法可以包括向细胞培养物添加转染混合物,实施温和离心(例如,约5分钟)以使聚合物
‑
核酸构建体复合物沉积在细胞上)并且除去转染混合物(含有游离聚合物,聚合物是相对小的并且通过离心基本不旋转沉淀)。在某些实施方式中,轻微离心可以包括约200g离心约5分钟。
[0387]
在本文所描述的方法的某些实施方式中,例如,可以在平底容器中进行转染,其中提高了试剂的量,将细胞密度调节至该提高的量并且根据细胞培养容器的表面积提高dna
的量。
[0388]
在本文所描述的方法的某些实施方式中,可以在转染期间在微载体(例如,微珠)上培养msc并因此可以混悬,任选地同时处于振荡或其它搅拌。在某些实施方式中,微载体可以包括微珠。在某些实施方式中,微载体可以包括1型猪胶原蛋白涂覆的微载体。在某些实施方式中,微载体可以包括3。在其中使用微载体的某些实施方式中,可以在振荡下进行转染并提高细胞密度,这可以允许大规模生产。另外,可以根据容器类型和所使用的微载体的密度/数目来调整振荡期间的rpm。
[0389]
使用平底容器和微载体的其它方法将遵照类似步骤,例如:步骤1
→
将msc暴露于包含与阳离子聚合物复合的核酸构建体的转染混合物;步骤2
→
将msc暴露于能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂;和步骤3
→
温育msc,从而提供用核酸构建体转染的msc。对每个容器调整要使用的每表面积cm2和细胞培养体积所添加的细胞数。对于微载体应用,将调节振荡速度以防止微载体聚集。
[0390]
在本文所描述的方法的某些实施方式中,在转染期间温育msc的步骤可以包括使生物反应器
‑
型搅拌(例如,使三角瓶旋转)、摇袋式生物反应器、旋转壁式生物反应器、搅拌罐式生物反应器或者振荡器
‑
型搅拌旋转至少一部分温育时间(对于其它实例,参见图52)。
[0391]
在任何上述方法的某些实施方式中,可以在转染的msc中瞬时表达,可以在转染的msc中稳定转染一种或多种功能基因,或它们的组合。
[0392]
在任何上述方法的某些实施方式中,可以用平均至少约1000、至少约2000、至少约3000、至少约4000、至少约5000、至少约6000、至少约7000、至少约8000、至少约9000或者至少约10000拷贝的核酸构建体各自转染转染的msc。
[0393]
在上述方法的某些实施方式中,通过转染转染的msc的多能性表型可以基本无变化。在某些实施方式中,多能性表型可以包括msc的肿瘤和/或癌症趋向性。在某些实施方式中,多能性表型可以包括其中在转染后cd表面标志物的表达可以基本无变化的免疫表型。在某些实施方式中,转染的msc可以是塑料
‑
附着的,可以表达cd105、cd73和cd90(>95%),可以缺乏cd45、cd34、cd14和hla
‑
dr表面分子的表达(<2%)并且可以能够体外分化为成骨细胞、脂肪细胞和成软骨细胞,从而满足国际细胞疗法协会(isct)所定义的免疫表型标准。已在上文中和以下实施例中详细描述了转染的细胞的表型。
[0394]
在任何上述方法的某些实施方式中,可以用核酸构建体转染至少约60%、至少约65%、至少约70%、至少约75%、至少约80%、至少约85%、至少约90%或者至少约95%的msc并且msc表达一种或多种功能基因。
[0395]
在任何上述方法的某些实施方式中,转染的msc的细胞存活力可以为至少约70%、至少约75%、至少约80%或者至少约85%。
[0396]
在任何上述方法的某些实施方式中,转染的msc可以是未分化的。
[0397]
在任何上述方法的某些实施方式中,方法可以不含基于病毒的转染载体材料。
[0398]
在任何上述方法的某些实施方式中,msc可以在转染后瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0399]
在某些实施方式中,所产生的msc可以对使用5
‑
氟胞嘧啶(5fc)或更昔洛韦(gcv)或两者的治疗敏感。在某些实施方式中,所产生的msc可以:a)将5fc转化为5
‑
氟尿嘧啶
(5fu)、5
‑
氟脲嘧啶单磷酸盐(fump)或两者;b)将更昔洛韦转化为更昔洛韦单磷酸盐;或者c)上述a)和b)的组合。
[0400]
在某些实施方式中,一种或多种功能基因可以包括荧光蛋白,并且方法还可以包括使用细胞分选或facs分离、选择或纯化转染的msc的步骤。可以实施该步骤,例如,当需要特别高的纯度时。如将理解的,这种分离、选择或纯化可以是任选的,因为在临床应用中,例如,考虑对治疗基因约≥70%阳性的群体可以是可接受的,并且在某些实施方式中,如本文所描述的,可以获得群体而无进一步的分离、选择或纯化步骤。
[0401]
在上述方法的另一实施方式中,本文提供了用从中表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的方法,方法包括:
[0402]
在生长培养基中培养msc;
[0403]
将包含与阳离子聚合物复合的核酸构建体的转染混合物添加至msc,同时不从msc除去生长培养基;
[0404]
将能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定msc的微管网络的第二试剂添加至msc;和
[0405]
温育msc,同时与转染混合物、第一试剂和第二试剂全部接触培养期。
[0406]
其中与转染混合物的添加同时,与转染混合物的添加顺序或者与转染混合物的添加组合,将第一试剂和第二试剂添加至msc;和
[0407]
其中在转染混合物的添加和温育期结束之间不对msc离心;
[0408]
从而提供用核酸构建体转染的msc。
[0409]
在某些实施方式中,在生长培养基中培养msc的步骤可以包括向细胞提供新鲜生长培养基(即用新鲜生长培养基替换用过或部分用过的生长培养基或者将新鲜生长培养基添加至用过或部分用过的生长培养基)。
[0410]
在某些实施方式中,温育期可以是至少约2小时。
[0411]
在某些实施方式中,温育期可以是约2小时、约3小时、约4小时、约5小时、约6小时、约7小时、约8小时、约9小时、约10小时、约11小时、约12小时、约13小时、约14小时、约15小时、约16小时、约17小时、约18小时、约19小时、约20小时、约21小时、约22小时、约23小时、约24小时、约25小时、约26小时、约27小时、约28小时、约29小时、约30小时、约31小时、约32小时、约33小时、约34小时、约35小时或者约36小时。
[0412]
在任何上述方法的某些实施方式中,方法可以产生如本文所描述的任何转染的msc。
[0413]
在另一实施方式中,本文提供了通过本文所描述的任何方法所产生的msc或多种msc。
[0414]
在另一实施方式中,本文提供了包含如本文所描述的任何一种或多种msc和药物可用的载体、稀释剂、赋形剂、细胞培养基或缓冲剂中的至少一种的组合物。
[0415]
在某些实施方式中,药物可用的载体、稀释剂、赋形剂、细胞培养基或缓冲剂可以包括(例如)任何适合的pbs缓冲液、冷冻保护载体、matrigel或水凝胶。在某些实施方式中,本文提供了包含如本文所描述的msc在pbs或另一种缓冲液或细胞培养基中的混悬液的组合物。在另一实施方式中,本文提供了包含用冷冻保护载体冷冻的如本文所描述的msc的组合物。
[0416]
在另一实施方式中,本文提供了包含本文所描述的任何一种或多种msc的治疗诊断试剂。举例来说,在某些实施方式中,治疗诊断试剂可以包括表达治疗或自杀基因和荧光蛋白两者的msc。msc可以具有癌症和/或肿瘤趋向性,并且可以通过荧光用于癌症或肿瘤细胞的指示位置,例如,在该点可以添加前体药物(在此使用自杀基因)以导致产生抗
‑
癌或抗
‑
肿瘤作用。
[0417]
在另一实施方式中,本文提供了用从中瞬时表达一种或多种功能基因的核酸构建体转染间质干细胞(msc)的试剂盒,试剂盒包含以下中的一种或多种:
[0418]
msc;
[0419]
设计用于瞬时表达一种或多种功能基因的核酸构建体;
[0420]
细胞培养基;
[0421]
阳离子聚合物;
[0422]
能够重定向来自胞内酸性区室的内吞核酸的第一试剂;
[0423]
能够稳定msc的微管网络的第二试剂;
[0424]
实施如本文所描述的方法的说明书;
[0425]
5fc;
[0426]
gcv;和/或
[0427]
5fu。
[0428]
在某些实施方式中,msc可以是如本文所描述的任何msc。在某些实施方式中,msc可以来源于脐血、新生儿
‑
相关组织、华通氏胶、脐带、脐带内膜、胎盘或其它msc细胞来源。在某些实施方式中,msc可以包括脂肪组织
‑
来源的msc(at
‑
msc)、骨髓
‑
来源的msc(bm
‑
msc)或者脐带
‑
来源的msc(uc
‑
msc)。在另一实施方式中,msc可以来源于人、狗、猫、马或其它物种。
[0429]
在某些实施方式中,核酸构建体可以是如本文所描述的任何核酸构建体并且一种或多种功能基因可以是如本文所描述的任何一种或多种功能基因。在某些实施方式中,核酸构建体可以包含无cpg的表达质粒或者其它无cpg的表达构建体、骨架/基质连接区(s/mar)、游离基因载体或者含有ebna
‑
1的构建体。
[0430]
在某些实施方式中,阳离子聚合物可以包括如本文所描述的任何阳离子聚合物。在某些实施方式中,阳离子聚合物可以包括直链或支链聚亚乙基亚胺(pei)、聚(酰胺胺)pamam或者另一种阳离子聚合物。在某些实施方式中,阳离子聚合物可以包括直链聚亚乙基亚胺(lpei)。在某些实施方式中,阳离子聚合物可以包括已鉴别为对msc具有低细胞毒性的阳离子聚合物。在某些实施方式中,阳离子聚合物可以具有约5kda至约200kda的大小。
[0431]
在某些实施方式中,第一试剂可以包括如本文所描述的任何适合的第一试剂。在某些实施方式中,第一试剂可以包括dopc、dppc或另一种促融脂质(fusogenic lipid)中的一种或多种。在某些实施方式中,第一试剂可以包括1,2
‑
二油酰基
‑
sn
‑
甘油基
‑3‑
磷酸乙醇胺(dope)/胆甾醇基半琥珀酸酯(chems)(dope/chems)、1,2
‑
二棕榈酰基
‑
sn
‑
甘油基
‑3‑
胆碱磷酸(dppc)或者另一种促融脂质(fusogenic lipid)或它们的任意组合。
[0432]
在某些实施方式中,第二试剂可以包括如本文所描述的任何适合的第二试剂。在某些实施方式中,第二试剂可以包括组蛋白脱乙酰酶抑制剂(hdaci),如saha(伏立诺他)。
[0433]
在某些实施方式中,一种或多种功能基因可以包括自杀基因;胞嘧啶脱氨酶
(cdy);胸苷激酶(tk);尿嘧啶转磷酸核糖基酶(uprt);可以或可以不作为融合构建体提供的cdy和uprt两者;荧光蛋白,如绿色荧光蛋白(gfp);cdy、uprt和gfp,其可以作为融合构建体提供;单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk);或其任意组合。
[0434]
在某些实施方式中,在试剂盒中阳离子聚合物与核酸构建体的比值可以为约1μg至约30μg阳离子聚合物每1μg核酸构建体。
[0435]
在某些实施方式中,试剂盒可以用于制备msc
‑
基抗癌剂。在某些实施方式中,试剂盒还可以包括用于实施如本文所描述的治疗癌症的方法的说明书和/或设备。在某些实施方式中,可以提供用于msc的肿瘤内或静脉内或皮下注射或输注的注射器或其它适合的注射装置。在某些实施方式中,可以用生物材料,如明胶海绵包埋msc以用于施用。
[0436]
在某些实施方式中,本文提供了用于癌症治疗的间质干细胞(msc)的可缩放非病毒基因修饰的方法。在某些实施方式中,如本文所描述的方法可以包括在存在转染增强剂制剂(trafen)的情况下,用一种或多种自杀基因转染msc。在某些实施方式中,这些方法可以包括使用能够重定向来自胞内酸性区室的内吞核酸的第一试剂和能够稳定其微管网络的第二试剂。在某些实施方式中,可以提供修饰的细胞的数目和表达的高效修饰以用于产生表达治疗基因,例如,自杀基因胞嘧啶脱氨酶(cd)的高效力msc。在某些实施方式中,可以将修饰的msc施用于具有肿瘤或癌症的受试者。在某些实施方式中,通过msc表达的治疗基因可以将前体药物转化为减小或消除肿瘤体积的毒性剂。在某些实施方式中,可以在用于治疗癌症和/或其它适应症的药剂的生产中使用本文所描述的方法。本文还描述了用于将基因材料递送至细胞中的方法和因此的试剂盒。在某些实施方式中,可以用通常任何适合的癌症靶向治疗基因和/或通常任何其它适合的治疗基因修饰msc以用于治疗通常任何其它适合的疾病和/或病症。
[0437]
使用转染的间质干细胞治疗疾病或病症,如癌症的使用和方法
[0438]
如本文详细描述的,提供了转染的msc和用于制备转染的msc的方法和试剂盒,其中转染的msc可以表达一种或多种功能基因。在某些实施方式中,一种或多种功能基因可以包括一种或多种治疗活性基因,从而产生(例如)一种或多种治疗活性的rna、肽、多肽或蛋白。如将理解的,本文所描述的msc因此可以用于治疗、预防或改善通常一种或多种功能基因对其具有治疗活性的任何疾病或病症中的使用。以下讨论主要涉及癌症治疗,然而技术人员通过考虑本文的教导内容将认识到在本文中还考虑了多种其它疾病或病症。
[0439]
在一个实施方式中,本文提供了如本文所描述的任何一种或多种msc用于杀伤癌细胞的使用。
[0440]
在一个实施方式中,本文提供了如本文所描述的任何一种或多种msc用于治疗对其有需要的受试者中的癌症的使用。
[0441]
在某些实施方式中,癌症可以包括多种类型的实体瘤中的任何一种或多种(如甲状腺癌、肉瘤、淋巴瘤、鳞癌等)。由于msc可以显示出强趋向性,因此考虑癌症的位置通常可以在任何地方并且位置可以不具有显著问题。此外,在某些实施方式中,考虑可以调整如本文所描述的msc和治疗以适合特定癌症,并且如本文所描述的msc前体药物策略通常可以对癌症类型是不可知的。
[0442]
在某些实施方式中,癌症可以(例如)包括淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或
胃肠癌或它们的任意组合。
[0443]
在某些实施方式中,受试者可以包括脊椎动物、哺乳动物或人。
[0444]
在某些实施方式中,msc可以用于与对要治疗的疾病或病症具有活性的一种或多种其它药物或治疗剂,如当疾病或病症是癌症时,一种或多种抗
‑
癌药物组合(同时、顺序或混合)使用。
[0445]
在某些实施方式中,具体地当通过转染的msc表达的一种或多种功能基因包括自杀基因或者一种或多种功能基因表达将前体药物转化为活性形式的酶时,msc可以与一种或多种相应前体药物组合(同时、顺序或混合)使用。举例来说,在某些实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy);胸苷激酶(tk);尿嘧啶转磷酸核糖基酶(5fu);可以或可以不作为融合构建体提供的cdy和uprt两者;单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk);或其任意组合,并且可以与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0446]
在另一实施方式中,本文提供了如本文所描述的任何一种或多种msc在生产癌症治疗药剂中的使用。在某些实施方式中,一种或多种msc可以与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0447]
在某些实施方式中,如本文所描述的一种或多种msc可以用于通过通常适合于要治疗的受试者和/或疾病的任何适合技术向受试者的施用。举例来说,在某些实施方式中,考虑可以向受试者全身(例如,通过静脉注射)或局部(例如,通过局部注射或植入)施用msc。在某些实施方式中,可以静脉内施用msc,如,例如,oncotarget 2017oct 6;8(46):80156
–
80166中所描述的,或通过颅内施用,如,例如,clin cancer res.2017jun15;23(12):2951
‑
2960中所描述的,以上每篇文献以其全部内容作为参考并入本文。在某些实施方式中,如本文所描述的一种或多种msc可以用于通过门静脉内、腹膜内、静脉内、肿瘤内、皮下、颅内注射或输注或者包埋在水凝胶或凝胶泡沫中用于向受试者施用或植入的施用向受试者的施用。
[0448]
在另一实施方式中,本文提供了治疗对其有需要的受试者中的癌症的方法,方法包括:
[0449]
将如本文所描述的一种或多种msc施用至受试者的癌细胞附近的区域,
[0450]
其中一种或多种msc中的一种或多种功能基因有助于对癌细胞的抗癌作用。
[0451]
在某些实施方式中,受试者可以包括脊椎动物、哺乳动物或人。
[0452]
在某些实施方式中,癌症可以(例如)包括淋巴瘤、明细胞癌、成胶质细胞瘤、替莫唑胺耐受性成胶质细胞瘤、肛周癌、口腔黑素瘤、甲状腺癌、软组织癌、癌症溃疡、鼻肿瘤或胃肠癌或它们的任意组合。
[0453]
在某些实施方式中,如本文所描述的一种或多种msc可以通过通常适合于要治疗的受试者和/或疾病的任何适合技术施用于受试者。举例来说,在某些实施方式中,考虑可以向受试者全身(例如,通过静脉注射)或局部(例如,通过局部注射、肿瘤内注射、皮下注射或者植入或输注)施用msc。在某些实施方式中,可以静脉内施用msc,如,例如,oncotarget2017oct 6;8(46):80156
–
80166中所描述的,或通过颅内施用,如,例如clin cancer res.2017jun 15;23(12):2951
‑
2960中所描述的,以上每篇文献以其全部内容作为参考并入本文。在某些实施方式中,如本文所描述的一种或多种msc可以用于通过静脉内、肿瘤内、皮下、颅内注射或输注或者包埋在水凝胶或凝胶泡沫中用于向受试者施用或植入
的施用向受试者的施用。
