用于制备高纯度的雌四醇的工业方法与流程
用于制备高纯度的雌四醇的工业方法
发明领域
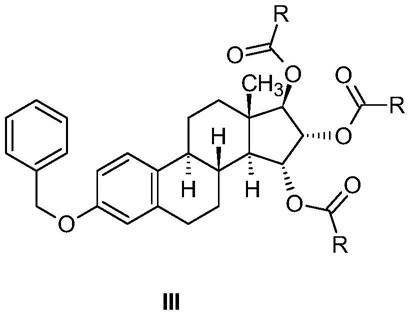
1.本发明涉及式(i)的雌四醇(雌-1,3,5(10)-三烯-3,15α,16α,17β-四醇)、通式(iii)的在3,15α,16α,17β-位被保护的其衍生物、通式(iv)的在15α,16α,17β-位被保护的其3-羟基衍生物的制备,以及在该制备方法中应用的通式(iii)和(iv)的中间体。本发明的另一方面是通过本发明的方法得到的式(i)的雌四醇在制备药物组合物中的用途。
2.发明背景
3.式(i)的雌酚(雌-1,3,5(10)-三烯-3,15α,16α,17β-四醇)是在人类妊娠期间由胎儿肝脏内源性产生的具有弱雌激素活性的化合物。
[0004][0005]
发现雌四醇在激素替代疗法、治疗阴道干燥的方法、治疗围绝经期症状(例如热潮红、盗汗)的方法、避孕的方法、增强性欲的方法、治疗皮肤和促进伤口愈合的方法、治疗或预防自身免疫病症、乳腺肿瘤、前列腺癌和结肠直肠肿瘤的方法和神经保护(例如新生儿脑病)的方法中有效(wo 02/094275 a1、wo 02/094276 a1、wo 02/094278 a1、wo 02/094279 a1、wo 03/041718 a1、wo 03/103684 a1、wo 03/103685 a1、wo 2004/006936 a1、wo 2004/037269 a1、wo 2007/081206 a1、wo 2008/085038 a2、wo 2013/021025 a1、wo 2013/156329 a1、wo 2018/024912 a1、wo 2018/065076 a1、wo 2019/025031 a1;2019年5月14日最近更新的雌四醇-https://adisinsight.springer.com/drugs/800044874;gaspard等人.,maturitas 124(2019)p.153abstract p09;apter等人.eur jcontracept reprod hc(2017)22(4):260-267;tskitishvili等人,jendocrinol.(2017)232(1):85-95;coelingh bennick等人,climacteric(2008)11(suppl1):47-58)。
[0006]
反应方案1中描述的雌四醇的合成是由fishman等人首次描述的(fishman,j和guzik,h.,tetrahedron letters,1967,30:2929-2932)。用四氢铝锂还原起始的15-烯-17-酮化合物,得到烯丙醇型化合物,由其形成二乙酸酯。在吡啶中用四氧化锇氧化所述二乙酸酯得到二乙酸雌四醇酯,将其在甲醇中与乙酸钾一起煮沸,得到雌四醇。该公布不包括收率和纯度数据,熔点(230-235℃),比旋度((etoh))和nmr(60mhz)数据作为鉴定数据的证据提供。
[0007][0008]
在suzuki等(suzuki,e.,namba,s.,kuruhara,h.,goto,j.,matsuki,y.,nambara,t.,steroids,1995,60,277-284)在反应方案2中描述的合成中,在吡啶的存在下,在苯中用等量的四氧化锇氧化15-烯-17-乙酰氧基化合物。通过柱色谱法分离所得到的二乙酸酯异构体,以46%的产率得到15α,16α,17β-二乙酸酯,以及以12%的产率得到15α,16α,17β-二乙酸酯异构体。
[0009]
由得到的产物的给定量计算的异构体比例为78.9/21.1(15α,16α/15β,16β)。
[0010]
将15α,16α,17β-二乙酸酯的碱水解得到雌四醇,产率为67%。没有提供纯度数据,233-235℃作为产物的熔点给出。
[0011][0012]