[0454]
如将理解的,在某些实施方式中,可以向受试者施用msc,从而使与疾病或病症有关的细胞(如癌细胞或肿瘤细胞)接触msc或处于msc适合的附近处,从而通过msc表达的一种或多种功能基因可以对与疾病或病症有关的细胞(直接或通过(例如)前体药物转化间接)发挥治疗效果。在某些实施方式中,msc可以对与疾病或病症有关的细胞具有趋向性,从而帮助msc在与疾病或病症有关的细胞的适合的附近处定位。在某些实施方式中,可以施用msc以直接接触与疾病或病症有关的细胞,或者使其处于距与疾病或病症有关的细胞约1
‑
3cm的附近处之内。在某些实施方式中,由于前体药物疗法可以导致旁观者效应,因此msc与疾病细胞(如癌细胞)的直接接触可以是不必要的。
[0455]
在某些实施方式中,具体地当通过转染的msc表达的一种或多种功能基因包括自杀基因或者一种或多种功能基因表达将前体药物转化为活性形式的酶时,msc可以与一种或多种相应前体药物组合(同时、顺序或混合)施用。举例来说,在某些实施方式中,一种或多种功能基因可以包括胞嘧啶脱氨酶(cdy);胸苷激酶(tk);尿嘧啶转磷酸核糖基酶(5fu);可以或可以不作为融合构建体提供的cdy和uprt两者;单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk);或其任意组合,并且可以与5fc、5fu、gcv或它们的任意组合进行组合使用。
[0456]
在某些实施方式中,可以设计msc的一种或多种功能基因和前体药物,从而除与疾病或病症有关的周围细胞(如癌细胞或肿瘤细胞,例如)之外,前体药物向活性形式的转化可以靶向或杀伤msc。因此,在其中msc将与另一种药物,如前体药物组合使用的某些实施方式中,可以期望维持msc和前体药物彼此分离直至首先将msc引入受试者的适当的细胞,从而在它们可以提供治疗效果之前不杀伤msc或者不使其失活。在某些实施方式中,msc可以通过趋向性花费至少一定时间来向肿瘤移动,并因此在某些实施方式中,例如,考虑可以延迟前体药物的施用直至msc处于肿瘤附近。
[0457]
在某些实施方式中,一种或多种msc可以在转染后,在向受试者施用后或两者瞬时表达一种或多种功能基因至少约7、至少约8、至少约9、至少约10、至少约11、至少约12、至少约13、至少约14、至少约15、至少约16或者至少约17天。
[0458]
在本文所描述的方法的某些实施方式中,方法还可以包括向受试者施用前体药物,如5fc、5fu、更昔洛韦或它们的任意组合的步骤,从而使一种或多种msc暴露于前体药物,如5fc、5fu、更昔洛韦或其组合,并且可以将前体药物转化为活性形式。
[0459]
在任何上述方法的某些实施方式中,方法还可以包括在施用一种或多种msc的步骤之前,使用如本文所描述的任何生产方法生产一种或多种转染的msc的步骤。
[0460]
在某些实施方式中,可以在转染之前进行msc的扩增和/或培养。举例来说,在某些实施方式中,对于大规模msc修饰,这种扩增可以是所期望的。
[0461]
实施例
[0462]
实施例1:用于癌症疗法的间质干细胞的非病毒修饰。用于基因导向的酶前体药物癌症疗法的工程化间质干细胞的有效非病毒方法
[0463]
已通过病毒和非病毒基因递送系统进行了用于前体药物基因疗法的间质干细胞(msc)的修饰。由于常规转染方法的低效率(0
‑
50%),病毒法已广泛用于临床前和临床研究。本文所描述的实施方式表明在存在一种或多种第一试剂,如促融脂质(fusogenic lipid)和第二试剂,如组蛋白脱乙酰酶6抑制剂(hdac6i)的情况下,基于聚亚乙基亚胺
(pei)的人体脂肪组织来源的msc(at
‑
msc)的修饰效率>90%。msc的细胞表型在修饰后保持不变,这是msc临床应用所期望的特征。通过配备这种方法,在神经胶质瘤、乳腺癌和胃癌细胞系中检验了产生融合的酵母胞嘧啶脱氨酶::尿嘧啶转磷酸核糖基酶(cdy::uprt)的修饰的msc的抗癌效力。通过5
‑
氟胞嘧啶的有效转换,cdy::uprt_at
‑
msc对人胃、乳腺和成胶质细胞瘤癌症体外显示出强细胞毒性效果。当与1%的治疗性cdy::uprt_at
‑
msc/5fc直接共培养时,在胃mkn1细胞系中观察到大于80%的抑制。通过无cpg表达质粒,将表达延长至转染后长达7天是可能的。转染后7天收集的cdy::uprt_at
‑
msc分别在胃mkn1和mkn28细胞系中显示出85%和95%的有效抑制。结果表明本发明所描述的方法可以在不使用病毒载体的情况下为msc
‑
基前体药物疗法提供替代方法。
[0464]
msc固有的肿瘤趋向性的累积证据已开拓了使用msc作为细胞载体来将抗癌剂特异性地递送至肿瘤和它们的转移位点的新兴平台[9,10]。最近,一些msc
‑
驱动的gdept临床试验已提供了有希望的结果,其保证了向ii期试验的进一步发展[7,11]。这种治疗方法使得能够在靶细胞附近局部且可控地酶促转化无毒前体药物。“旁观者效应”提高了对靶细胞的细胞毒性[7]。已在广谱实体癌[7,8],包括胃癌[12
‑
14]、乳腺癌[15,16]和成胶质细胞瘤[17
‑
19]中证实了产生cd的msc的抗癌潜能。临床前研究已证明胞嘧啶脱氨酶/5
‑
氟胞嘧啶(cd/5fc)是非常稳健的,其中肿瘤块中低至4%的cd阳性细胞足以完全消除肿瘤[20
‑
22]。使用cd/5fc系统的显著进展在于包括尿嘧啶转磷酸核糖基酶(uprt)、直接将5fu转化为5
‑
氟脲嘧啶单磷酸盐(fump)的嘧啶补救酶,因此绕过了限速酶二氢嘧啶脱氢酶(dpd)和乳清酸转磷酸核糖基酶(oprt)[23
‑
26]。与cd/5fc和5fu相比,cd::uprt/5fc将5fc向其活性代谢产物的转化提高了30
‑
1500倍[24,27]。
[0465]
瞬时转染可以具有单位细胞的高有效负荷,从而避免了可能引起细胞衰老的抗生素选择和数周的处理工作[40]并且降低了肿瘤的趋向性[41]以及与病毒诱导的msc转化有关的安全性问题[42]。尽管非病毒方法在易于生产、低成本和安全性谱方面具有优于病毒载体的优势[43],但是缺乏对msc修饰的广泛使用主要是由于通常遇到的低转染效率(0
‑
35%)所造成的[44,45]。例如,由于化学品基转染方法的较差表现(<5%的效率)[46],已通过反转录病毒转导工程化人脂肪组织来源的msc(at
‑
msc)来表达cd::uprt[47,48]。
[0466]
在本研究中,有可能使用阳离子聚合物结合trafen以高效率修饰at
‑
msc及其它msc来源,从而使得能够开发产生cdy::uprt的治疗性msc,而不需要使用病毒或建立稳定细胞系。
[0467]
结果
[0468]
用于at
‑
msc修饰的有效非病毒lpei基转染方法
[0469]
用编码gfp报告基因的质粒在24
‑
孔组织培养容器中转染at
‑
msc(年龄组18
‑
30)以评价lpei和lipofectamine 3000(l3k)的转染效率。尽管,存在更多使用lpei转染的细胞,但是贴壁细胞数目小于当使用l3k时的贴壁细胞数目(图8a)。尽管转染后保持高细胞存活力,但是当与未转染的对照相比时,在lpei介导的转染之后,贴壁细胞数目显著降低。通过使用提高的量的pdna,进一步降低了贴壁细胞的数目(图28),这与先前观察结果一致(swiech等人,bmc biotechnology,11(114),2011;mccall等人,frontiers in moleuclar neuroscience,5,(2012);ho等人,bioscience reports,38,2018;madeira等人,journal of biotechnology,151,130
‑
136,2011)。通过使用较低的量的聚合物,以200ng pdna转染
at
‑
msc来获得高贴壁细胞的尝试仅导致转染效率显著降低(图8b)。
[0470]
然后,我们研究了增强子()35与较低的量的pdna(200ng)一起以及多种dna:聚合物的比值用于增强转染的使用(图8b和29)。转染了超过80%的at
‑
msc细胞(图8b和29),且与未转染对照相当的贴壁细胞数和存活力(图29)。我们然后扩大研究以包括分离自另一供体(年龄组31
‑
45)的其它at
‑
msc。使用相同规程,转染效率高达90%的转染的细胞(图9)。
[0471]
为了建立at
‑
msc基因修饰的可靠规程,我们测试并比较了以多种量的dna,lpei和lipofectamine 3000的转染。用编码gfp报告基因的质粒在24
‑
孔组织培养容器中转染at
‑
msc(年龄组18
‑
30)以监测多种参数的效率(图8a)。明显地,at
‑
msc难以被lipofectamine 3000转染。尽管在转染效率方面lpei优于lipofectamine 3000,但是观察到显著降低的细胞数目,这表明了潜在的细胞负担。
[0472]
然而,以较低量的pdna和聚合物转染at
‑
msc以降低细胞负担的尝试显著降低了转染效率(图8b)。我们先前阐明多聚复合物的胞内运输途径的研究导致开发了trafen基方法[54]。trafen可以包括组蛋白脱乙酰基抑制剂(hdaci)和促融脂质(fusogenic lipid)。在本文的研究中,我们检验了这些试剂在使得能够以较低的量的pdna和聚合物进行有效转染中的潜能。转染了超过80%的at
‑
msc细胞,且无细胞负担(图8b)。我们然后扩大研究以包括分离自另一供体(年龄31
‑
45)的其它at
‑
msc。使用相同规程,实现了接近90%的转染效率(图9)。
[0473]
人脂肪组织来源的间质干细胞(at
‑
msc,roosterbio)分离自女性供体(lot00088,年龄18
‑
30)。在hmsc高效基础培养基(roosterbio)中维持at
‑
msc。根据生产商的说明书,培养和维持乳腺癌细胞系mda
‑
mb
‑
231(htb
‑
26,atcc)和原代人皮肤成纤维细胞(atcc,pcs
‑
201
‑
012)。paula lam(duke nus medical school)友好提供了神经胶质瘤细胞系u
‑
251mg。在补充有10%胎牛血清(fbs,biowest)的dmem(杜尔贝科改良的伊戈尔培养基)中培养u
‑
251mg细胞系。yong wei peng博士(新加坡国立大学癌症研究所(national university cancer institute,singapore))友好提供了胃癌细胞系mkn1和mkn28。在补充有10%fbs的rpmi(roswell park memorial institute medium,thermo scientific)中培养胃癌细胞系。将细胞保持在37℃,加湿气氛和5%co2中。
[0474]
治疗诊断性cdy::uprt_at
‑
msc的鉴定,和cdy::uprt_at
‑
msc的功能性的确定
[0475]
为了产生表达融合的胞嘧啶脱氨酶和尿嘧啶转磷酸核糖基酶的at
‑
msc(cdy::uprt_at
‑
msc),按照离心规程用lpei转染at
‑
msc。在存在trafen的情况下,基于用cdy::uprt::gfp转染的at
‑
msc的gfp分析,接近于80
±
2.3%的转染效率是可达到的(图1a)。如流式细胞术分析中所示,在存在trafen的情况下,大部分转染的细胞表达高水平的gfp。尽管提高dna的量适度改善了lpei和lipofectamine 3000的转染效率,但是它们的表现仍不令人满意(图1b)。这些数据表明需要trafen帮助多聚复合物在at
‑
msc中的胞内运输[49]。将cdy::uprt表达的持续时间确认为在转染后维持至少7天(图1c,10)。
[0476]
基于免疫细胞化学分析,在存在增强子的情况下,以较低的量的pdna(200ng)显著改善了转染。在不存在增强子的情况下,提高pdna的量适度提高了lpei和lipofectamine 3000的转染效率(图1)。扩大该观察结果,我们构建了编码胞嘧啶脱氨酶、尿嘧啶转磷酸核糖基酶和绿色荧光蛋白(cdy::uprt:gfp)的融合基因以用于直接显象和定量。在存在增强子的情况下,与单独的lpei的使用相比,转染效率显著提高(~80%),且存活力无显著变化
(图30)。值得注意地,用cdy::uprt:gfp或cdy::uprt修饰的at
‑
msc的抗
‑
癌效率无显著差异(图31)。总体而言,结果表明通过增强子显著改善了at
‑
msc的转染,这可能与辅助pdna在bm
‑
msc中的胞内运输的机制类似(ho等人,nucleic acids research,45(38),2017)。
[0477]
在3
‑
5代,将所关心的转基因引入at
‑
msc。对于每个孔(6
‑
孔板形式),将5mg/ml lpei(pei max,polyscience)以不同的pdna和peimax的比值加入至无血清dmem中的pdna中。以100μl的总体积,将混合物在室温下温育15min。根据pdna的量,μg:1mg/ml lpei的体积,μl计算pdna:lpei的比值。然后,将lpei/pdna复合物添加至无血清dmem培养基(1:20)以制备转染混合物。除去培养基并用转染混合物替换,然后以200g轻轻离心5min。在离心后,除去转染混合物并用补充或未补充trafen的完全培养基替换。trafen由dope/chems(polar avanti lipid)和伏立诺他(saha,bio vision)组成。分析前,将细胞温育24h。
[0478]
如前,实施流式细胞术、免疫印迹和免疫细胞化学[49]。简要地,流式细胞术:通过attune nxt流式细胞仪系统(thermofisher scientific)定量荧光阳性细胞百分比并使用invitrogen attune nxt软件(thermofisher scientific)分析原始数据。成像:使用配备有用于观察dapi(ex357/em447)、gfp(ex470/em510)荧光的3个荧光立方体的evos fl细胞成像系统(thermofisher scientific)采集细胞图象。免疫印迹:分别用绵羊抗
‑
cdy(pa185365,thermofisher scientific)和单克隆抗
‑
β
‑
肌动蛋白(a2228,sigma
‑
aldrich),通过免疫印迹技术分析样品。免疫细胞化学:用绵羊抗
‑
cdy和alexa fluor 488驴抗
‑
绵羊荧光第二抗体(a11015,thermofisher scientific)标记样品。使用evos fl细胞成像系统进行图象采集。使用相同光学设置采集所有图像。
[0479]
cdy::uprt的表达使得at
‑
msc对前体药物5fc敏感(cdy::uprt修饰的at
‑
msc对5fc的暴露随时间降低了细胞存活力)(图2a),从而表明了cdy转基因的转磷酸核糖基酶域的功能性。此外,cdy::uprt_at
‑
msc表明在存在活性细胞毒性药物5fu的情况下,自杀作用敏感性提高。这种作用可能是由于uprt转基因的活性,其催化5
‑
fu向5
‑
氟脲嘧啶单磷酸盐转化[25]。这与其它研究中的观察结果一致[19,55]。
[0480]
lpei基转染方法不影响at
‑
msc的表型特征。
[0481]
对于未来的临床使用,期望确保at
‑
msc的质量在基因修饰后保持无变化。为了排除可以以at
‑
msc的质量为代价实现at
‑
msc的高转染效率的可能性,根据国际细胞疗法协会(isct)所定义的主要标准评价特征[51]。的确,为了研究高转染可能改变at
‑
msc表型的可能性,使用如国际细胞疗法协会(isct)所定义的标志物,通过标准facs分析进行cdy::uprt_at
‑
msc的免疫分型[51]。以未修饰的at
‑
msc作为参照,我们通过标准facs分析对于表面标志物表达中的潜在变化分析了cdy::uprt_at
‑
msc的免疫表型。与未修饰的at
‑
msc相比,cdy::uprt_at
‑
msc显示出相同的谱图。据发现两种细胞类型对于cd90、cd73和cd105是阳性的,而对于cd14、cd20、cd34、cd45和hla
‑
dr是阴性的(图3a,27b)。cdy::uprt的表达不影响at
‑
msc向成骨(图3b,27a)和生脂谱系(图3c,27a)的分化能力。明显地,表达cdy::uprt::gfp的at
‑
msc中的油滴的存在为转染后at
‑
msc的分化潜能提供了直接证据(图11)。油滴表明分化成生脂谱系的潜能不受转染和转基因表达的影响(图11)。在另外的研究中,使用这种方法,在转染后软骨形成分化也不受影响(未显示)。
[0482]
为了检验生产cdy::uprt的at
‑
msc的表型,根据生产商的说明书,用msc分型试剂盒标记细胞,试剂盒由抗体cd73、cd90、cd105、cd14、cd20、cd34、cd45和hla
‑
dr组成
(miltenyi biotech)。此后,用facs分析标志物的表达。高质量msc群体包括>95%的cd90、cd105和cd73阳性细胞。表达cd14、cd20、cd34、cd45和hla
‑
dr的群体可以小于1%[51]。通过其分化为成骨和生脂谱系的能力确认了at
‑
msc的多潜能性[52,53]。用stempro
tm
成骨分化试剂盒和stempro
tm
脂肪形成分化试剂盒(thermofisher scientific)诱导at
‑
msc的分化。将未修饰的at
‑
msc用作对照。产生cdy::uprt的at
‑
msc的表型和分化潜能可以与未修饰的at
‑
msc无显著差异。
[0483]
cdy::uprt_at
‑
msc保留了对癌细胞系的体外趋向性
[0484]
对于成功的肿瘤细胞靶向,at
‑
msc对癌细胞释放的细胞因子的趋化性反应是所期望的[10]。因此,期望基因修饰不改变at
‑
msc对癌细胞的趋向性。在本文中,使用侵袭测定检验lpei基转染对at