在专利申请wo2004/041839a2(pantarhei)中-反应方案3-从保护为3-苄基醚的雌酮开始,通过几个已知的步骤形成δ15-雌二醇-苄基醚17-乙酸酯,通过用庚烷-乙酸乙酯溶剂混合物处理用聚合物结合的四氧化锇氧化,得到粗产物,然后,将其在三元溶剂混合物(庚烷-乙酸乙酯-乙醇)中结晶,以43%的产率得到纯度为98.7%(异构体纯度:99.5%)的雌四醇-苄基醚-17-乙酸酯。通过催化氢化(92%产率)和碱水解(92.5%产率)脱保护,得到雌四醇化合物。给出关于产物纯度的数据为99.5%。同样的方案也公开在专利申请wo2013/012328a1(donesta)中。
[0013]
根据该描述,在高损失下,同时在从技术上不利的三元溶剂混合物中进行结晶,得到纯中间体。这种方案也产生了经济问题。
[0014]
反
[0015]
专利申请wo2013/050553a1(estetra)-反应方案4-也描述了高锰酸钾作为氧化剂,但是没有给出异构体比例、纯度和产率数据。
[0016][0017]
在专利申请wo2013/034780a2(crystal pharma)的实施例中-反应方案5,使用与聚(4-乙烯基吡啶)(pvp)结合的5-四氧化锇作为氧化剂,用于在55-60℃温度下进行顺式羟基化。在四氧化锇氧化δ
15-17-乙酰氧基衍生物的情况下,测定反应混合物中15α,16α/15β,16β异构体比例为80/20,以88%产率得到的产物,但没有给出纯度数据。
[0018][0019]
在δ
15-17β-羟基化合物的情况下,得到62%产率和90/10的15α,16α/15β,16β异构体比例,但是没有报道纯度数据:
[0020][0021]
在苄基醚的情况下,以99%的产率得到了比例90/10的15α,16α/15β,16β异构体混合物,但没有给出纯度数据:
[0022][0023]
在3-苯甲酰基化合物的情况下,以92%产率得到了比例90/10的15α,16α/15β,16β异构体混合物,但是没有给出纯度数据::
[0024][0025][0026]
在叔丁基-二甲基甲硅烷基醚化合物的情况下,以101%产率得到了比例90/10的15α,16α/15β,16β异构体混合物,但是没有给出纯度数据:
[0027][0028]
在(1-丁氧基乙基)-醚化合物的情况下,以96.5%产率得到了比例90/10的15α,16α/15β,16β异构体混合物,但是没有给出纯度数据:
[0029][0030]
wo2013/034780a2(crystal pharma)没有提供关于其中公开的式(i)的3-oh保护的雌四醇衍生物的纯度信息,没有公开由其制备雌四醇,并且其教导不允许实现以活性物质纯度制备雌四醇。
[0031]
专利申请wo2015/040051a1(crystal pharma)证明通过具有相同或不同保护基的δ
15-3,17β-二羟基的衍生物的顺式羟基化。描述了用高锰酸钾氧化剂的极低(1-9%)转化率。使用四氧化锇-pvp氧化剂,以良好的转化率获得了在3位和17位的羟基上被保护的雌四醇衍生物。脱保护分别得到了比例为98/2-99/1的15α,16α/15β,16β异构体混合物形式的雌四醇。说明书中没有给出纯度数据。此外,说明书没有包含关于如何从所获得的中间体制备活性物质纯度的雌四醇的任何信息。
[0032]
从所有这些,可以得出结论,要么没有解决制备药物级纯度的雌四醇的方法,要么可以以不利的产率和低经济性解决纯活性物质的制备。
[0033]
现有技术的药典要求目前规定了许多测试方法,例如高效液相色谱纯度的测试方
法,以及规定和限制杂质的数目和数量。在甾体活性物质的情况下,通常的要求是应用0.5%总杂质限度和0.10%的单一杂质限度。为了满足目标产物质量的要求,制备合适纯度的关键中间体是有利的,这对于具有不利结晶和纯化性质的化合物(例如雌四醇)尤其准确。
[0034]
鉴于以上所述,仍存在未满足的需求以提供用于制备雌四醇的替代工业方法,所述方法允许以高纯度制备雌四醇并且可经由具有有利性质(例如结晶、纯化、可分离、收率)的中间体进行。
[0035]
发明简述
[0036]