‑
msc的肿瘤趋向性的潜在影响。研究了在存在癌细胞的情况下,at
‑
msc通过胞外基质的载体迁移(vectorial migration)。at
‑
msc通过胞外基质的方向性迁移表明了at
‑
msc对癌细胞的趋向性。通过mda
‑
231
‑
mb、u251
‑
mg和mkn1显著诱导了at
‑
msc通过胞外基质的侵袭,但是通过hek293t不行(图4a)。该观察结果与将hek293t用作非癌性细胞系对照的其它研究一致[56,57]。迁移的at
‑
msc和cdy::uprt_at
‑
msc的相当的数目表明肿瘤归巢能力不受lpei介导的转染和cdy::uprt在at
‑
msc中的表达的影响。为了进一步确认at
‑
msc的迁移取决于癌细胞分泌的特异性趋化因子,我们假设更多的癌细胞数目应导致迁移性at
‑
msc的增加。的确,显著更高数目的cdy::uprt_at
‑
msc朝u
‑
251mg和mkn1迁移。另一方面,在使用mda
‑
mb
‑
231细胞系的条件下发现迁移的at
‑
msc的适度增加(图4)。通过胞外基质侵袭的cdy::uprt_at
‑
msc的数目取决于癌细胞的数目,其中朝u
‑
251mg和mkn1迁移的细胞数目较多,但朝mda
‑
mb
‑
231细胞系迁移的较少(图4)。
[0485]
对于每个处理,将at
‑
msc、mkn1、mkn45、mda
‑
mb
‑
231(10,000个细胞每孔)和u
‑
251mg(4000个细胞每孔)的四个重复在96
‑
孔板中铺板。24小时后,将培养基更换为含有不同浓度的5
‑
氟胞嘧啶(5
‑
fc,invivogen)或者5
‑
氟尿嘧啶(5fc,invivogen)的培养基。1至5天后,对板进行celltiter 96aqueous one solution细胞增殖测定(promega)。在490nm,通过分光光度法测量比色读数。结果表示为相对于无5
‑
fc或5
‑
fu的条件中的细胞(设置为100%)的细胞存活力百分比。
[0486]
细胞侵袭测定的示例性方法如下所示。使用bd biocoat
tm matrigel侵袭室(bd biosciences)确定at
‑
msc的肿瘤趋向性。将癌细胞系或hek293t细胞加载至24
‑
孔板的下孔中。24小时后,将无血清dmem中的未修饰的和产生cdy::uprt的at
‑
msc添加至侵袭室上。用1
×
pbs清洗下孔,充满无血清dmem,组装用于侵袭测定。在24h温育后,从小室(insert)内部除去非侵袭细胞和matrigel。用hoechst 33342(thermofisher scientific)对侵袭细胞染色并通过成像系统照相。对3帧中的细胞个数计数。
[0487]
cdy::uprt_at
‑
msc/5fc介导的体外细胞毒性
[0488]
cdy::uprt_at
‑
msc对靶细胞的细胞毒性作用的证实对于在用于前体药物癌症疗法的治疗诊断性msc的产生中lpei基转染/trafen的采用是重要的。通常通过mts测定评价胞嘧啶脱氨酶/5fc在增殖抑制中的影响。我们首先比较了cdy::uprt_at
‑
msc/5fc和5fu在神经胶质瘤、乳腺癌和胃癌细胞系中的抗
‑
癌效率(图12)。cdy::uprt_at
‑
msc/5fc和5fu相当的抗癌作用表明了在5fc向细胞毒性药物的转化中的高效率。以cdy::uprt_at
‑
msc比癌细胞的1:1比值,抗
‑
癌作用与5fu的直接药理学作用相当。为了进一步检验cdy::uprt_at
‑
msc/5fc的治疗潜能,以多种msc比癌细胞比值,将细胞与靶癌细胞直接共培养(图5a)。即使以1:50的cdy::uprt_at
‑
msc/5fc比u251
‑
mg、mda
‑
mb
‑
231和mkn1的共培养比值,分别可以观察到几乎57%、69%和89%的增殖抑制。混合培养的这种比值代表了癌细胞内2%的治疗性细胞。当使用10%的治疗性细胞时,可以在所有癌细胞中获得大于86%的增殖抑制。值得注意地,在mkn1群体中仅用1%的治疗性细胞观察到了85%的增殖抑制。在无5fc的共培养中未观察到增殖抑制,表明at
‑
msc缺乏抗
‑
癌性(图5b)。
[0489]
直接共培养方法的示例性方法如下所示。将胃癌细胞系和乳腺癌细胞系(5000个细胞)和u
‑
251mg(2000个细胞)的四个重复在96
‑
孔板中铺板。5小时后,以1个at
‑
msc比1、5、10、50和100个癌细胞的比值,将提高数目的未修饰的或者产生cdy::uprt的at
‑
msc加入至癌细胞培养中。1天后,将培养基更换为补充有2%fbs,具有或不具有5
‑
fc(0
‑
150μg/ml)的dmem。5天后,通过增殖测定测量细胞存活力。将无5
‑
fc条件设置为100%。
[0490]
参考到其中治疗性细胞可能不直接体内接触癌细胞的情况,使用间接共培养实验来评价cdy::uprt_at
‑
msc/5fc的细胞毒性作用。在mda
‑
mb
‑
231对cdy::uprt_at
‑
msc/5fc暴露后4天,观察到接近90%的增殖抑制(图5c)。在不存在细胞
‑
细胞接触的情况下,cdy::uprt_at
‑
msc/5fc的抗癌效率与直接共培养模型高度相当。总体上,这些数据表明当治疗性细胞接触或接近靶细胞时,可以发挥有效的细胞毒性抗癌作用。我们然后扩大研究以比较正常混合的胃细胞(hs738
‑
未转化的人胎儿胃/肠细胞)对5种胃癌细胞系的敏感性。cdy::uprt_at
‑
msc/5fc对胃癌细胞系选择性发挥细胞毒性抗癌作用(图13),这表明了治疗性细胞/5fc对癌细胞的特异性靶向,但是对正常细胞不行。
[0491]
间接共培养方法的示例性方法如下所示。将mb
‑
mda
‑
231细胞在24
‑
孔板上铺板(5
×
104个细胞每孔)。将at
‑
msc或者cdy::uprt_at
‑
msc(5
×
104个细胞每孔)在转板(corning,c05/3422)上铺板。培养6h后,将具有治疗性细胞的小室转移至具有mb
‑
mda
‑
231细胞系,具有或不具有5fc的孔中。在温育4天后评价细胞毒性作用。除去转板,将培养基替换为含有1μg/ml hoechst 3222的1
×
pbs。使用synergy h1酶标仪,以分别为358nm和461nm的激发和发射波长分析染色的细胞。以80的增益设置,记录9个细胞培养区域的rfu。
[0492]
lpei/trafen增强子产生了高效cdy::uprt_at
‑
msc
[0493]
我们假设自杀基因的高表达在产生治疗性at
‑
msc的过程中可以是重要的。然后,我们比较了使用已通过用pcmv
‑
gfp(图8)或者编码cdy::uprt的无cpg质粒(图1)转染at
‑
msc测试的不同规程处理的治疗性细胞的效力。如所期望的,使用不同规程制备的治疗性细胞的抗癌效率高度依赖于每个规程的转染效率(图6)。在存在trafen(增强子)的情况下产生的cdy::uprt_at
‑
msc的抗癌效率显著超过其它方法,特别是在mb
‑
mda
‑
231和u251
‑
mg中。这两种细胞系表现出对5fu毒性的不良敏感性(图12)并且需要高浓度的5fu来抑制它们的增殖。以1个msc比10个癌细胞的比值,在与在存在trafen的情况下产生的cdy::uprt_at
‑
msc共培养的所有癌细胞系中观察到了完全的增殖抑制。值得注意地,本文所描述的当前的方法适用于得自多种来源的msc(图14)和其它自杀基因,如单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)(图15)。不考虑细胞来源(脂肪细胞、骨髓或脐带来源的msc;图14),使用增强子的转染规程产生了具有类似效力的修饰的msc。此外,我们具有用另一种自杀基因,如单纯疱疹病毒
‑
1胸苷激酶(hsv
‑
tk)成功转染的msc(图15)。
[0494]
通过瞬时转染,cdy::uprt在at
‑
msc中的长期表达
[0495]
基于msc的体内生物分布的证据,基于施用途径和肿瘤位置,预计msc需要1至4天来在残余肿瘤中铺展,并且归巢至远端肿瘤病灶[29,58,59]。为了确保自杀基因的持续表达,在用于前体药物疗法的修饰的msc的制备中采用了病毒转导和连续抗生素选择[19,29,60]。我们已证实cdy::uprt在转染后继续表达长达7天(图1c)。为了验证修饰的at
‑
msc在适合于显著抗癌作用的持续时间内保持功能性,在平行研究中确定了在转染后第1天和第7天采集的修饰的msc的癌症杀伤效率。通过在转染后第1天和第7天收获的cdy::uprt_at
‑
msc获得了相当的增殖抑制(图7)。
[0496]
在本研究中,故意选择无cpg质粒,因此使用无cpg质粒观察到了增强和延长的表达[61,62]。另外,在本研究中使用的质粒主链含有基质附着区(mar)以改善基因表达的稳定性[63]。的确,通过不具有这两个特征的表达质粒,cdy::uprt的表达在转染后第3天剧烈降低。如所期望的,在转染后第7天收集的修饰的at
‑
msc的抗癌效率比在转染后第1天收集的其对应物差400倍(图16)。尽管在存在trafen的情况下修饰的at
‑
msc显示出比其它方法强的抗癌效力,但是增殖抑制效率随时间降低,这表明瓶颈在于成功基因递送后的事件。值得注意地,trafen使得能够具有优良的抗
‑
癌效力,尽管修饰的at
‑
msc的功能性随时间减小。
[0497]
的确,通过瞬时转染,cdy::uprt在at
‑
msc中的延长表达是可能的。为了研究修饰的at
‑
msc中转基因的表达和功能的持续时间,检验了在转染后第1天和第7天收集的修饰的msc的抗癌效率。明显地,转基因cdy::uprt的表达在转染后7
‑
天的时间段内是显著的(图7c),这与使用cd::uprt::gfp的观察结果一致(图10)。通过在转染后第1天(图7a)或第7天(图7b)收获的cdy::uprt_at
‑
msc观察到了相当的癌细胞的增殖抑制。
[0498]
施用msc
[0499]
msc的施用方式可以取决于癌症类型和/或具体应用。本文描述了产生用于自杀前体药物疗法的治疗性msc的上游过程开发。可以(例如)静脉内施用修饰的msc以用于腺癌治疗(如treat
‑
me phase 1 trial,oncotarget.2017 oct 6;8(46):80156
–
80166中)。在某些实施方式中,可以进行颅内施用,例如,如在肿瘤切除或活组织检查期间,经历cd
‑
nsc的颅内施用的复发性高分级神经胶质瘤患者中的(clin cancer res.2017 jun 15;23(12):2951
‑
2960)。
[0500]
前体药物/基因组合。
[0501]
本研究进一步涉及胞嘧啶脱氨酶/尿嘧啶转磷酸核糖基酶和5
‑
氟胞嘧啶系统的融合基因。我们还测试了胸腺嘧啶核苷激酶/更昔洛韦系统。类似地,trafen方法在抗
‑
癌效率中优于其它非病毒方法(图14b)。本发明所描述的方法可以用作用于本领域中已知的自杀基因/前体药物系统的多种msc类型的非病毒修饰平台,如j clin invest.2000 may 1;105(9):1161
–
1167中的表3中所列的那些,该文献以其全部内容作为参考并入本文。
[0502]
非病毒与病毒转染的比较
[0503]
试剂制剂trafen(如pct/sg2013/000464中所描述的)使得在本文所描述的研究中具有高拷贝胞内质粒dna的转基因能够生产性表达,从而在本研究中导致产生了具有高治疗有效负荷的msc。trafen系统相对于病毒方法的显著优势在于转基因表达的早期发生(图23a)和单位细胞显著更高的表达(图23b)。这些特征可以使得能够缩短细胞制备过程并且使得单位细胞的自杀基因的有效负荷更高,从而潜在降低了用于治疗的生产成本和msc数
目。
[0504]
稳定细胞系的产生。
[0505]
稳定细胞系的产生设法获得高转染细胞数目。抗生素选择是高度劳动密集的(2
‑
3周)并且可能潜在损害msc质量[88],引起细胞衰老[88]并降低肿瘤趋向性[89]以及与病毒诱导的msc转化有关的安全性问题[90]。在不进行抗生素选择的情况下,不良的转染效率可能无法满足临床应用。考虑到使用本文所开发和描述的方法的成功的高转染,不再需要产生稳定细胞系。通过亚克隆至无cpg载体中的治疗基因,cduprt在7天时间段内的高表达在消除癌细胞中与转染后第1天的那些同样有效(图7a,7b)。尽管考虑到选择与本文所描述的转染方法相容,但是由于效率>80%,因此在本文中未选择转染的msc。在某些实施方式中,考虑对于疗法,稳定细胞系的产生是不必需的。
[0506]
cd::uprt_at
‑
msc介导的肿瘤体内生长抑制
[0507]
为了体内检验方法,将cd::uprt_at
‑
msc直接注射到皮下(s.c.)肿瘤中。对于未修饰的msc用于实验性癌症治疗的使用,规模相当的研究提出了正相反的结果(christodoulou等人,stem cell res ther,9(336),2018)。为了确保抗
‑
肿瘤作用是由于cd::uprt的表达,除了前体药物对照组(5fc)外,使用了细胞对照组(msc加5fc)。在当前研究中,msc加5fc不发挥显著的促肿瘤或抗
‑
肿瘤作用。在处理组中观察到了肿瘤生长的显著抑制(图32)。通过一个治疗循环,在最后一次5fc施用后3天,在处理组中观察到了肿瘤大小平均45%的减小(图32a中的第7天)。处理组中的整体肿瘤大小显著小于前体药物和细胞对照组(图32b)。
[0508]
这些结果提供了cdept在皮下小鼠模型
‑
成胶质细胞瘤细胞系u251mg中的效力的体内证据。
[0509]
讨论
[0510]
已在多项临床前[9,20,21]和临床试验[7,9]中证明了通过msc介导的gdept的治疗价值。由于替代性、非基于病毒的方法缺乏效力,因此将病毒用作msc基因修饰的方式。本研究证实了在不需要病毒的情况下,产生具有用于前体药物癌症疗法的高有效负荷的治疗诊断at
‑
msc的制剂和规程的使用。有效转染方法的有益结果在于产生cdy::uprt的at
‑
msc的抗
‑
癌效力的显著功能性改善。本文所描述的方法不改变at
‑
msc的质量和特征并且适用于多种类型的msc和治疗基因。
[0511]
基因递送方法在msc驱动的前体药物基因疗法的开发过程中起到重要作用。通常在临床前研究和临床试验[28
‑
31]中使用病毒载体以修饰msc用于gdept。经常地,在表达cdy的at
‑
msc的产生中使用反转录病毒[19,47,48,60,64]。简要地,连续3天用含有反转录病毒的培养基转导at
‑
msc三次。在使用之前,在细胞前,在存在抗生素g418的情况下进一步扩增细胞10天。替代方法是慢病毒基因递送系统。已报道通过以150的moi,在at
‑
msc中单次转导实现了高达80%的效率[65,66]。不幸地,聚凝胺可以潜在抑制msc增殖[67]。
[0512]
与病毒基因递送系统相比,我们报道了容易的pei基转染方法,该方法快速、简单、成本
‑
有效、无需抗生素选择或多次转染。由于可以用数千个dna拷贝转染每个细胞[35,36],发现大部分细胞表达高水平的所关心的基因(图1,8b)。这可以潜在解释cdy::uprt_at
‑
msc的高效力。的确,通过pei+增强子所产生的cdy::uprt_at
‑
msc显示出与5fu相当的抗癌效率,从而表明了前体药物5fc向毒性剂的有效转换(图13)。在转染后未进一步纯化或抗
生素选择的情况下,md
‑
mba
‑
231、u251
‑
mg、mkn45和mkn1细胞系的完全抑制是通过10%的治疗性at
‑
msc可实现的,at
‑
msc是在存在trafen的情况下通过pei基转染产生的(图6c)。另一方面,在at
‑
msc中单独的pei和lipofectamine3000较差的转染效率(图1)导致了低抗
‑
癌效率,特别是在md
‑
mba
‑
231和u251
‑
mg细胞系中(图6)。
[0513]
将使用本文所报道的方法制备的治疗性at
‑
msc的表型和特征很好地与其它研究所提供的数据进行比较,其中用反转录病毒修饰at
‑
msc以表达用于前体药物癌症疗法的cdy或cdy::uprt[19,47,48,60,64]。我们参考未修饰的at
‑
msc,评价了cd::uprt_at
‑
msc的表型标志物、分化潜能和肿瘤趋向性,从而确认我们的方法不影响at
‑
msc的质量(图3,4)。这是修饰的at
‑
msc的治疗诊断应用所期望的特征[51]。与其它报道一致[60,68,69],cdy::uprt_at
‑
msc能够在间接共培养实验中发挥细胞毒性(图5c),从而进一步确认旁观者效应不需要细胞
‑
与
‑
细胞的接触。类似地,值得注意的是治疗性msc对cdy::uprt/5fc系统敏感(图2),因此防止治疗性细胞的长时间存活;从而满足了作为细胞载体所期望的要求[69]。以1个治疗性msc比10个癌细胞的比例,kucerova等人证实了在存在通过反转录病毒转导所产生的cdy_at
‑
msc的情况下,mda
‑
mb
‑
231显著的40%的增殖抑制[47]。有趣地,通过cdy::uprt的at
‑
msc的修饰不会改善抗
‑
癌效力[16],从而导致产生了有关cdy::uprt/5fc和hsv
‑
tk/gcv的组合前体药物治疗的工作。意外地,我们观察到以10%的治疗性细胞,接近于91%的mda
‑
mb
‑
231增殖抑制(图5a),这可能是由于单位细胞的自杀基因的高表达水平所造成的。
[0514]
在cd表达神经干细胞的临床试验中,对复发性神经胶质瘤患者施用了11天的治疗方案[70]。在体内动物研究中,基于癌症类型,治疗持续时间在6[28,29,47]至23天[29]的范围内。通常,在3周的持续时间内,每周施用修饰的msc。在本研究中,我们成功证实在用pei加trafen转染的at