本发明涉及式(i)的雌四醇、通式(iii)的在3,15α,16α,17β-位被保护的其衍生物、通式(iv)的在15α,16α,17β-位被保护的其3-羟基衍生物的制备方法,以及在所述方法中应用的通式(iii)和(iv)的中间体。本发明的工业方法是从式(ii)的化合物开始制备式(i)的雌四醇。
[0037]
式(ii)的在3位被保护的三醇衍生物可以根据专利申请wo2013/034780a2(crystal pharma)中描述的方法-反应方案7-由3-苄氧基-雌-1,3,5(10),15-四烯-17-醇开始制备。通式(iii)的化合物是通过酰化式(ii)的化合物及其纯化获得的。
[0038]
通式(iv)的化合物是通过使通式(iii)的化合物脱苄基化获得的。
[0039]
式(i)的雌四醇是通过碱性水解通式(iv)的化合物制备的。
[0040]
本发明还涉及上述方法的通式(iii)和(iv)的中间体。
[0041]
本发明进一步涉及通过本发明所述的方法得到的式(i)的雌四醇在制备药物组合物中的用途。
[0042]
发明详述
[0043]
本发明涉及式(i)的雌四醇、通式(iii)的在3,15α,16α,17β-位被保护的其衍生物和通式(iv)的在15α,16α,17β-位被保护的其3-羟基衍生物的制备方法,以及在所述方法中应用的通式(iii)和(iv)的中间体。本发明的工业方法是根据以下反应方案从式(ii)的化合物开始制备式(i)的雌四醇,其中r表示甲基或氢:
[0044]
[0045]
式(ii)的在3位被保护的三醇衍生物可以根据专利申请wo2013/034780a2(crystal pharma)中描述的方法制备,即其中公开的示例化合物7,任选地在共氧化剂如三烷基胺n-氧化物,如三甲基胺或三乙胺n-氧化物的存在下,在水混溶性溶剂如2-丁酮、丙酮、四氢呋喃、叔丁醇,优选2-丁酮中,用氧化剂如锇酸钾或四氧化锇氧化3-苄氧基-雌-1,3,5(10),15-四烯-17-醇。
[0046]
步骤(a)15,16,17-三醇衍生物的酰化
[0047]
在根据本发明方法的步骤a)中,由通式(iii)表示的在3,15α,16α,17β位保护的雌四醇衍生物是通过在合适的溶剂中,使用合适的反应物,在分离或不分离下,酰化式(ii)的在3位保护的15,16,17-三醇衍生物获得的。
[0048]
酰化中使用的溶剂是选自脂族和芳族烃、卤代烃、酯和醚,优选与水不混溶的溶剂,如甲苯、二氯甲烷或乙酸乙酯。
[0049]
在一个实施方案中,当r=甲基(乙酰化)时,用于酰化的反应物优选为乙酸酐、乙酰氯或乙酰溴。
[0050]
在另一个实施方案中,当r=氢(甲酰化)时,用于酰化的反应物优选为乙酸-甲酸混合酸酐。
[0051]
酰化在胺碱,优选吡啶或4-二甲基氨基吡啶存在下进行。
[0052]
酰化反应在惰性气氛下进行,优选在n2气氛下进行。
[0053]
在一个实施方案中,酰化步骤进一步包括从c
1-3
醇,优选甲醇中结晶得到的式(iii)化合物。
[0054]
在另一个实施方案中,步骤(a)可以与上述二羟基化依次进行,然后酰化,无需纯化和/或分离中间体-式(ii)化合物-,同时仍然以良好的产率获得高纯度的终产物。这在工业应用中是特别有利的,其中减少制备步骤的数量产生经济优势和工艺简化,因为如两个步骤之间的纯化和/或分离的步骤将不再是必需的。
[0055]
本发明提供通式(iii)化合物,其中r为甲基或氢,即(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯(实施例1)和(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三甲酸酯(实施例3)。
[0056]
步骤(b)3,15,16,17-保护的衍生物的脱苄基化
[0057]
在根据本发明的方法的步骤b)中,通过转移或催化氢化除去通式(iii)表示的衍生物的3位苄基保护基,得到通式(iv)的在15α,16α,17β位被保护的3-羟基雌四醇衍生物。
[0058]