‑
msc中,在无抗生素选择的情况下,cdy::uprt延长表达长达7天(图1,7)。瞬时转染的at
‑
msc中自杀基因的表达在整个治疗方案所要求的持续时间内是可持续的[70]。这保证了非病毒基因递送系统在干细胞驱动的前体药物疗法的开发过程中的采用。明显地,用cd::uprt::gfp修饰的at
‑
msc显示出与cd::uprt相当的抗
‑
癌效率(数据未显示)。代替可以潜在影响msc质量的抗生素选择[40],可以将gfp标签用于cd::uprt阳性at
‑
msc的流式细胞术分离,这进一步使治疗诊断性细胞产生的工作流程流水线化。
[0515]
越来越多的基因和细胞疗法临床试验表明确保用于先前缺少治疗选择的疾病的这种治疗范式的出现的激动人心的时代。这种趋势已导致产生了对临床级病毒生产能力的更大需求。病毒产量不足已成为细胞和基因疗法发展和商业化的瓶颈[71]。本研究描述了用于聚合物基离体msc修饰的有用工具,其可以是高度可缩放且成本
‑
有效的。所提议的工作流程绕过了病毒载体供应中的限制并且促进了msc修饰,同时不会损害治疗诊断性msc的质量和抗
‑
癌效力。通过具有有效的非病毒基基因修饰工作流程,我们注意到这种方法可以用于容易地通过多个抗癌基因的共转染实现进一步治疗效果,其可以显著拓宽用于癌症治疗的治疗策略的范围。
[0516]
最近对于msc在肿瘤抑制和生长中的作用,出现了相矛盾的报道。据信这些矛盾很大程度上是由于技术差异和固有的生物不均一性所造成的。无论如何,认为基因修饰的msc可以提供更适合的癌症疗法策略,因为它们通常比不稳定且不均一的未处理过的msc更安全且更有效。
[0517]
本研究在不使用病毒的情况下,以高效率实现了用于产生用于前体药物癌症疗法的治疗诊断性at
‑
msc的at
‑
msc的成功修饰。用可商购的聚合物(pei
‑
max)转染了约一半的细胞群体并且在存在增强子的情况下,以低毒性显著改善了效率(图1a)。这种修饰方法不需要对于>70%的cd表达细胞的高表达msc进行纯化,也不需要抗生素选择,这与用于人临床试验的放行测试(release testing)一致。由于低转染效率或高细胞毒性,先前以有限的成功完成了开发新型阳离子聚合物和脂质以修饰msc的工作。最近,据报道聚(β
‑
氨基
‑
酯)(pbae)聚合物结构以高效率和低毒性转染msc。尽管很好地修饰了细胞,但是显著影响了迁移能力。
[0518]
为了使at
‑
msc用作用于疗法的靶向药物递送载体,期望用于修饰它们的方法不显著改变它们的表型特征和行为,包括它们的多潜能性和它们的迁移和侵袭能力。在修饰和天然at
‑
msc的表型标志物表达和分化潜能中无显著性差异(图3),这是修饰的at
‑
msc的治疗诊断学应用的基本准则。固有的肿瘤趋向性是作为用于治疗剂递送的细胞载体的msc的归巢/迁移性的重要特征。尽管转基因高度过表达,但是在体外存在癌细胞的情况下,修饰的细胞的迁移能力与天然msc相当(图4)。
[0519]
正在研究一些gdept系统用于癌症的治疗以改善常规癌症化疗的效力和安全性。在近期研究中所测试的酶/前体药物系统中,cd::uprt是最有效的并且已与干细胞一起用于临床试验。在本研究中,使用低至10%的治疗性at
‑
msc,cd::uprt修饰细胞有效抑制md
‑
mba
‑
231、u251
‑
mg、mkn45和mkn1细胞系的生长。值得注意地,以1个治疗性msc比10个癌细胞的比值,将mda
‑
mb
‑
231的增殖抑制了~90%(图5a)。以类似比值,kucerova等人证实当使用通过反转录病毒转导修饰的at
‑
msc时,相同细胞类型的增殖抑制仅为40%(kucerova等人,j gene med,10,1071
‑
1082,2008)。另一项研究报道在mda
‑
mb
‑
231与病毒转导的cdy::uprt_msc的共培养(以1个msc比4个癌细胞的比值)中细胞数目降低~60%(kucerova等人,stem cell research,8,247
‑
258,2012)。cd::uprt_at
‑
mscs/5fc的瘤内施用显示用非病毒方法修饰的msc能够发挥更高的抗
‑
癌作用(图32)。通过相当的研究设计,kwon等人(kwon等人,clinical and experimental otorhinolaryngology,6,176
‑
183,2013)和nouri等人(nouri等人,journal of controlled release:official journal of the controlled release society,200,179
‑
187,2015)分别报道以1和6个剂量的cd_msc/5fc治疗循环,肿瘤生长抑制,但不消退。不希望受理论束缚,考虑使用本文所开发的方法的修饰可以导致有效负荷提高,从而导致癌细胞更有效的杀伤。
[0520]
msc介导的cd/5fc治疗已建议作为克服5
‑
fu全身性毒性的策略(you等人,journal of gastroenterology and hepatology,24,1393
‑
1400,2009;kwon等人,clinical and experimental otorhinolaryngology,6,176
‑
183)。在本文的整个体内研究中,我们未观察到如其它研究中已显示的受试者体重的显著变化或其它直接副作用(数据未显示)(you等人,journal of gastroenterology and hepatology,24,1393
‑
1400,2009;kwon等人,clinical and experimental otorhinolaryngology,6,176
‑
183)。由于全身性毒性的减轻,cd
‑
msc的反复注射可以有可能提高抗肿瘤活性。另外,值得注意的是治疗性msc对cdy::uprt/5fc系统敏感(图2),因此限制了治疗性细胞的存活;从而满足了“击中即跑(hit and run)”策略的基本要求,不会留下细胞载体的痕迹(mohr等人,cancer letters,414,239
‑
249,2018)。
cancer institute,singapore))友好提供了胃癌细胞系mkn1和mkn28。在补充有10%fbs的rpmi(roswell park memorial institute medium,thermo scientific)中培养胃癌细胞系。将细胞保持在37℃,加湿气氛和5%co2中。
[0528]
含有cd::uprt的无cpg表达质粒的构建
[0529]
表达融合的胞嘧啶脱氨酶和尿嘧啶转磷酸核糖基酶的质粒dna(pdna)(4265bp pselect
‑
zeo
‑
fcyfur(https://www.invivogen.com/pselect
‑
zeo
‑
fcyfur))购自invivogen。通过如所描述的交叉铺设体外组装(cliva)克隆技术进行cd::uprt的无cpg表达质粒的构建[50]。简要地,使用pselect
‑
zeo
‑
fcyfur作为聚合酶链反应(pcr)中的模板,用cd::uprt替代质粒pcpgfree
‑
lucia(invivogen)中的lucia(https://www.invivogen.com/pcpgfree)。如所说明的,在抗生素zeocin的选择下,在大肠杆菌(escherichia coli)dh5αgt115株(invivogen)中增殖所有pdna。根据生产商的说明书,通过e.z.n.a.endo
‑
free plasmid maxi试剂盒(omega bio
‑
tek)纯化质粒。
[0530]
转染程序
[0531]
一般离心方法:
[0532]
在3
‑
5代,将所关心的转基因引入at
‑
msc。对于每个孔(6
‑
孔板形式),将5mg/ml lpei(pei max,polyscience)以不同的pdna和peimax的比值加入至无血清dmem中的pdna中。在某些实施方式中,基于所选择的聚合物,n/p比可以在5
‑
100的范围内。以100μl的总体积,将混合物在室温下温育15min。根据pdna的量,μg:1mg/ml lpei的体积,μl计算pdna:lpei的比值。然后,将lpei/pdna复合物添加至无血清dmem培养基(1:20)以制备转染混合物。除去培养基并用转染混合物替换,然后以200g轻轻离心5min。在离心后,除去转染混合物并用补充或未补充trafen的完全培养基替换。trafen由dope/chems(polar avanti lipid)和伏立诺他(saha,bio vision)组成。比值为9:2,并且以1.25μm使用saha。分析前,将细胞温育24h。
[0533]
上述规程通常是用于图1
‑
16、20和23中所提供的实验的方法的示例/代表,其允许基于具体实验进行改变和修改。
[0534]
一般非离心方法:
[0535]
细胞制备:将细胞扩增
‑
培养基从冰箱中取出并在室温下加温。msc小瓶(0.5m细胞/小瓶)得自液氮杜瓦瓶并通过剧烈搅拌在37℃水浴中快速融化。注意:在某些实施方式中,msc可以得自不同来源/物种/供体。在转移至生物安全柜之前,对小瓶孔喷雾70%的乙醇。将细胞无菌转移至15ml离心管。将4ml培养基缓慢添加(滴加)至细胞。以200
×
g离心5min。在不扰动细胞小粒的情况下,小心除去上清液。将细胞在10ml培养基中再混悬。良好混合并将细胞接种到1
×
t75容器中。在收获前,使用显微镜观察细胞生长以确保培养达到>80%汇合。2天后,细胞随时可使用。
[0536]
细胞制备:细胞生长
‑
为了收获细胞,将容器转移至生物安全柜并除去用过的培养基。除去培养基并用3ml 1
×
pbs漂洗一次。吸出清洗溶液。将2ml accutase加入至烧瓶,并在37℃温育(3
‑
5min)。轻敲以用于从烧瓶表面去除保留的细胞。添加4ml新鲜培养基以使accutase活性淬灭。将细胞混悬液转移至15ml离心管。以200
×
g离心5min。吸出上清液并将细胞与10ml新鲜培养基再混悬。良好混合并将0.1ml细胞转移至微量离心管以用于细胞计数。细胞计数应在0.1
‑1×
106个细胞/ml的范围内。可以将细胞混悬液继代培养,冷冻或用
于产生表达cd::uprt的msc。对于继代培养,将5000
‑
7000个细胞/cm2的细胞培养物表面接种。因此,加满新鲜培养基。细胞可以继代培养多达6代。对于冷冻保存,将细胞以200
×
g离心5min。吸出上清液,并将细胞以1m个细胞/ml在kbm banker2中再混悬。将500μl混悬液细胞等分至每个小瓶。
[0537]
表达cd::uprt的msc的产生:细胞接种
‑
msc的最优汇合是~60%。在转染前24小时接种细胞。注意:trafen
tm
试剂在存在血清和抗生素的情况下是稳定的。可以在整个实验期间使用标准培养基。对于10m细胞的制备,转染前24小时在至少2
×
t175中接种的建议细胞数目:
[0538]
细胞类型
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
细胞密度
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
培养基
[0539]
任何msc类型
ꢀꢀꢀꢀꢀꢀꢀꢀ1×
t175中2
‑
3m
ꢀꢀꢀꢀꢀꢀꢀꢀ
20ml
[0540]
*由于不同的msc生长速率,细胞数目可以不同。
[0541]
表达cd::uprt的msc的产生:dna转染规程
[0542]
试剂的制备:
[0543]
促融脂质(fusogenic lipid),第一试剂:作为1
×
工作储液提供。hdaci,第二试剂:将hdaci溶解在dmso之中。等分并将稀释溶液储存在
‑
20℃。
[0544]
步骤1:复合
[0545]
将9
‑
50μg的dna在1500μl的复合缓冲液中稀释。涡旋5秒以混合。将阳离子聚合物添加至稀释的dna。涡旋5秒以混合。*将1.5
‑
30μg聚合物添加至1μg dna。将转染混合物在室温下温育15min。
[0546]
步骤2:trafen混合物的制备
[0547]
在转染混合物的温育期间,将0.2
‑
1mg第一试剂和第二试剂合并。通过移液快速混合。不要涡旋。在室温下温育10
‑
20min。
[0548]
步骤3:转染
[0549]
将2500μl新鲜培养基添加至转染试剂/dna混合物。将4200μl转染试剂/dna混合物滴加至培养容器。在添加转染试剂/dna之前,不从细胞除去生长培养基。
[0550]
将trafen混合物滴加至培养容器。
[0551]
前后并从一侧到另一侧轻轻摇动培养容器以混合。
[0552]
将培养容器放回温育箱。
[0553]
转染后24小时使用细胞。
[0554]
表达cd::uprt的msc的产生:细胞收获
‑
[0555]
为了收获细胞,将容器转移至生物安全柜并除去用过的培养基。除去培养基并用10ml 1
×
pbs漂洗一次。吸出清洗溶液。将5ml accutase加入至烧瓶,在37℃温育(3
‑
5min)。轻敲以从烧瓶表面去除保留的细胞。添加10ml新鲜培养基以使accutase活性淬灭。将细胞混悬液转移至15ml离心管。良好混合并将0.1ml的细胞转移至微量离心管以用于细胞计数
‑
每个烧瓶的总细胞数目可以为~5m。以200
×
g离心5min。吸出上清液并将细胞与10ml 1
×
pbs再混悬。以200
×
g离心5min。吸出上清液并将细胞与10ml 1
×
pbs再混悬。以200
×
g离心5min。吸出上清液。将细胞在剩余的pbs中再混悬。细胞随时可使用。
[0556]
上述规程通常是用于图17
‑
19、21
‑
22、24和26
‑
27中所提供的实验的方法的示例/代表,其允许基于具体实验进行改变和修改。通常在约1.9至约75cm2的培养容器中进行所
指明的附图中所提供的实验。
[0557]
在某些实施方式中,如以上所描述的那些的非离心方法可以适合于大规模msc修饰。例如,约175cm2表面积的大规模操作可以适应于这些非离心方法。
[0558]
表达分析
[0559]
如前所描述,实施流式细胞术、免疫印迹和免疫细胞化学[49]。
[0560]
流式细胞术:通过attune nxt流式细胞仪系统(thermofisher scientific)定量荧光阳性细胞百分比并使用invitrogen attune nxt软件(thermofisher scientific)分析原始数据。
[0561]
成像:使用配备有用于观察dapi(ex357/em447)、gfp(ex470/em510)荧光的3个荧光立方体的evos fl细胞成像系统(thermofisher scientific)采集细胞图象。
[0562]
免疫印迹:分别用绵羊抗
‑
cdy(pa185365,thermofisher scientific)和单克隆抗
‑
β
‑
肌动蛋白(a2228,sigma
‑
aldrich),通过免疫印迹技术分析样品。
[0563]
免疫细胞化学:用绵羊抗
‑
cdy和alexa fluor 488驴抗
‑
绵羊荧光第二抗体(a11015,thermofisher scientific)标记样品。使用evos fl细胞成像系统进行图象采集。使用相同光学设置采集所有图像。
[0564]
产生cdy::uprt的at
‑
msc的表征和分化潜能
[0565]
为了检验生产cdy::uprt的at
‑
msc的表型,根据生产商的说明书,用msc分型试剂盒标记细胞,试剂盒由抗体cd73、cd90、cd105、cd14、cd20、cd34、cd45和hla
‑
dr组成(miltenyi biotech)。此后,用facs分析标志物的表达。高质量msc群体包括>95%的cd90、cd105和cd73阳性细胞。表达cd14、cd20、cd34、cd45和hla
‑
dr的群体将小于1%[51]。通过其分化为成骨和生脂谱系的能力确认了at
‑
msc的多潜能性[52,53]。用stempro
tm
成骨分化试剂盒和stempro
tm
脂肪形成分化试剂盒(thermofisher scientific)诱导at
‑
msc的分化。将未修饰的at
‑
msc用作对照。产生cdy::uprt的at
‑
msc的表型和分化潜能不应与未修饰的at
‑
msc显著不同。
[0566]
细胞存活力测定
[0567]
对于每个处理,将at
‑
msc、mkn1、mkn45、mda
‑
mb
‑
231(10,000个细胞每孔)和u
‑
251mg(4000个细胞每孔)的四个重复在96
‑
孔板中铺板。24小时后,将培养基更换为含有不同浓度的5
‑
氟胞嘧啶(5
‑
fc,invivogen)或者5
‑
氟尿嘧啶(5fc,invivogen)的培养基。1至5天后,对板进行celltiter 96 aqueous one solution细胞增殖测定(promega)。在490nm,通过分光光度法测量比色读数。结果表示为相对于无5
‑
fc或5
‑
fu的条件中的细胞(设置为100%)的细胞存活力百分比。
[0568]
体外药物敏感性
[0569]
对于每个处理,将at
‑
msc、mkn1、mkn45、mda
‑
mb
‑
231(10,000个细胞每孔)和u
‑
251mg(5,000个细胞每孔)的四个重复在96
‑
孔板中铺板。24小时后,将培养基更换为含有不同浓度的5
‑
氟胞嘧啶(5
‑
fc,invivogen)或者5
‑
氟尿嘧啶(5fc,invivogen)的培养基。1至5天后,对板进行celltiter 96 aqueous one solution细胞增殖测定(promega)。在490nm,通过分光光度法测量比色读数。结果表示为相对于无5
‑
fc或5
‑
fu的条件中的细胞(设置为100%)的细胞存活力百分比。
[0570]
产生cdy::uprt的at