在一个实施方案中,通过用氢气催化氢化进行脱苄基化,其中催化剂选自钯或在载体(碳、氧化铝等)上的钯。催化剂优选为pd/c。用于催化氢化的溶剂选自醇、酯和酮,优选乙酸乙酯。
[0059]
在另一个实施方案中,通过使用环己烯试剂的转移氢化进行脱苄基化。用于转移氢化的溶剂是醇,优选乙醇。
[0060]
脱苄基化步骤进一步包括从酯、烃、醇或其混合物中,优选从乙酸乙酯/正庚烷的混合物中结晶得到的通式(iv)化合物。
[0061]
本发明提供通式(iv)的化合物,其中r为氢,即(15α,16α,17β)-3-羟基雌-1,3,5(10)-三烯-15,16,17-三基三甲酸酯(triyl triformiate)(实施例4)。
[0062]
步骤(c)15,16,17-酰基保护的衍生物的水解
[0063]
在根据本发明的方法的步骤c)中,通过在合适的溶剂中用碱金属碳酸盐或碱金属氢氧化物在碱性介质中使通式(iv)的衍生物脱保护来制备式(i)的雌四醇。
[0064]
水解中使用的溶剂选自水、醇类溶剂或其混合物,优选c
1-3
醇,更优选甲醇和水的混合物。
[0065]
在一个实施方案中,水解是在碱金属碳酸盐或碱金属碳酸氢盐,优选碳酸钾的存在下进行的。
[0066]
在另一个实施方案中,水解是在碱金属醇盐或碱金属氢氧化物,优选氢氧化钠或氢氧化锂的存在下进行的。
[0067]
基于上述,本领域技术人员可以容易地选择试剂、溶剂、温度、压力和其它反应条件。本发明的方法中使用的原料、试剂和溶剂是可商购的和/或可以由本领域技术人员容易地制备的。实施例中公开的产物的纯度通过本领域技术人员已知的高效液相色谱分离技术测定,使用最广泛使用的硅胶(例如ascents,kintex)作为固定相和具有线性梯度设置的常用洗脱剂(例如水、甲醇、乙腈)的多组分混合物。
[0068]
虽然文献中描述的式(i)的化合物和式(ii)的在3位被保护的15α,16α,17β-三醇具有不利的结晶性质,但是出人意料地,通式(iii)和(iv)的化合物结晶良好,可以以高产率纯化并且可以高选择性地从异构体副产物中分离。
[0069]
就实施本发明而言,当r表示甲基时,步骤(a)至(c)是更优选的。
[0070]
本发明的另一个实施方案是通过上述方法得到的式(i)的雌四醇在制备药物组合物中的用途。
[0071]
术语“药物组合物”(或“组合物”)指包含治疗有效量的活性成分(例如式(i)化合物)和可药用赋形剂的混合物或溶液,所述活性成分例如施用于需要其的患者,例如人,优选绝经前或绝经后的妇女(wo2016/203006a1、wo2016/203009a1、wo2016/203044a1)。
[0072]
本发明的药物组合物可以配制成各种剂型,例如固体或液体剂型。优选地,药物组合物是固体口服剂型,例如片剂(例如,口含片、舌下片、泡腾片、可咀嚼片、口分散片)。
[0073]
本发明的式(i)化合物可以与可药用赋形剂以单剂量或多剂量共同施用。
[0074]
本发明还涉及药物组合物,其包含式(i)的化合物与一种或多种、优选一种其它活性成分的联合。该联合组合物包含在单一剂型中的式(i)的化合物和一种或多种其它活性成分连同可药用赋形剂。其它活性成分优选是孕激素化合物,例如但不限于孕酮、左炔诺孕酮、诺孕酯、炔诺酮、地屈孕酮、屈螺酮、3-β-羟基-去氧孕烯、依托孕烯、17-脱乙酰基-诺孕酯、19-去甲孕酮、乙酰氧基孕烯醇酮、烯丙雌醇、阿那孕酮、氯地孕酮、环丙孕酮、地美孕酮、去氧孕烯、地诺孕酮、二氢孕酮、二甲炔酮、乙炔睾酮、双醋炔诺醇、醋酸氟孕酮、孕三烯酮(gastrinone)、孕二烯酮、孕三烯酮(gestrinone)、羟甲基孕酮、羟基孕酮、利奈孕酮、美罗孕酮、甲羟孕酮、甲地孕酮、美仑孕酮、诺美孕酮、异炔诺酮、甲基炔诺酮(包括d-甲基炔诺酮和d1-甲基炔诺酮)、诺孕烯酮(norgestrienone)、甲基诺龙(normethisterone)、奎孕醇(quingestanol)、(17α)-17-羟基-11-亚甲基-19-去甲孕甾(norpregna)-4,15-二烯-20-炔-3-酮、替勃龙、曲美孕酮、醋苯阿尔孕酮、奈甾酮(nestorone)、普美孕酮、17-羟孕酮酯、19-去甲-17-羟孕酮、17α-乙炔基-睾酮、17α-乙炔基-19-去甲睾酮、d-17β-乙酰氧基-13β-乙基-17α-乙炔基-甾(gon)-4-烯-3-酮肟。更优选地,孕激素试剂是屈螺酮。其它活性成分也可以是钙或维生素,优选例如和维生素d。
[0075]
达到期望的治疗效果所需的剂量可以在很宽的范围内变化,并且在每种情况下将适应个体的需要,考虑到疾病的严重性、待治疗患者的状况和体重、对活性成分的敏感性、给药途径和每日治疗的次数。含有根据本发明的式(i)的活性成分的药物组合物通常每剂量单位包含0.01至20mg,优选1.5mg至15mg,更优选15mg的活性成分。当组合物还含有屈螺酮作为其它活性成分时,组合物通常每剂量单位含有0.01至10mg,优选1.5mg至5mg,更优选3mg的屈螺酮。该联合组合物还可含有维生素d。
[0076]
本发明的药物组合物可以通过本身已知的方法制备,例如通过制粒(湿法或干法)或通过压制。本发明的药物组合物可以使用一种或多种生理(或药学)可接受的赋形剂以常规方式配制。可以使用本领域公知的任何技术和赋形剂,这样的赋形剂选自下列种类,例如但不限于片剂赋形剂、片剂粘合剂、释放改性剂、崩解剂、助流剂、润滑剂、甜味剂、调味剂、调味品或包衣材料。合适的药物赋形剂的实例是淀粉、微晶纤维素、滑石、葡萄糖、乳糖、明胶、二氧化硅、硬脂酸镁、硬脂酸钠、单硬脂酸甘油酯、纤维素衍生物、氯化钠、甘油、丙二醇、水、乙醇等。上述赋形剂和各种制备方法仅是代表性的实例。也可以使用本领域已知的其它材料和工艺技术。
[0077]
本发明的一个实施方案是通过上述方法得到的式(i)的雌四醇在制备用于激素替代疗法的药物、治疗阴道干燥的方法、治疗围绝经期症状(例如潮热、盗汗)的方法、避孕的方法、增强性欲的方法、治疗皮肤和促进伤口愈合的方法、治疗或预防自身免疫病症、乳腺肿瘤、前列腺癌和结肠直肠肿瘤的方法、或用于神经保护的用途。
[0078]
本发明的另一个实施方案优选地是通过上述方法得到的式(i)的雌四醇在制备用于避孕药物(更优选地与屈螺酮组合(wo2019/154899a1))中的用途。
[0079]
本发明的另一个实施方案优选是通过上述方法得到的式(i)的雌四醇在制备用于激素替代疗法的药物中的用途。
[0080]
本发明的另一个实施方案优选地是通过上述方法得到的式(i)的雌四醇在制备用于神经保护(例如,新生儿脑病)的药物中的用途。
[0081]
本发明的另一个实施方案是通过上述方法得到的式(i)的雌四醇在制备用于治疗围绝经期症状的药物(更优选与屈螺酮和维生素d组合)中的用途。
[0082]
参照实施例
[0083]
根据wo2013/034780a2(crystal pharma)制备((15ξ,16ξ,17β)-雌-1,3,5(10)-三烯-3,15,16,17-四醇)的雌四醇异构体混合物
[0084]
a)顺式羟基化
[0085]
(15α,16α,17β)-和(15β,16β,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇
[0086]
在20-25℃下,在n2气氛下,将20.0g(55.5mmol)的3-苄氧基-雌-1,3,5(10),15-四烯-17-醇(wo2004/041839(pantarhei),实施例7)溶于1400ml的四氢呋喃中,然后,向反应混合物中加入14ml的2w/v%在叔丁醇中的四氧化锇(oso4)(280mg oso4含量)和11g的n-甲基吗啉n-氧化物和150ml水的溶液,并在20-25℃下,在n2气氛下搅拌24小时。通过tlc(正庚烷:丙酮1:1)监测反应。
[0087]
后处理:向该溶液中滴加140ml的5%na2s2o5溶液,并向其中加入100mg的活性炭,搅拌混合物30分钟,通过硅藻土垫过滤该混合物。从滤液中蒸馏出有机溶剂,加入400ml的