‑
msc的体外抗癌效力
[0571]
直接共培养:将胃癌细胞系和乳腺癌细胞系(5000个细胞)和u
‑
251mg(2000个细胞)的四个重复在96
‑
孔板中铺板。5小时后,以1个at
‑
msc比1、5、10、50和100个癌细胞的比值,将提高数目的未修饰的或者产生cdy::uprt的at
‑
msc加入至癌细胞培养中。1天后,将培养基更换为补充有2%fbs,具有或不具有5
‑
fc(0
‑
150μg/ml)的dmem。5天后,通过增殖测定测量细胞存活力(商品化测定
‑
https://www.promega.sg/products/cell
‑
health
‑
assays/cell
‑
viability
‑
and
‑
cytotoxicity
‑
assays/celltiter
‑
96
‑
non_radioactive
‑
cell
‑
proliferation
‑
assay
‑
_mtt_/?catnum=g4000)。将无5
‑
fc条件设置为100%。
[0572]
间接共培养:将mb
‑
mda
‑
231细胞在24
‑
孔板上铺板(5
×
104个细胞每孔)。将at
‑
msc或者cdy::uprt_at
‑
msc(5
×
104个细胞每孔)在转板(corning,c05/3422)上铺板。培养6h后,将具有治疗性细胞的小室转移至具有mb
‑
mda
‑
231细胞系,具有或不具有5fc的孔中。在温育4天后评价细胞毒性作用。除去转板,将培养基替换为含有1μg/ml hoechst 3222的1
×
pbs。使用synergy h1酶标仪,以分别为358nm和461nm的激发和发射波长分析染色的细胞。以80的增益设置,记录9个细胞培养区域的rfu。将相对于对照(未转染的at
‑
msc和mb
‑
mda
‑
231细胞的共培养)计算处理后的增殖抑制。
[0573]
产生cdy::uprt的at
‑
msc的体内抗癌效力
[0574]
5至6
‑
周大的雌性裸鼠购自invivos并且在iacuc批准的规程下用于体内研究(r18
‑
1383)。通过异氟烷吸入使小鼠麻醉,并将在100μl dmem(50%matrigel)中混悬的5
×
106个替莫唑胺耐受性u
‑
251mg细胞s.c.注射在背侧区域(每只小鼠一个肿瘤)。通过数字卡尺监测肿瘤生长。当肿瘤测量的平均体积为80
‑
200mm3时,开始治疗。将所有小鼠随机分布到3个组中,每组包含5只小鼠。前体药物对照组每天接受前体药物的注射。细胞对照组接受1
×
106个msc的瘤内注射加前体药物的每天注射。处理组接受1
×
106个cd::uprt_at
‑
msc的瘤内注射加前体药物的每天注射。在第0天瘤内施用修饰或未修饰的msc(单一剂量)。1天后,小鼠接受500mg/kg 5fc的i.p.施用,连续4天。在细胞注射前(第0天)和msc施用后第7、11和15天,测量肿瘤尺寸和体重。
[0575]
细胞侵袭测定
[0576]
使用bd biocoat
tm matrigel侵袭室(bd biosciences)确定at
‑
msc的肿瘤趋向性。将癌细胞系或hek293t细胞加载至24
‑
孔板的下孔中。24小时后,将无血清dmem中的未修饰的和产生cdy::uprt的at
‑
msc添加至侵袭室上。用1
×
pbs清洗下孔,充满无血清dmem,组装用于侵袭测定。在24h温育后,从小室(insert)内部除去非侵袭细胞和matrigel。用hoechst 33342(thermofisher scientific)对侵袭细胞染色并通过成像系统照相。对3帧中的细胞个数计数。
[0577]
统计分析
[0578]
当使用学生t检验时,使用未配对的双尾检验,并假定读数变化呈正态分布。
[0579]
缩写
[0580]
msc:间质干细胞;pei:聚亚乙基亚胺;hdac6i:组蛋白脱乙酰酶6抑制剂;cd::uprt:融合的酵母胞嘧啶脱氨酶::尿嘧啶转磷酸核糖基酶;cd:胞嘧啶脱氨酶;gdept:基因
‑
导向的酶前体药物疗法;uprt:尿嘧啶转磷酸核糖基酶;fump:5
‑
氟脲嘧啶单磷酸盐;5fc:5氟胞嘧啶;5fu:5氟尿嘧啶;dpd:二氢嘧啶脱氢酶;oprt:乳清酸转磷酸核糖基酶;gfp:绿色荧光蛋白;at
‑
msc:人脂肪组织来源的msc;dmem:杜尔贝科改良的伊戈尔培养基;fbs:胎牛
血清;pdna:质粒dna;moi:感染复数;hsv
‑
tk:单纯疱疹病毒
‑
1胸苷激酶。
[0581]
实施例2
‑
用于msc高效转染的可缩放方法
[0582]
省略离心的方法可以改善可缩放性。然而,这可能导致温育时间延长并且如果未适当选择阳离子聚合物,则会出现困难。可以期望方法开发以对于每个msc供体/类型优化规程。在本研究中,我们已鉴别了使得能够产生用于治疗目的的临床有用的修饰的msc的重要步骤(图17)。表5中详细说明了规程所期望的特征的实例。
[0583]
表5:所期望的特征的实例
[0584][0585]
可缩放性
[0586]
msc前体药物基因递送的临床应用的重要方面在于基因递送方法的生产可缩放性。先前提高基于聚合物转染的离心的trafen的报道具有有限的可缩放性。本研究描述了多聚复合物和trafen向任何培养容器的直接添加,而不需要离心。本发明的发展的具体差异在于对聚合物和trafen的暴露持续时间。在这些研究中,可以将适合的聚合物和trafen制剂与细胞温育而无需离心。这可以提供生产缩放的方式,因为对大型容器离心的需要既不必需,也不方便(图24a
‑
b)。值得注意地,不考虑细胞培养的表面积,转染效率保持无变化,从而表明了将trafen的增强作用转化至临床前和临床规模的可行性。
[0587]
为了在这些高度转染的msc的生产中缩放,实施了阳离子聚合物和trafen的相容性并且相容性是所期望的。在示例性方法开发中(图17),评价了分子量(mw)范围在<4至200kda的多种直链(lpei)和支链(bpei)在不需要离心的情况下的高转染效率和低细胞毒性。基于msc类型、供体和培养条件,可以选择最适合的组合物,然后使用trafen进行增强(如图20中,例如)。因此,重要步骤在于可以通过trafen增强的这些相容性聚合物的鉴别。
[0588]
在某些实施方式中,可以对具体细胞类型选择或调整聚合物类型、聚合物结构(直链、支链)和/或聚合物尺寸。举例来说,图20中的结果表明图20a中的msc优选大聚合物,而图20b中的msc优选小聚合物。在某些实施方式中,msc可以包括uc
‑
msc,并且聚合物可以包括大于约50kda、约50至约200kda之间或者大于约200kda的聚合物。在某些实施方式中,msc可以包括bm
‑
msc,并且聚合物可以包括小于约50kda、约50kda至5kda之间或者小于约5kda的聚合物。在某些实施方式中,聚合物可以包括lpei。
[0589]
低毒性和高效率。
[0590]
参考图26,对于msc在前体药物疗法中的成功使用,细胞的健康是所期望的以确保msc在治疗期间的功能性。以小于350ng的dna的量,实现了最大转染效率(>90%),同时细胞
存活力无显著降低,如通过碘化丙啶排除测定所指示的(图26)。该数据表明了工作流程的实施方式在以高效率且同时不会在msc培养中引入显著的细胞毒性的msc修饰中的稳健性。
[0591]
dna优化(图17中的1)
[0592]
dna的量
[0593]
高dna的量可以导致细胞毒性。对于1.9cm2的表面积,显示用于msc转染的dna的范围在100至500ng的范围内。如图18所示,由于高细胞毒性,提高dna的量无益。
[0594]
dna载体设计
[0595]
使用适合的dna载体设计可以延长转基因表达。稳定细胞系的产生的目标是获得高转染细胞数。抗生素选择是高度劳动密集的(2
‑
3周)并且可能潜在损害msc质量[17],引起细胞衰老[17]并降低肿瘤趋向性[18]以及与病毒诱导的msc转化有关的安全性问题[19]。在不进行抗生素选择的情况下,传统方法的不良转染效率可能无法满足临床应用。
[0596]
考虑到使用本文所开发和描述的方法的成功的高转染,可以不必需产生稳定细胞系。通过亚克隆至无cpg载体中的治疗基因,cdy::uprt在7天时间段内的高表达在消除癌细胞中与转染后第1天的那些的有效性相当(图7)。使用无离心的转染规程观察到了延长表达(图19)。cdy::uprt的表达随时间显著降低,从而导致修饰的msc的抗
‑
癌效力减小(图16)。
[0597]
除了从质粒构建体除去一个或多个cpg岛之外,本发明的方法与本领域中已知的其它质粒的使用相容,例如,以下文献中所列的载体:jackson da等人,designing nonviral vectors for efficient gene transfer and long
‑
term gene expression,molecular therapy,14:613
‑
26,以上文献以其全部内容作为参考并入本文。适合的载体可以包括已显示导致延长表达的那些,如:骨架/基质连接区(s/mar)、附加型载体和含有ebna
‑
1的载体。
[0598]
聚合物优化(图17中的2)
[0599]
在本发明的实施方式中,可以将适合的聚合物与细胞温育而无需离心。这可以提供生产缩放的方式,因为对大型容器离心的需要既不必需,也不方便。将细胞和转染混合物的暴露和温育时间延长大于20分钟可以引入细胞毒性。某些聚合物可以在一定条件下显示出细胞毒性。例如,ho等人,enhanced transfection of a macromolecular lignin
‑
based dna complex with low cellular toxicity,biosci.rep.(2018)38:1
‑
9遇到了使用木质素
‑
pgea
‑
pegma的毒性。
[0600]
相容性聚合物与细胞类型的选择可以是所期望的以确保更高的质量和更高的转染效率。评价了分子量(mw)范围在2至200kda的多种直链(lpei)和支链(bpei)在不需要离心的情况下的高转染效率和低细胞毒性。基于msc类型、供体和培养条件,选择最适合的组合物,然后使用trafen进行增强。例如,图19显示<200kda和<5kda的lpei分别与脐带msc(uc
‑
msc)和bm
‑
msc相容。
[0601]
除聚合物的相容性之外,聚合物的量的优化可以提高细胞存活力。细胞毒性与聚合物和dna的量高度相关,并且量的增加导致了更高的毒性,如通过碘化丙啶(pi)阴性细胞的低%所指示的。当商品化聚合物的含量和浓度未知时(例如,使用试剂,如turbofect、polyfect和transficient),通过提高聚合物的量观察到了细胞存活力的降低。对于pei基聚合物,我们对1μg的dna测试了1μg
‑
30μg的范围内的聚合物的量(图20,21)。
[0602]
这些研究导致产生了可以包括以下步骤的msc高效转染的实例方法。
[0603]
细胞制备
[0604]
细胞扩增:将培养基从冰箱中取出并在室温下使其加温。从液氮杜瓦瓶获得msc小瓶(0.5m细胞/小瓶)并通过剧烈搅拌在37℃水浴中快速融化。msc可以得自不同来源/物种/供体。在转移至生物安全柜之前,对小瓶孔喷雾70%的乙醇。将细胞无菌转移至15ml离心管。将4ml培养基缓慢添加(滴加)至细胞。以200
×
g离心5min。在不扰动细胞小粒的情况下,小心除去上清液。将细胞在10ml培养基中再混悬。良好混合并将细胞接种到1
×
t75容器中。在收获前,使用显微镜观察细胞生长以确保培养达到>80%汇合。2天后,细胞随时可使用。
[0605]
细胞生长:为了收获细胞,将容器转移至生物安全柜并除去用过的培养基。除去培养基并用3ml 1
×
pbs漂洗一次。吸出清洗溶液。将2ml accutase加入至烧瓶,在37℃温育(3
‑
5min)。轻敲以从烧瓶表面去除保留的细胞。添加4ml新鲜培养基以使accutase活性淬灭。将细胞混悬液转移至15ml离心管。以200
×
g离心5min。吸出上清液并将细胞与10ml新鲜培养基再混悬。良好混合并将0.1ml细胞转移至微量离心管以用于细胞计数。细胞计数应在0.1
‑1×
106个细胞/ml的范围内。可以将细胞混悬液继代培养,冷冻或用于产生表达cd::uprt的msc。对于继代培养,将5000
‑
7000个细胞/cm2的细胞培养物表面接种。因此,加满新鲜培养基。细胞可以继代培养多达6代。对于冷冻保存,将细胞以200
×
g离心5min。吸出上清液,并将细胞以1m个细胞/ml在kbm banker 2中再混悬。将500μl混悬液细胞等分至每个小瓶。
[0606]
表达cd::uprt的msc的产生
[0607]
细胞接种:msc的最优汇合是~60%。在转染前24小时接种细胞。注意:trafen
tm
试剂在存在血清和抗生素的情况下是稳定的。可以在整个实验期间使用标准培养基。
[0608]
dna转染规程
[0609]
试剂的制备:促融脂质(fusogenic lipid),第一试剂:作为1
×
工作储液提供。hdaci,第二试剂:将hdaci溶解在dmso之中。等分并将稀释溶液储存在
‑
20℃。
[0610]
步骤1:复合
[0611]
将9
‑
50μg的dna在1500μl的复合缓冲液中稀释。涡旋5秒以混合。将阳离子聚合物添加至稀释的dna。涡旋5秒以混合。*将1.5
‑
30μg聚合物添加至1μg dna。将转染混合物在室温下温育15min。
[0612]
步骤2:trafen混合物的制备
[0613]
在转染混合物的温育期间,将0.2
‑
1mg第一试剂和第二试剂合并。通过移液快速混合。不要涡旋!在室温下温育10
‑
20min。
[0614]
步骤3:转染
[0615]
将2500μl新鲜培养基添加至转染试剂/dna混合物。将4200μl转染试剂/dna混合物滴加至培养容器。在添加转染试剂/dna之前,不从细胞除去生长培养基。将trafen混合物滴加至培养容器。前后并从一侧到另一侧轻轻摇动培养容器以混合。将培养容器放回温育箱。细胞在转染后24小时随时可使用。
[0616]
细胞收获:为了收获细胞,将容器转移至生物安全柜并除去用过的培养基。除去培养基并用10ml 1
×
pbs漂洗一次。吸出清洗溶液。将5ml accutase加入至烧瓶,在37℃温育(3
‑
5min)。轻敲以从烧瓶表面去除保留的细胞。添加10ml新鲜培养基以使accutase活性淬灭。将细胞混悬液转移至15ml离心管。良好混合并将0.1ml的细胞转移至微量离心管以用于
细胞计数
‑
每个烧瓶的总细胞数目应为~5m。以200
×
g离心5min。吸出上清液并将细胞与10ml 1
×
pbs再混悬。以200
×
g离心5min。吸出上清液并将细胞与10ml 1
×
pbs再混悬。以200
×
g离心5min。吸出上清液。将细胞在剩余的pbs中再混悬。细胞随时可使用。
[0617]
实施例3:开发用于任何多种msc细胞类型的转染方法和过程
[0618]
如图17中所描述的,一步或多步的过程优化可以是所期望的以通过经验鉴别trafen增强msc转染的条件。我们发现方法在以>70%的效率修饰的msc的修饰后,提供了提高的表达持续时间,可缩放性和msc质量。
[0619]
可以有效转染不同的msc类型。多种类型的msc已用于癌症疗法(表2)。已在用于前体药物基因疗法的多种msc类型(骨髓、脂肪来源的、脐带)中证实了当前的方法(图14b)。本文所描述的方法的实施方式可以用于任何msc类型,如脂肪msc、bm msc(如roosterbio)、uc msc(如atcc、cell applications)、脐带内膜msc(如cell research corporation)。
[0620]
用于任何多种msc的方法开发(图25)
[0621]
细胞差异度
[0622]
参考图14b,显示了来自不同来源的干细胞中相当的转染效率和抗癌效率。在存在trafen的情况下,用离心规程转染脂肪组织(at,roosterbio)、骨髓(bm,roosterbio)和uc(脐带,atcc)来源的msc。转染后24小时、将细胞胰蛋白酶化并采集用于免疫印迹分析(图14a)。将细胞裂解以用于使用靶向cdy和肌动蛋白的抗体的免疫印迹法分析。在相同实验中,收获细胞以用于与多种癌细胞系以1个msc比50个癌细胞的比值的共培养研究(图14b)。将细胞在含有100μg/ml 5fc的培养基中共培养5天。在温育结束时,通过以波长ex340/em488测量用hoechst 33342染色的细胞的rfu,通过分光光度法评价剩余的细胞数。将使用未修饰的msc的条件用作对照。因此计算增殖抑制百分比。图片表示从四个重复收集的数据,平均值+sem。
[0623]
trafen选择
[0624]
在某些实施方式中,trafen选择可以基于[1]转染效率、[2]细胞存活力或两者。
[0625]
在某些实施方式中,可以通过筛选聚合物文库来选择/优化trafen相容性转染试剂,聚合物文库可以包括可商购的聚合物,如turbofect(thermoscientific)、jetprime(poplyplus transfection)。pei是本文所识别的聚合物的实例。
[0626]
核酸构建体选择
[0627]
可以基于具体应用,选择和优化dna设计和量。在某些实施方式中,dna设计可以基于[1]表达持续时间、[2]细胞存活力或两者。可以基于[1]转染效率、细胞存活力或两者优化dna的量。
[0628]
阳离子聚合物选择
[0629]
可以基于具体应用,选择和优化阳离子聚合物。在某些实施方式中,可以通过筛选尺寸范围在约4至约200kda的可用的聚合物文库来选择和优化阳离子聚合物。
[0630]
可以基于具体应用,选择和优化聚合物的比例。在某些实施方式中,可以通过在约5
‑
100的n/p范围内测试来选择和优化聚合物比例。
[0631]
在某些实施方式中,可以基于转染效率和细胞存活力的平衡来选择聚合物类型和比例。
[0632]
在某些实施方式中,对于具体的基因
‑
基应用,可以进行选择和/或优化并且它们
可以包括聚合物筛选、dna的量的筛选和聚合物/dna比值的筛选。基于[1]转染效率>70%,[2]细胞存活力>70%的结果,筛选结果可以确定规程/工作流程。
[0633]
优化
[0634]
培养条件
[0635]
细胞生长(接种):msc的最优汇合可以为~60%。可以在转染前24小时接种msc。注意:trafen
tm
试剂在存在血清和抗生素的情况下是稳定的。可以在整个实验期间使用标准培养基。
[0636]
细胞健康:转染后,细胞存活力优选地为>约70%,如通过碘化丙啶测定所定义的。优选地,就分化潜能和表型标志物而言,应不显著损害细胞的量。
[0637]
细胞密度:在某些实施方式中,细胞密度通常可以在约60
‑
90%的范围内。可以(例如)以约60%、约70%、约870%或约90%的密度进行优化以检验转染。
[0638]
传代:在某些实施方式中,对于1
‑
25代的msc,效率可以是一致的,只要msc不衰老。
[0639]
dna转染优化:
[0640]
复合:举例来说,将9
‑
50μg dna在1500μl复合缓冲液中稀释。涡旋5秒以混合。将阳离子聚合物添加至稀释的dna。涡旋5秒以混合。*将1.5
‑
30μg聚合物添加至1μg dna。将转染混合物在室温下温育15min。可以对dna的量进行优化。对于约1.9cm2的表面积,dna的量可以在约100至约500ng之间改变。在某些实施方式中,在某些实例中,高于约500ng的dna的量可以对干细胞是有毒的。
[0641]
trafen混合物的制备:在转染混合物的温育期间,例如,将0.2
‑
1mg第一试剂和第二试剂合并。如将理解的,在某些实施方式中,第一和第二试剂的量和/或比例可以基于所使用的细胞类型而改变。例如,在某些实施方式中,促融脂质(fusogenic lipid)和辅助脂质的比例可以是不同的。第二试剂可以包括,例如,hdaci,其可以靶向hdac6。第一和第二试剂可以包括wo2014/070111中所描述的那些,该专利以其全部内容作为参考并入本文。
[0642]
转染培养基:举例来说,将2500μl新鲜培养基添加至转染试剂/dna混合物。转染培养基可以用于dna
‑
聚合物复合物的制备。可以将dna和聚合物添加至转染培养基,并且例如,温育约10至约45分钟。温育后,可以将转染混合物直接添加至细胞培养物。
[0643]
转基因
‑
载体设计:转基因载体可以是不同的。通常,转基因载体可以包括启动子和转基因。对于扩大或延长的表达,可以使用模块的添加,如除去cpg岛的密码子优化、s/mar和/或启动子优化。
[0644]
温育期:在某些实施方式中,可以在约37摄氏度进行温育约2至约48小时、或者约2至约24小时、例如。
[0645]
因此可以确定多种条件,包括dna的量、聚合物、细胞密度和trafen制剂的范围,并且可以对于msc来源进行调节。在方法优化后,产品可以包括具体的工作流程/规程,和在某些实施方式中,可以配制成试剂盒形式的优化试剂(质粒dna、聚合物、trafen)。这种方法可以在其它实验室和环境中提供方法的稳健性和/或有助于转染结果的优良可重复性。
[0646]
实施例4
‑
不旋转离心的规程的其它结果
[0647]
实施了对cd::uprt_msc对替莫唑胺耐受性成胶质细胞瘤模型的抗癌效率的研究(体外和体内数据)。msc来源是来自roosterbio的脂肪组织来源的msc(at
‑
msc)。
[0648]
与病毒修饰的msc用于癌症治疗的使用(eu临床试验登记号:2012
‑
003741
‑
15)的
放行标准一致,对于任何非病毒基因递送策略,相当水平的细胞修饰(>75%)和细胞存活力(>80%)是高度期望的。使用trafen介导的转染规程已显示出超过90%的转染效率以及超过80%的细胞存活力(图33a&33b,注意根据以上所描述的非离心规程制备修饰的msc,尽管可以使用离心和非离心规程两者)。这高于基于近期在临床试验中所使用的treatme
‑
1规程的80%的细胞存活力的所指明的阈值(niess,2015;von elnem,2019)。这在转染后2天实现,无需抗生素选择。另外,结果表明了转染效率与修饰的msc杀伤癌细胞的能力之间的相关性(图33c&33d)。这确认trafen可以用于产生成本
‑
有效、非专门设计类型(off
‑
the shelf
‑
style)、同种异体的msc
‑
gdept。
[0649]
一旦获得大量cd:uprt:gfp_msc,我们评价了修饰后的干细胞性质。按照isct所定义的特征(dominici,cytotherapy 8,315
‑
317,2006),修饰的msc应保留cd标志物表达和分化潜能。对于cd标志物的鉴定,超过95%的cd:uprt:gfp_msc表达cd73、cd90和cd105,并且小于2%的细胞表达cd14、cd20、cd34和cd45,这类似于未处理过的msc(图34a)。一旦诱导,表达cd:uprt:gfp的细胞能够分化成成骨和生脂谱系两者(图34b),这是与未处理过的msc类似的特征。通过at
‑
msc修饰,我们观察到在未处理过的和转染的at
‑
msc之间肿瘤趋向性无显著性差异(图34c)。发现相对于非癌性细胞,即成纤维细胞,at
‑
msc特异性迁移至癌细胞系。该数据表明作为肿瘤靶向的细胞载体,trafen介导的转染不影响msc表型。
[0650]
由于高复发性gbm概率和替莫唑胺(tmz)耐受性,多形性胶质母细胞瘤(gbm)的治疗是非常困难的并且是尚未实现的需要。可以潜在地作为tmz无反应者的第二线疗法提供cdept。在本文中,我们确定了非病毒cd:uprt:gfp_msc/5
‑
fc系统对替莫唑胺(tmz)耐受性神经胶质瘤细胞系是否有效。亲代和tmz耐受性u251
‑
mg细胞系两者对这些修饰的msc的敏感性与5
‑
fc相同(图35a)。共培养的细胞存活力以msc
‑
剂量依赖性方式升高。通过msc与癌症比例的降低,观察到了更高的细胞存活力,表明杀伤较少。在u87
‑
mg和u87
‑
mgtmzr40中发现了类似的观察结果(图35b)。然后,我们确定了这对于先前报道为tmz耐受性的hgcc患者来源的神经胶质瘤细胞系是否有效。类似地,我们观察到了hgcc神经胶质瘤细胞系的有效杀伤(图35c)。为了确定系统对非癌细胞的毒性,我们在成纤维细胞培养中检验了cd:uprt:gfp_msc/5
‑
fc的细胞毒性。有趣地,成纤维细胞较不敏感,即使以1:1的msc比成纤维细胞的比例,结果为47.78
±
2.93%(图35d)。
[0651]
然后,我们使用tmz耐受性细胞u251
‑
mg
tmzr40
研究了前体药物疗法在皮下小鼠模型中的体内细胞毒性作用。小鼠的cd:uprt:gfp/msc组显示通过单次瘤内施用细胞,肿瘤体积显著减小。早至处理后7天,处理的修饰的细胞和未处理过的细胞对照组之间的差异是显著的并且在15天的时间段内保持显著(图36a)。在处理后15天收获肿瘤,并且在未处理过的和修饰的msc之间观察到了肿瘤重量的显著性差异(图36b)。有趣地,在使用修饰的细胞时的剂量升高(0.5
×
106相对于1
×
106)显示出类似的肿瘤体积变化(图36a,灰色和浅蓝色圆形,和图36b)。本研究中的重要发现在于似乎无可观察的全身毒性,如通过动物体重变化所确定的(图36c)。在3个循环
‑
治疗完成后,观察到了长期肿瘤抑制(图37)。
[0652]
实施例5
‑
大型动物研究
‑
多点研究性研究
[0653]
实施了大型动物研究。所实现的本研究的主要目标如下所示:
[0654]
1)无显著副作用
[0655]
2)肿瘤尺寸减小
[0656]
3)生活质量改善
[0657]
4)寿命延长
[0658]
根据以上所描述的非离心规程,放大到500cm2培养容器中的20
‑
30m个细胞,制备修饰的msc细胞。
[0659]
动物参与不考虑肿瘤类型或狗的年龄。入选标准包括具有活组织检查数据的动物。排除标准包括不可以采样的肿瘤或者患者患有全身性疾病(如高热、免疫抑制、器官衰竭)。已在表现出多种癌症的狗和猫中证实了cdept的抗
‑
癌效力;包括淋巴瘤(淋巴结肿大)、甲状腺癌、黑素瘤、肛周癌、软组织肉瘤、鼻癌、胃肠癌、淋巴瘤(血液传播)。在所有情况下,在每个循环完成前后进行血液测试,bun和alkp值无显著变化。整体上,临床表现表明了如本文所描述的cd::uprt_msc/5fc的临床益处和优良的安全性谱。
[0660]
本文所提供的数据是每种癌症类型的代表性病例。图38显示了肛周癌数据,图39显示了口腔黑素瘤数据,图40显示了甲状腺癌数据,图41显示了软组织的软组织肉瘤(癌症溃疡)数据,图42显示了鼻肿瘤数据,并且图43显示了胃肠癌数据。
[0661]
图38显示了肛周癌治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2020年1月):存活,未报道复发。图39显示了口腔黑素瘤治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2020年1月):存活。图40显示了甲状腺癌治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2019年6月):存活。图41显示了软组织肉瘤(癌症溃疡)治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2018年11月):存活,未报到复发。14
‑
11
‑
2018的超声报告显示在左侧肛围区存在轮廓分明的低回声圆形块,测量为4
×3×
2cm。与周围或更深的器官无粘连。不存在转移,特别是在腰下淋巴结中。膀胱中存在少量1.5mm尿石,少数存在于前列腺部。其它器官正常。目前已完全缓解。图42显示了鼻肿瘤治疗数据。施用途径为狗cd::uprt::gfp_msc的瘤内注射。最近一次修订(2020年1月):存活。图43显示了胃肠癌治疗数据。施用途径为狗cd::uprt::gfp_msc的静脉内输注。最近一次修订(2019年7月):存活。根据超声报告,尽管存在二次生长,但是原始生长显著减小。这些研究的详细情况如以下治疗计划中,其中对于猫患者的差异在于使用了修饰的人msc。用提取自狗供体的脐带内膜msc或脂肪组织来源的msc处理所有狗患者。
[0662]
1.1cd::uprt::gfp_msc的剂量、时间表和施用途径的调整
[0663]
在动物研究中所研究的剂量在10
×
106至40
×
106个修饰的msc的范围内,并且所有剂量都良好耐受。对于身体
‑
表面肿瘤,通过直接瘤内注射安全施用了5
‑
20
×
106个治疗性细胞的剂量。对于内部块,通过直接静脉注射安全施用了30
×
106的剂量。用最大10
×
106(瘤内施用)和20
×
106治疗性msc(静脉内施用)组合处理具有身体
‑
表面和内部块两者的受试者。到目前为止,在研究性研究中未鉴别最大耐受剂量。
[0664]
本研究对于瘤内和静脉内施用分别使用了多至1ml和10ml的体积。
[0665]
1.2 5fc的剂量、时间表和施用途径的调整
[0666]
所研究的5fc剂量在20mg/kg/天至50mg/kg/天的范围内,并且所有剂量都良好耐受。本研究对于4
‑
天口服5fc疗程,使用了35
‑
50mg/kg/天的剂量。这对每个治疗循环重复。对受试者的主人描述了有关口服5fc制剂的信息。
[0667]
1.3研究持续时间和随访
[0668]
对接受治疗的受试者进行随访。跟踪本研究所招募的所有患者的存活。
[0669]
1.4研究终止的标准
[0670]
发起人和受试者的主人保留随时终止研究的权利。
[0671]
1.5治疗时间表和随访计划
[0672]
每次治疗的3个循环的持续时间为21天。
[0673]
1.6治疗指南
[0674]
由于受试者可能在多个位点(内部或表面)具有不同尺寸的实体瘤,因此研究者可以选择表1中的任何修饰的msc的剂量。当选择治疗时,研究者应考虑受试者的肿瘤尺寸和位点的临床记录。
[0675]
表:根据肿瘤的位点和尺寸,不同受试者的建议剂量
[0676][0677][0678]
1.7程序:瘤内注射
[0679]
1.用消毒剂小心擦拭身体表面10秒并风干。
[0680]
2.外科医生将约0.5ml治疗性细胞吸入注射器。将使用特细ii号短针(ultra
‑
fine ii short needle)(30g,胰岛素注射器)。注意:对于含有大量脓的肿瘤块,在开始注射治疗性细胞之前尽可能多的除去脓。
[0681]
3.将内容物完全从eppendorf管吸入注射器。
[0682]
4.对于身体
‑
表面肿瘤,通过向肿瘤多次注射施用治疗性细胞。尽可能地,以0.8cm的深度垂直于肿瘤块进行注射。注射如下所示的管的全部内容物:进行了多至5次约100μl的注射(总计0.5ml)。在~10秒内,缓慢注射适当体积的载体,并且在移除前,将针留在原位20
‑
25秒。缓慢移除针并重复注射,注意在整个肿瘤块上分布注射。
[0683]
1.8程序:静脉内施用
[0684]
以10ml,30min的流速进行静脉内施用。以5
‑
10ml的总体积制备治疗性细胞
[0685]
1.9 5fc的口服施用
[0686]
以胶囊剂的形式,每天两次口服施用5fc。
[0687]
研究中的程序和评价
[0688]
2.2.身体检查
[0689]
身体检查包括心和肺的听诊、腹部检查和淋巴结扪诊。如果有临床表现,则应进行其它系统的检查。
[0690]
2.3.生命体征
[0691]
每次访问时应记录体温和体重。
[0692]
2.4常规实验室评价
[0693]
实施具有分类计数和血小板计数以及化学组(包括电解质、bun、肌酐、估计的gfr[筛选时]、总胆红素、碱性磷酸酶、alt和ast、ldh和尿酸)的cbc。
[0694]
2.5.潜在不利作用的监测
[0695]
在每次回顾期间,应将有关不利作用的病征的问卷发布给受试者的主人。例如,应监测受试者的毛发脱落、皮疹、恶心、呕吐腹泻和食欲不振。
[0696]
实施例6
‑
trafen转染方法对msc来源不可知
[0697]
在本实施例中,在以下msc来源中证实了trafen对转染增强的作用:人:脐带来源、脐带内膜来源、脂肪组织来源和骨髓来源的msc。结果如图44所示。用含有gfp转基因的载体修饰来自不同商品化来源的msc类型。图片中的柱显示了如通过流式细胞术所测量的gfp+群体的%;在图45中,显示了狗脐带内膜
‑
来源和脂肪组织
‑
来源的msc的结果。成功修饰了来自不同来源的msc以表达cd::uprt::gfp。
[0698]
使用如以上所描述的非离心规程进行本研究。
[0699]
如图44所示,确定了多种msc类型中trafen的作用。(a)使用非离心规程,在存在或不存在trafen的情况下,通过pmaxgfp转染uc
‑
msc(cell applications)、ad
‑
msc和bm
‑
msc(roosterbio)。转染后1天,采集荧光图像。显示了代表性图象。(b)使用非离心规程,在存在trafen的情况下,通过pmaxgfp转染得自脐带内膜(cell research corp)、胎儿组织(bti collaborator)、脐带(cell applications)、狗脂肪(ivet animal hospital)、人脂肪和骨髓(roosterbio)的msc。转染后2天,收获细胞以用于facs分析。图片显示了表达gfp的细胞的百分比。
[0700]
图45显示了使用人脐带内膜(cell research corp)和人脂肪组织来源的(hayandra)结果。使用非离心规程,在存在trafen的情况下通过cd::uprt::gfp表达载体转染msc。转染后2天,采集明视野和荧光图像。此后,收获细胞以用于facs分析。facs谱证实了在这两种类型中不同的表达水平。
[0701]
实施例7
‑
修饰的msc细胞的放大产生
‑
平底和微珠培养系统
[0702]
在本实施例中,研究了修饰的msc细胞的产生的放大选择。开发了两种方法,第一种是平板系统,而第二种是微珠
‑
基培养系统。
[0703]
在平板转染系统中,使用相容性阳离子聚合物和增强子(例如,trafen)开发了转染方法以有效转染msc。通过提高烧瓶表面积,研究了该方法在修饰的细胞的生产中的可缩放性。据发现t175烧瓶(面积175cm2)的放大的线性度与ad
‑
和uc
‑
msc数目高度相关,其r2接近于1(图46a)。根据所获得的结果,通过t175烧瓶,可以获得接近3百万个gfp+细胞。此外,随着培养容器尺寸的增加,转染效率在大于90%保持无变化(图46b)。通过所建立的线性度,10
‑
层培养室(10
‑
stack chamber,6,360cm2)的尺寸应产生~1.1
×
108个细胞,这对于单次人患者治疗是足够的。为了证实这些发现,我们在
的一个室中进行转染(图46c)。明显地,放大转染以产生足够用于(例如)临床前或临床研究的修饰的细胞是可行的。
[0704]
使用如以上所描述的非离心规程进行本研究。
[0705]
图46显示了ad
‑
msc和uc
‑
msc在平底表面上的放大的线性度。使用非离心方法,在存在trafen的情况下,通过cd::uprt::gfp表达载体转染ad
‑
msc和uc
‑
msc。在不更换培养基的情况下,转染后2天收获细胞。(a)将转染的活细胞个数对容器表面积作图。(b)来自ad和uc
‑
msc的facs分析的gfp+%的代表性图象。(c)将狗脐带内膜msc(cell research corp)在不同容器中以15000至20000个细胞/cm2铺板。1天后,在存在trafen的情况下,通过cd::uprt::gfp多聚复合物转染细胞。在24h温育后,收获细胞。通过自动细胞计数器nc
‑