二氯甲烷。分离各相。用200ml的10%盐酸和200ml的饱和氯化钠溶液洗涤有机相,然后干燥,并浓缩。将浓缩的残余物溶于在200ml的甲醇中,并在0-5℃下滴加到2l的水中,搅拌1小时,过滤并在过滤器上用20ml的水洗涤晶体。将该物质在40℃的真空下干燥至恒重。得到19.68g(89.86%)的黄白色晶体。
[0088]
纯度(hplc):85.18%ααβ-异构体,5.43%βββ-异构体(面积)(比例94.0:6.0)
[0089]
在有机溶剂中,通常在烃、醚、酯、醇或其混合物中重结晶期间,(15ξ,16ξ,17β)-雌-1,3,5(10)-三烯-3,15,16,17-四醇粘结(晶体粘在一起,从而防止物质被过滤和回收,从而防止被纯化)。通常,该化合物仅可从水或可与水可混溶的溶剂(通常为醇)的混合物中结晶,但是在这种情况下,关于异构体比例的改善,不可能实现任何显著的变化。
[0090]
b)氢化
[0091]
(15α,16α,17β)-,和(15β,16β,17β)-雌-1,3,5(10)-三烯-15,16,17-三醇
[0092]
在20-25℃下,在n2气氛下,将19.5g(15ξ,16ξ,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇溶于400ml的甲醇中。将2.0g的10%pd/c催化剂悬浮在100ml的深冷甲醇中,然后加入到所述溶液中。将n2气氛改变为h2气氛,并在大气压下于20-25℃下搅拌反应混合物6小时。
[0093]
后处理:过滤出催化剂,将反应混合物在减压下浓缩至45ml,加入45ml的水,并在0-5℃下搅拌该混合物1小时,然后过滤并在过滤器上用20ml的水洗涤两次,干燥至恒重,从而得到14.5g(96.67%)的白色结晶产物。
[0094]
纯度(hplc):87.53%ααβ-异构体,5,46%βββ-异构体(面积)(比例94.13:5.87)
[0095]
本发明通过以下非限制性实施例进一步说明。从上述描述和实施例,本领域技术人员可以确定本发明的必要特征,并且在不背离本发明的精神和范围的情况下,进行某些改变和修饰以使本发明适应各种应用和环境。因此,本发明不限于如下所述的示例性实施例,而是限于所附权利要求书的范围。
实施例
[0096]
实施例1
[0097]
(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯方法a(分离的)
[0098]
a.)顺式羟基化
[0099]
(15α,16α,17β)-,和(15β,16β,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇
[0100]
在20-25℃下,在n2气氛下,将40mg的锇酸钾二水合物(k2oso4·
2h2o)悬浮在100ml的2-丁酮(甲基乙基酮)中,加入7.7ml的纯净水和1.1g的三甲胺n-氧化物二水合物。将2.0g(5.5mmol)的3-苄氧基-雌-1,3,5(10),15-四烯-17-醇(wo2004/041839(pantarhei),实施例7)溶于40ml的2-丁酮中,并滴加到反应混合物中。然后,在n2气氛下,在20-25℃下搅拌该反应混合物28小时。通过tplc(正庚烷:丙酮1:1)监测反应。
[0101]
后处理:将25ml的10%na2s2o5溶液加入到该混合物中,接着,加入100mg的活性炭,然后,搅拌1小时。通过硅藻土垫过滤,然后,加入etoac和10%hcl溶液。分离各相,用etoac萃取水相。用饱和nacl和10%na2s2o5溶液洗涤合并的有机相。经na2so4干燥,过滤,然后浓
缩。因此,得到1.8g(81.8%)的产物。
[0102]
纯度(hplc):85.0%ααβ-异构体,9.9%βββ-异构体(面积)(比例89.6:10.4)
[0103]
b)酰化
[0104]
(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇三乙酸酯
[0105]