3000确定收集的细胞总数。用流式细胞术分析gfp表达。图片提供了从培养容器收集的多种细胞数目中的gfp阳性细胞的%。
[0706]
当msc对表面贴壁时,生物反应器/摇瓶中的生长可以使用微载体进行附着。在本实施例中所描述的微载体
‑
基(例如,微珠
‑
基)转染系统中,研究了一些微载体来确定对msc生长的相容性。这些微载体具有如所总结的不同性质,如它们的直径、密度、涂层和电荷(图47a)。通过测量细胞存活力,实施研究以鉴别不同微载体对于msc生长的相容性。未涂覆的微载体1和p plus 102
‑
l上的生长获得了最少数目的细胞(图47b),使用1,无活的msc,这与无微载体对照样品类似。msc在涂覆了1型猪胶原蛋白的微载体3上良好生长,从而在第5天产生了最高数目的细胞。因此,选择3用于进一步研究。转染细胞的数目与微载体的总表面积线性相关(图48)。有趣地,当使msc适应于混悬时,随着细胞数目的增加,gfp+表达%减少。由于烧瓶振荡速度可以影响可以物理阻挡对多聚复合物的暴露的细胞聚集,因此一旦接种,我们提高了msc的搅拌速度以使细胞更好地散布。将振荡速度提高至70rpm,转染了更多的细胞(>90%的表达gfp的细胞)。有趣地,在低细胞数目(1
×
106至3
×
106)的情况下,提高速度降低了细胞存活力,这可能是由于剪切应力的提高所造成的。这通过接种时提高细胞数目(4
×
106个细胞)得到减轻(图49)。在本研究中,从125ml三角瓶中的~150cm2的微载体上的50ml体积获得了~4
×
106个gfp+细胞。这些结果证实如果我们使用具有在1.25l体积中的~3750cm2的尺寸的4l三角瓶,则有可能将细胞密度提高至~1
×
108个细胞。
[0707]
使用如以上所描述的非离心规程,通过另外的振荡程序进行本研究。该规程的更多细节如下所描述:
[0708]
用于微载体培养的规程
[0709]
根据生产商的说明书,在多种微载体中培养ad
‑
msc(roosterbio),即c
‑
gen 102、pro
‑
f 102、p plus 102
‑
l(thermofisher)、1、微载体珠、3微载体珠(ge healthcare's life sciences)和ii微载体(corning)。简要地,在使用121℃高压灭菌30min之前,将微载体在pbs(20mg/ml)中水合。将1.9cm2的微载体表面用于24
‑
孔板。在培养ad
‑
msc之前,在使用前将微载体在完全培养基中在37℃平衡1h。然后,培养ad
‑
msc并接种到微载体上,以50或70rpm的搅拌速度用于生长和转染研究。
[0710]
图47显示了在ad msc中使用不同微载体的结果。(a)所使用的微载体的描述,(b)
将不同天,在不同微载体上生长的活细胞数作图。
[0711]
图48显示了从板上的微载体放大到烧瓶中的结果。根据总表面积,将人ad
‑
msc在微载体上铺板。随着培养中微载体数目的提高,获得了更大的表面积。将细胞以20
‑
40
×
10^3个细胞/cm3在微载体上铺板。1天后,在存在trafen的情况下,通过pmaxgfp多聚复合物转染细胞。转染后1天,收获细胞用于细胞计数和facs分析。(a)将转染的活细胞个数对容器表面积作图。(b)以4
×
放大倍数采集转染细胞的代表性图象。
[0712]
对于微载体转染,在转染前将ad
‑
msc以不同接种密度接种到24
‑
孔非贴壁板中的1.9cm23微载体上,并以50rpm的搅拌速度培养24h。类似于平底转染,在温育15min后,使用滴加方式将聚合物和dna复合物加入至细胞培养物。类似地,在多聚复合物添加前,将转染增强子补充至完全培养基。对于微载体(大规模)转染,将ad
‑
msc以多种表面积的优化细胞密度(20,000至40,000/cm2),并因此在125ml三角瓶中接种到3上,并对于转染使用50或70rpm的搅拌速度。在修饰的msc的温育和生产期间,搅拌速度是恒定的。
[0713]
实施例8
‑
通过非病毒方法修饰的msc显示出比通过慢病毒修饰的msc更高的抗
‑
癌效力
[0714]
本实施例旨在比较通过慢病毒和trafen介导的转染所产生的cd::uprt::gfp_msc的癌症杀伤效率。为此,在慢病毒感染后,通过抗生素选择产生了稳定表达cd::uprt::gfp的msc。荧光图像和流式细胞术分析(图50a,50b)表明在转导的msc(稳定表达cd::uprt::gfp)中转基因的整体表达水平显著较低。明显地,19%和25.7%的trafen修饰的群体分别表达中(蓝色)和高(橙色)水平的cd::uprt::gfp(50b)。在转导的msc群体中,<20%的群体表达中等水平的cd::uprt::gfp。因此,转导的msc具有较低的癌症杀伤效率,特别是以1个msc比50和100个癌细胞的比例。
[0715]
图50显示了通过慢病毒或者trafen介导的转染方法修饰的at
‑
msc的cd::uprt::gfp表达和抗癌效率的比较结果。(a)感染后3天,对msc进行1μg/ml嘌罗霉素选择2
‑
周。在建立稳定表达cd::uprt::gfp的msc后,设立另一组实验以通过trafen介导的转染产生cd::uprt::gfp_msc。转染后2天,采集修饰的msc的荧光图像。(b)此后,收获两个培养物并进行(b)facs分析和(c,d)共培养研究。图片中的柱表示通过mts测定获得的在1个msc比1、5、50、100个癌细胞的多种比值下的癌症杀伤效率。通过双尾学生t检验评价通过慢病毒相对于trafen方法产生的cd::uprt::gfp_msc的癌症杀伤效力的显著性差异;**,p
‑
值<0.005;*,p
‑
值<0.05。在共培养实验结束时,采集明视野图象。
[0716]
方法:
[0717]
非病毒msc:如使用如以上所描述的非离心规程所制备的。
[0718]
慢病毒:通过具有cd::uprt::gfp的慢病毒载体感染msc。在存在8μg/ml聚凝胺的情况下,以moi5通过慢病毒转导msc。感染后1天,用含有1μg/ml嘌罗霉素的新鲜培养基替换培养基1周。收获细胞以用于facs分析和共培养研究。
[0719]
表达谱。
[0720]
使用流式细胞术分析,通过cd::uprt::gfp_慢病毒和cd::uprt::gfp_trafen修饰的msc的表达谱是不同的。facs谱上的3种标志物表明了高(橙色)、中(蓝色)、低(醋栗色
(currant))表达。更高的表达表明更高的cd::uprt::gfp的有效负荷。通过trafen方法(非离心)修饰的msc导致产生了具有高cd::uprt::gfp表达(19%)的群体,但是慢病毒修饰的msc不行。更高的有效负荷可以导致更高的癌症杀伤效率。
[0721]
癌症杀伤效率
[0722]
与通过慢病毒修饰的msc相比,在使用通过trafen方法修饰的msc的细胞治疗中观察到了显著更高的癌症杀伤效率。
[0723]
实施例9
‑
病例研究:猫淋巴瘤,静脉注射表达cd::uprt::gfp的人脂肪组织来源的msc
[0724]
如下所示进行治疗性细胞的制备:转染人脂肪组织来源的msc)并冷冻保存。在施用当天,融化冷冻的cd::uprt::gfp_msc并在低温溶液中配制用于静脉内施用。由于以下理解:淋巴瘤具有潜在的骨髓参与,但没有可测量的明显物质,因此决定改善的优良指示将是pcv(细胞压积值)。在治疗持续期间,患者需要的输血次数从每周3次减少为每周0或1次。随着pcv值的升高,表明贫血症不太严重。另外,通过主人报告以下观察结果:
[0725]
1)更活跃
[0726]
2)恢复先前习惯,如在门口欢迎主人和与主人互动。
[0727]
3)食欲的改善。
[0728][0729]
本研究的治疗计划如以上所列的狗的治疗规程中所描述的。msc如以上所描述,使用非离心方法,在存在trafen的情况下修饰
[0730]
实施例10
‑
同情性使用:46岁复发性明细胞癌患者中表达cd::uprt::gfp的msc的瘤内注射
[0731]
对患有复发性明细胞癌的46岁患者进行同情性使用治疗。通过如本文所描述的表达cd::uprt::gfp的msc的瘤内注射治疗受试者。结果如图51中所示。
[0732]
从通过抽脂术获得的患者组织提取脂肪组织msc。在msc扩增后,在hayandra peduli's gmp机构中产生细胞库。优化转染规程。在存在trafen的情况下,在t225烧瓶中转染6m msc并且可以实现多至75%的转染效率。将trafen规程和试剂转移至gmp机构以用于产生用于同情性使用的cd::uprt::gfp_msc。
[0733]
在治疗当天收获修饰的msc并在2ml plasmalyte中制备。将20至50m个细胞瘤内注射到肿瘤块外周的20个位点。在msc施用后1天,通过口服向患者施用总计2000mg 5fc/天的5fc(每天4
×
500mg 5fc丸剂)。施用5fc 4天。
[0734]
每周重复msc和5fc施用循环。数据显示每个治疗循环后1周的肿瘤块照片以及疼
痛水平。
[0735]
实施例11
‑
离心相对于非离心方法
[0736]
图52提供了与非离心/旋转离心转染方法的实例(底部)相比,典型的离心/旋转离心
‑
基转染方法(顶部)的示意图。
[0737]
在离心/旋转离心
‑
基转染中,将细胞接种到容器中并添加dna/聚合物复合物。在所显示的实施例中,如所指示的,实施旋转离心步骤约5
‑
10分钟。然后除去培养基,并添加增强子(即trafen)。还提供了从以这种方式处理的脂肪组织来源的msc收集的数据。
[0738]
为了比较,在图52的底部显示了非离心/旋转离心转染方法。对这种方法显示了多种选择。在一个实施方式中,将细胞接种到容器中,添加dna/聚合物复合物并且还添加了增强子(例如,trafen),然后温育至少约24小时(在本文中未实施旋转离心/离心)。然后除去培养基,从而提供了转染的msc。显示了进行这种处理的脂肪组织
‑
来源的msc的数据。
[0739]
图52(底部)显示了非离心/旋转离心转染方法的另一种实施方式。在该实施方式中,接种细胞(上图
‑
平底培养容器;下图
‑
微珠),并且添加dna/聚合物复合物以及增强子(例如,trafen)。在振荡的同时,温育进行至少约24小时(还显示了其它选择,如生物反应器
‑
类型,包括(例如)转瓶、摇袋式生物反应器系统、旋转壁式生物反应器设计和搅拌罐式生物反应器设计)。然后,通过收集实施收获,接着旋转离心(例如,在所示实施例中,以约300g进行3mins)、添加1
×
pbs,然后再一次旋转离心(例如,在所示实施例中,以约300g进行3mins),然后添加胰蛋白酶,以100rpm振荡并用培养基淬灭。进行过滤,同时进行清洗(在所示实施例中,用1
×
pbs)。进行旋转离心(例如,在所示实施例中,以约300g进行3mins),并因此获得了用于下游分析的细胞小粒。显示了以这种方式处理的细胞的数据,并且提供了仅聚合物相对于聚合物+trafen的比较。
[0740]
实施例12
‑
冷冻保存的修饰的msc(使用trafen产生)是活的并且是功能性的
[0741]
图53提供了冷冻保存修饰的msc(使用trafen制备)以允许其长期储存的工作流程的示意图。如所示的,可以将修饰的msc置于冷冻保存储存。当需要时,可以从储存中移除细胞并通过在低温溶液中融化来准备用于使用。
[0742]
图54显示了如以上所描述的图53所示的冷冻保存,然后融化的修饰的msc的细胞存活力、表达水平和功能活性的结果。如所示的,在冷冻保存并在低温溶液中融化和维持最多72h之后,修饰的msc保留了高细胞存活力和表达水平。
[0743]
将细胞在低温溶液(hypothermosol)中融化并在4℃储存多至3天。每天测量细胞存活力和cd::uprt::gfp+细胞%。
[0744]
已通过举例说明描述了一种或多种说明性实施方式。本领域技术人员将理解可以在不背离如权利要求中所定义的本发明的范围的情况下做出一些改变和修改。
[0745]
参考文献
[0746]
1.sinden,j.d.,et al.,human neural stem cell therapy for chronic ischemic stroke:charting progress from laboratory to patients.stem cells and development,2017.26(13):p.933
‑
947.
[0747]
2.lalu,m.m.,et al.,safety of cell therapy with mesenchymal stromal cells(safecell):a systematic review and meta
‑
analysis of clinical trials.plos one,2012.7(10):p.e47559.
systemically delivered mesenchymal stem cells:tropism for brain tumors and biodistribution.int j nanomedicine,2016.11:p.13
‑
23.
[0804]
59.xie,c.,et al.,systemically infused mesenchymal stem cells show different homing profiles in healthy and tumor mouse models.stem cells transl med,2017.6(4):p.1120
‑
1131.
[0805]
60.cavarretta,i.t.,et al.,adipose tissue
‑
derived mesenchymal stem cells expressing prodrug
‑
converting enzyme inhibit human prostate tumor growth.mol ther,2010.18(1):p.223
‑
31.
[0806]
61.wongrakpanich,a.,et al.,the absence of cpg in plasmid dna
‑
chitosan polyplexes enhances transfection efficiencies and reduces inflammatory responses in murine lungs.mol pharm,2014.11(3):p.1022
‑
31.
[0807]
62.moritz,b.,p.b.becker,and u.gopfert,cmv promoter mutants with a reduced propensity to productivity loss in cho cells.sci rep,2015.5:p.16952.
[0808]
63.zhao,c.p.,et al.,matrix attachment region combinations increase transgene expression in transfected chinese hamster ovary cells.sci rep,2017.7:p.42805.
[0809]
64.altanerova,v.,et al.,human adipose tissue
‑
derived mesenchymal stem cells expressing yeast cytosinedeaminase::uracil phosphoribosyltransferase inhibit intracerebral rat glioblastoma.int j cancer,2012.130(10):p.2455
‑
63.
[0810]
65.meyerrose,t.e.,et al.,lentiviral
‑
transduced human mesenchymal stem cells persistently express therapeutic levels of enzyme in a xenotransplantation model of human disease.stem cells,2008.26(7):p.1713
‑
22.
[0811]
66.liu,y.,et al.,lentiviral
‑
mediated gene transfer into human adipose
‑
derived stem cells:role of nell1 versus bmp2 in osteogenesis and adipogenesis in vitro.acta biochim biophys sin(shanghai),2012.44(10):p.856
‑
65.
[0812]
67.lin,p.,et al.,polybrene inhibits human mesenchymal stem cell proliferation during lentiviral transduction.plos one,2011.6(8):p.e23891.
[0813]
68.khatri,a.,et al.,combination of cytosine deaminase with uracil phosphoribosyl transferase leads to local and distant bystander effects against rm1 prostate cancer in mice.j gene med,2006.8(9):p.1086
‑
96.
[0814]
69.abrate,a.,et al.,mesenchymal stemcells expressing therapeutic genes induce autochthonous prostate tumour regression.eur j cancer,2014.50(14):p.2478
‑
88.
[0815]
70.portnow,j.,et al.,neural stem cell
‑
based anticancer gene therapy:a first
‑
in
‑
human study in recurrent high
‑
grade glioma patients.clin cancer res,2017.23(12):p.2951
‑
2960.
[0816]
71.rader,r.cell and gene therapies:industry faces potential capacity shortages.2017.37.
[0817]
72.niess,h.,et al.,treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells(treat
‑
me1):study protocol of a phase i/ii clinical trial.bmc cancer,2015.15(1):p.237.
[0818]
73.aboody,k.s.,et al.,neural stem cell
‑
mediated enzyme/prodrug therapy for glioma:preclinical studies.sci transl med,2013.5(184):p.184ra59.
[0819]
74.tate,c.c.,et al.,human mesenchymal stromal cells and their derivative,sb623 cells,rescue neural cells via trophic support following in vitro ischemia.cell transplant,2010.19(8):p.973
‑
84.
[0820]
75.pollock,k.,et al.,human mesenchymal stem cells genetically engineered to overexpress brain
‑
derived neurotrophic factor improve outcomes in huntington's disease mouse models.mol ther,2016.24(5):p.965
‑
77.
[0821]
76.kaestner,l.,a.scholz,and p.lipp,conceptual and technical aspects of transfection and gene delivery.bioorganic&medicinal chemistry letters,2015.25(6):p.1171
‑
1176.
[0822]
77.charrier,s.,et al.,quantification of lentiviral vector copy numbers in individual hematopoietic colony
‑
forming cells shows vector dose
‑
dependent effects on the frequency and level of transduction.gene therapy,2011.18(5):p.479
‑
487.
[0823]
78.cooper,m.j.,et al.,safety
‑
modified episomal vectors for human gene therapy.proceedings of the national academy of sciences of the united states of america,1997.94(12):p.6450
‑
6455.
[0824]
79.batard,p.,m.jordan,and f.wurm,transfer of high copy number plasmid into mammalian cells by calcium phosphate transfection.gene,2001.270(1
‑
2):p.61
‑
8.
[0825]
80.cohen,r.n.,et al.,quantification of plasmid dna copies in the nucleus after lipoplex and polyplex transfection.journal of controlled release:official journal of the controlled release society,2009.135(2):p.166
‑
174.
[0826]
81.van der loo,j.c.m.,et al.,scale
‑
up and manufacturing of clinical
‑
grade self
‑
inactivatingγ
‑
retroviral vectors by transient transfection.gene therapy,2012.19(3):p.246
‑
254.
[0827]
82.vicente,t.,et al.,virus production for clinical gene therapy.methods mol biol,2009.542:p.447
‑
70.
[0828]
83.van der loo,j.c.and j.f.wright,progress and challenges in viral vector manufacturing.hum mol genet,2016.25(r1):p.r42
‑
52.
[0829]
84.kolata,g.,gene therapy hits a peculiar roadblock:a virus shortage.2017,the new york times.
[0830]
85.stanton,d.
‘
dramatic’gene therapy demand drives viral vector delays.2018.
[0831]
86.mammoser,g.virus shortage impacting gene therapy experiments.2017.
[0832]
87.challener,c.a.cell and gene therapies face manufacturing challenges.2017.
[0833]
88.kallifatidis,g.,et al.,improved lentiviral transduction of human mesenchymal stem cells for therapeutic intervention in pancreatic cancer.cancer gene ther,2008.15(4):p.231
‑
40.
[0834]
89.kyriakou,c.,et al.,factors that influence short
‑
termhoming of human bone marrow
‑
derived mesenchymal stemcells in a xenogeneic animal model.haematologica,2008.93(10):p.1457
‑
65.
[0835]
90.gwendal,l.and y.l.paula,recent discoveries concerning the tumor
‑
mesenchymal stem cell interactions.biochim biophys acta,2016.1866(2):p.290
‑
299.
[0836]
91.ramamoorth,m.and a.narvekar,non viral vectors in gene therapy
‑
an overview.j clin diagn res,2015.9(1):p.ge01
‑
6.
[0837]
92.baek,k.,et al.,gene transfection for stem cell therapy.current stem cell reports,2016.2(1):p.52
‑
61.
[0838]
93.haleem
‑
smith,h.,et al.,optimization of high
‑
efficiency transfection of adult human mesenchymal stem cells in vitro.mol biotechnol,2005.30(1):p.9
‑
20.
[0839]
94.kim,s.m.,et al.,in vivo near
‑
infrared imaging for the tracking of systemically delivered mesenchymal stem cells:tropism for brain tumors and biodistribution.int j nanomedicine,2016.11:p.13
‑
23.
[0840]
95.xie,c.,et al.,systemically infused mesenchymal stem cells show different homing profiles in healthy and tumor mouse models.stem cells transl med,2017.6(4):p.1120
‑
1131.
[0841]
96.cavarretta,i.t.,et al.,adipose tissue
‑
derived mesenchymal stem cells expressing prodrug
‑
converting enzyme inhibit human prostate tumor growth.mol ther,2010.18(1):p.223
‑
31.
[0842]
97.altaner,c.,et al.,complete regression of glioblastoma by mesenchymal stemcells mediated prodrug gene therapy simulating clinical therapeutic scenario.int j cancer,2014.134(6):p.1458
‑
65.
[0843]
98.wongrakpanich,a.,et al.,the absence of cpg in plasmid dna
‑
chitosan polyplexes enhances transfection efficiencies and reduces inflammatory responses in murine lungs.mol pharm,2014.11(3):p.1022
‑
31.
[0844]
99.moritz,b.,p.b.becker,and u.gopfert,cmv promoter mutants with a reduced propensity to productivity loss in cho cells.sci rep,2015.5:p.16952.
[0845]
100.zhao,c.p.,et al.,matrix attachment region combinations increase transgene expression in transfected chinese hamster ovary cells.sci rep,2017.7:p.42805.
targeting suicide genes in cancer.journal of cellular physiology 233,3831
‑
3845,doi:10.1002/jcp.26094(2018).
[0864]
119.zhang,j.,kale,v.&chen,m.gene
‑
directed enzyme prodrug therapy.aaps j 17,102
‑
110,doi:10.1208/s12248
‑
014
‑
9675
‑
7(2015).
[0865]
120.altaner,c.prodrug cancer gene therapy.cancer letters 270,191
‑
201,
[0866]
doi:10.1016/j.canlet.2008.04.023.
[0867]
121.niu,y.,li,j.s.&luo,x.r.enhancement of expression of survivin promoter
‑
driven cd/tk double suicide genes by the nuclear matrix attachment region in transgenic gastric cancer cells.gene 534,177
‑
182,doi:10.1016/j.gene.2013.10.064(2014).
[0868]
122.you,m.h.et al.cytosine deaminase
‑
producing human mesenchymal stem cells mediate an antitumor effect in a mouse xenograft model.journal of gastroenterology and hepatology 24,1393
‑
1400,doi:10.1111/j.1440
‑
1746.2009.05862.x(2009).
[0869]
123.qiao,l.,xu,z.l.,zhao,t.j.,ye,l.h.&zhang,x.d.dkk
‑
1 secreted by mesenchymal stem cells inhibits growth of breast cancer cells via depression of wnt signalling.cancer letters 269,67
‑
77,doi:10.1016/j.canlet.2008.04.032(2008).
[0870]
124.matuskova,m.et al.combined enzyme/prodrug treatment by genetically engineered at
‑
msc exerts synergy and inhibits growth of mda
‑
mb
‑
231 induced lung metastases.j exp clin cancer res 34,33,doi:10.1186/s13046
‑
015
‑
0149
‑
2(2015).
[0871]
125.jung,j.h.et al.three
‑
dimensional assessment of bystander effects of mesenchymal stem cells carrying a cytosine deaminase gene on glioma cells.american journal of cancer research 5,2686
‑
2696(2015).
[0872]
126.altaner,c.et al.complete regression of glioblastoma by mesenchymal stem cells mediated prodrug gene therapy simulating clinical therapeutic scenario.int j cancer 134,1458
‑
1465,doi:10.1002/ijc.28455(2014).
[0873]
127.karjoo,z.,chen,x.&hatefi,a.progress and problems with the use of suicide genes for targeted cancer therapy.adv drug deliv rev 99,113
‑
128,doi:10.1016/j.addr.2015.05.009(2016).
[0874]
128.shi,d.z.et al.pharmacokinetics and the bystander effect in cd::uprt/5
‑
fc bi
‑
gene therapy of glioma.chin med j(engl)122,1267
‑
1272(2009).
[0875]
129.xing,l.et al.expression of the bifunctional suicide gene cduprt increases radiosensitization and bystander effect of 5
‑
fc in prostate cancer cells.radiother oncol 92,345
‑
352,doi:10.1016/j.radonc.2009.04.003(2009).
[0876]
130.gopinath,p.&ghosh,s.s.implication of functional activity for determining therapeutic efficacy of suicide genes in vitro.biotechnol lett 30,1913
‑
1921,doi:10.1007/s10529
‑
008
‑
9787
‑
1(2008).
[0877]
131.niess,h.et al.treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells(treat
‑
me1):study protocol of a phase i/ii clinical trial.bmc cancer 15,237,doi:10.1186/s12885
‑
015
‑
1241
‑
x(2015).
[0878]
132.aboody,k.s.et al.neural stem cell
‑
mediated enzyme/prodrug therapy for glioma:preclinical studies.sci transl med 5,184ra159,doi:10.1126/scitranslmed.3005365(2013).
[0879]
133.pollock,k.et al.human mesenchymal stem cells genetically engineered to overexpress brain
‑
derived neurotrophic factor improve outcomes in huntington's disease mouse models.mol ther 24,965
‑
977,doi:10.1038/mt.2016.12(2016).
[0880]
134.ciuffi,a.the benefits of integration.clinical microbiology and infection 22,324
‑
332,doi:https://doi.org/10.1016/j.cmi.2016.02.013(2016).
[0881]
135.thomas,c.e.,ehrhardt,a.&kay,m.a.progress and problems with the use of viral vectors for gene therapy.nat rev genet 4,346
‑
358,doi:10.1038/nrg1066(2003).
[0882]
136.dewey,r.a.et al.chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus
‑
mediated gene therapy:implications for clinical trials.nat med 5,1256
‑
1263,doi:10.1038/15207(1999).
[0883]
137.vicente,t.,peixoto,c.,carrondo,m.j.&alves,p.m.virus production for clinical gene therapy.methods mol biol 542,447
‑
470,doi:10.1007/978
‑1‑
59745
‑
561
‑
9_24(2009).
[0884]
138.van der loo,j.c.&wright,j.f.progress and challenges in viral vector manufacturing.hum mol genet 25,r42
‑
52,doi:10.1093/hmg/ddv451(2016).
[0885]
139.charrier,s.et al.quantification of lentiviral vector copy numbers in individual hematopoietic colony
‑
forming cells shows vector dose
‑
dependent effects on the frequency and level of transduction.gene therapy 18,479
‑
487,doi:10.1038/gt.2010.163(2011).
[0886]
140.batard,p.,jordan,m.&wurm,f.transfer of high copy number plasmid into mammalian cells by calciumphosphate transfection.gene 270,61
‑
68(2001).
[0887]
141.cohen,r.n.,van der aa,m.a.e.m.,macaraeg,n.,lee,a.p.&szoka,f.c.quantification of plasmid dna copies in the nucleus after lipoplex and polyplex transfection.journal of controlled release:official journal of the controlled release society 135,166
‑
174,doi:10.1016/j.jconrel.2008.12.016(2009).
[0888]
142.hamann,a.,nguyen,a.&pannier,a.k.nucleic acid delivery to mesenchymal stem cells:a review of nonviral methods and applications.j biol eng 13,7,doi:10.1186/s13036
‑
019
‑
0140
‑
0(2019).
[0889]
143.ramamoorth,m.&narvekar,a.non viral vectors in gene therapy
‑
an overview.j clin diagn res 9,ge01
‑
06,doi:10.7860/jcdr/2015/10443.5394(2015).
[0890]
144.baek,k.,tu,c.,zoldan,j.&suggs,l.j.gene transfection for stem cell therapy.current stem cell reports 2,52
‑
61,doi:10.1007/s40778
‑
016
‑
0029
‑
5(2016).
[0891]
145.abdul halim,n.s.,fakiruddin,k.s.,ali,s.a.&yahaya,b.h.a comparative study of non
‑
viral gene delivery techniques to human adipose
‑
derived mesenchymal stem cell.int j mol sci 15,15044
‑
15060,doi:10.3390/ijms150915044(2014).
[0892]
146.ho,y.k.,zhou,l.h.,tam,k.c.&too,h.p.enhanced non
‑
viral gene delivery by coordinated endosomal release and inhibition of beta
‑
tubulin deactylase.nucleic acids research 45,e38,doi:10.1093/nar/gkw1143(2017).
[0893]
147.zou,r.,zhou,k.,stephanopoulos,g.&too,h.p.combinatorial engineering of 1
‑
deoxy
‑
d
‑
xylulose 5
‑
phosphate pathway using cross
‑
lapping in vitro assembly(cliva)method.plos one 8,e79557,doi:10.1371/journal.pone.0079557(2013).
[0894]
148.dominici,m.et al.minimal criteria for defining multipotent mesenchymal stromal cells.the international society for cellular therapy position statement.cytotherapy 8,315
‑
317,doi:10.1080/14653240600855905(2006).
[0895]
149.swiech,k.et al.transient transfection of serum
‑
free suspension hek293 cell culture for efficient production of human rfviii.bmc biotechnology 11,114,doi:10.1186/1472
‑
6750
‑
11
‑
114(2011).
[0896]
150.mccall,j.,nicholson,l.,weidner,n.&blesch,a.optimization of adult sensory neuron electroporation to study mechanisms of neurite growth.frontiers in molecular neuroscience 5,doi:10.3389/fnmol.2012.00011(2012).
[0897]
151.ho,yoon k.et al.enhanced transfection of a macromolecular lignin
‑
based dna complex with low cellular toxicity.bioscience reports 38,bsr20181021,doi:10.1042/bsr20181021(2018).
[0898]
152.madeira,c.et al.gene delivery to human bone marrow mesenchymal stem cells by microporation.journal of biotechnology 151,130
‑
136,doi:10.1016/j.jbiotec.2010.11.002(2011).
[0899]
153.bourbeau,d.,lavoie,g.,nalbantoglu,j.&massie,b.suicide gene therapy with an adenovirus expressing the fusion gene cd::uprt in human glioblastomas:different sensitivities correlate with p53 status.j gene med 6,1320
‑
1332,doi:10.1002/jgm.611(2004).
[0900]
154.marofi,f.,vahedi,g.,biglari,a.,esmaeilzadeh,a.&athari,s.s.mesenchymal stromal/stem cells:a new era in the cell
‑
based targeted gene therapy of cancer.frontiers in immunology 8,1770,doi:10.3389/fimmu.2017.01770
vitro gene transfer with cationic molecules up to 1000
‑
fold.gene ther 3,1074
‑
1080(1996).
[0923]
177.christodoulou,i.et al.mesenchymal stem cells in preclinical cancer cytotherapy:a systematic review.stem cell research&therapy 9,336,doi:10.1186/s13287
‑
018
‑
1078
‑
8(2018).
[0924]
178.niess,h.et al.treatment of advanced gastrointestinal tumors with genetically modified autologous mesenchymal stromal cells(treat
‑
me1):study protocol of a phase i/ii clinical trial.bmc cancer 15,237
‑
237,doi:10.1186/s12885
‑
015
‑
1241
‑
x(2015).
[0925]
179.von einem,j.c.et al.treatment of advanced gastrointestinal cancer with genetically modified autologous mesenchymal stem cells:results from the phase 1/2 treat
‑
me1 trial.international journal of cancer 0,doi:10.1002/ijc.32230(2019).
[0926]
180.dominici,m.et al.minimal criteria for defining multipotent mesenchymal stromal cells.the international society for cellular therapy position statement.cytotherapy 8,315
‑
317,doi:10.1080/14653240600855905(2006).
[0927]
181.xie,y.et al.the human glioblastoma cell culture resource:validated cell models representing all molecular subtypes.ebiomedicine 2,1351
‑
1363,doi:10.1016/j.ebiom.2015.08.026(2015).
[0928]
182.schweizer,m.t.et al.a phase i study to assess the safety and cancer
‑
homing ability of allogeneic bone marrow
‑
derived mesenchymal stem cells in men with localized prostate cancer.stem cells translational medicine 8,441
‑
449,doi:10.1002/sctm.18
‑
0230(2019).
[0929]
183.ferrari,c.et al.limiting cell aggregation during mesenchymal stem cell expansion on microcarriers.biotechnology progress 28,780
‑
787,doi:10.1002/btpr.1527(2012).
[0930]
184.takahashi,i.,sato,k.,mera,h.,wakitani,s.&takagi,m.effects of agitation rate on aggregation during beads
‑
to
‑
beads subcultivation of microcarrier culture of human mesenchymal stem cells.cytotechnology 69,503
‑
509,doi:10.1007/s10616
‑
016
‑
9999
‑
5(2017).
[0931]
185.rawat,j.&gadgil,m.shear stress increases cytotoxicity and reduces transfection efficiency of liposomal gene delivery to cho
‑
s cells.cytotechnology 68,2529
‑
2538,doi:10.1007/s10616
‑
016
‑
9974
‑
1(2016).
[0932]
在本文中以及说明书中的其它处所引用的所有参考文献以其全部内容作为参考并入本文。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1