在n2气氛下,将1.0g(2.53mmol)的(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇溶于15ml的二氯甲烷中。加入1.5ml的三乙胺、6.0ml的乙酸和72mg的4-二甲基氨基吡啶,并搅拌2小时。通过tlc(甲苯:丙酮4:1)监测反应。
[0106]
后处理:将3ml的乙醇滴加到混合物中,搅拌30分钟,然后,加入10%的nahco3溶液,并搅拌另外的30分钟。分离各相,用10%的nahco3溶液洗涤有机相两次,然后,用饱和的盐水洗涤。经na2so4干燥,过滤,并将溶剂转换成meoh,从其中结晶。在过滤和干燥之后,得到1.2g的物质。为了获得合适的异构体比例,将产物从甲醇中再重结晶两次,因此,得到1.1g(84.46%)的产物。
[0107]
纯度(hplc):99.2%ααβ-异构体,0.14%βββ-异构体(面积)。
[0108]
方法b(没有分离)
[0109]
a)顺式羟基化
[0110]
3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇的(15α,16α,17β),(15β,16β,17β)异构体混合物
[0111]
在20-25℃下,在n2气氛下,将30.03g(83.3mmol)的3-苄氧基-雌-1,3,5(10),15-四烯-17-醇(wo4004/041839(pantarhei),实施例7)溶于480ml的2-丁酮(甲基乙基酮)中,接着,加入600mg的锇酸钾二水合物(k2oso4.2h2o)、48.0ml的纯净水和16.5g的三甲胺n-氧化物二水合物。然后,在n2气氛下,在40-45℃下,搅拌该反应混合物7小时。通过tlc(正庚烷:丙酮1:1)监测反应。
[0112]
后处理:在40-45℃下,将300ml的10%(w/v)偏亚硫酸氢钠溶液(焦亚硫酸钠)滴加到反应混合物中,并搅拌1小时。然后,将浆液通过硅藻土垫过滤,并用2-丁酮洗涤过滤器。然后,通过蒸馏从滤液中除去2-丁酮。将600.0ml的乙酸乙酯和300ml的10%(w/v)碳酸氢钠溶液(30g nahco3)加入到残余物中,在强力搅拌几分钟之后沉降,分离各相。用乙酸乙酯将水相洗涤两次。用1%(w/v)的edta-四na盐溶液和饱和盐水的混合物洗涤合并的有机相。在相分离之后,将乙酸乙酯有机相浓缩至最终体积为450ml,从而也脱水。产物不进行分离,而是进一步转移至酰化反应。
[0113]
b)酰化
[0114]
(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯
[0115]
将72.0ml的乙酸酐、48ml的三乙胺和1.8g的4-二甲基氨基吡啶加入到步骤a)中得到的乙酸乙酯溶液中,接着,在n2气氛下,在35-40℃下,搅拌3小时。通过tlc(甲苯:丙酮4:1)监测反应。
[0116]
后处理:将24ml的乙醇滴加到混合物中,搅拌30分钟,然后冷却至20-25℃,接着加入240ml的纯化水和60ml的10%(w/v,d=1.047,17.88g的浓hcl)盐酸溶液,在强力搅拌几分钟之后,沉降,分离各相。用乙酸乙酯萃取水相。用10%(w/v)的碳酸氢钠溶液和饱和盐水的混合物洗涤合并的有机相,并分离各相。将有机相用na2so4干燥,用氧化铝、硅胶和活性炭澄清,并在20-25℃下搅拌1小时。然后,过滤出澄清剂,并用乙酸乙酯洗涤过滤器。
[0117]
在减压下浓缩该滤液,然后浓缩溶剂并蒸馏以将溶剂改变为甲醇,最后从纯甲醇中结晶物质。将得到的粗产物重结晶,而无需干燥。
[0118]
c)重结晶
[0119]
将步骤b)中得到的粗产物溶于二氯甲烷中,蒸馏出甲醇,最后从纯甲醇中结晶。再重复该操作一次。因此,得到30.4g(69.8%)的白色晶体。
[0120]
纯度(hplc):99.2%ααβ-异构体,0.14%βββ-异构体(面积)。
[0121]
mp.:156.5-157.5℃.
[0122]
ei-hrms:c
31h36
o7[m
+
]的理论值:520.24555;实测值:520.24459;δ=-1.86ppm.
[0123]1h nmr(499.9mhz,cdcl3)δ=5.39(1h,dd,j=8.4hz,j=6.6hz,h-16),5.16(1h,dd,j=10.4hz,j=8.4hz,h-15),5.01(1h,d,j=6.6hz,h-17),2.08(3h,s,17-乙酰基),2.06(3h,s,15-乙酰基),2.04(3h,s,16-乙酰基),0.94(3h,s,h-18)
[0124]
13
c nmr(125.7mhz,cdcl3)δ=169.8(17-乙酰基co c-20),169.0(15-乙酰基co),168.7(16-乙酰基co),83.1(c-17),72.5(c-16),69.8(c-15),51.4(c-14),39.2(c-13),19.9(17-乙酰基-ch3),19.7(15-乙酰基-ch3),19.6(16-乙酰基-ch3),13.5(c-18)
[0125]
实施例2
[0126]
(15α,16α,17β)-3-羟基雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯
[0127]
方法a
[0128]
在20-25℃下,在n2气氛下,将25.7g(49.36mmol)的(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯(实施例1)溶于315ml的乙酸乙酯中。将770mg 10%钯碳催化剂悬浮于19ml的深冷乙酸乙酯中,然后加入到溶液中。将n2气氛改变为h2气氛,并在大气压下下,在20-25℃下,搅拌该反应混合物3小时。
[0129]
后处理:过滤出催化剂,用乙酸乙酯洗涤,并在减压下浓缩至终体积,然后加入正庚烷,并将悬浮液保持在0-5℃下1小时,然后过滤,并在过滤器上用正庚烷洗涤结晶产物,在40℃下真空干燥至恒重。因此,得到19.88g(93.55%)的白色结晶产物。
[0130]
纯度(hplc):99.42%ααβ-异构体,0.04%βββ-异构体(面积)。
[0131]
方法b
[0132]
在20-25℃下,将0.5g的(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯(实施例1)悬浮于14ml的乙醇中,然后加入0.5ml的环己烯和38mg的10%pd/c催化剂,接着在回流温度搅拌1小时。通过tlc(甲苯:丙酮4:1)监测反应。
[0133]
后处理:从反应混合物中过滤出催化剂,并将混合物浓缩至干。因此,得到0.41g(99.17%)的白色结晶产物。
[0134]
纯度(hplc):97.99%ααβ-异构体,0.14%βββ-异构体(面积)。
[0135]
mp.:181.5-185.5℃
[0136]
ei-hrms:c
24h30
o7[m
+
]的理论值:430.19860;实测值:430.19927;δ=1.55ppm.
[0137]1h nmr(499.9mhz,cdcl3)δ=5.41(1h,dd,j=8.4hz,j=6.6hz,h-16),5.18(1h,dd,j=10.5hz,j=8.4hz,h-15),5.03(1h,d,j=6.6hz,h-17),(3h,s,17-乙酰基),2.10(3h,s,15-乙酰基),2.07(3h,s,16-乙酰基),1.77(1h,t,j=11.1hz,h-14),0.95(3h,s,h-18)
[0138]
13
c nmr(125.7mhz,cdcl3)δ=170.9(17-乙酰基co),170.1(15-乙酰基co),169.8
(16-乙酰基co),84.1(c-17),73.5(c-16),70.8(c-15),52.4(c-14),40.2(c-13),20.9(17-乙酰基-ch3),20.7(15-乙酰基-ch3),20.6(16-乙酰基-ch3),14.5(c-18)
[0139]
实施例3
[0140]
(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三基三甲酸酯
[0141]
将5.00g的(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三醇(实施例1,方法“a”,步骤a))溶于73ml的吡啶中,并冷却至0℃,然后在约0-10℃之间,在约25分钟,经由加料漏斗加入由冷却至0℃的49ml的甲酸和18.3ml的乙酸的酸酐制成的混合酸酐的混合物。在搅拌1小时之后,将305ml的水加入到反应混合物中,过滤出得到的白色沉淀,并用水洗涤。干燥的粗产物称重为5.65g(93.23%)。
[0142]
将粗产物-根据实施例1的方法b步骤c)-从甲醇中重结晶,得到3.92g(69.4%)呈白色晶体的纯的标题产物。
[0143]
纯度(hplc):99.2%ααβ-异构体,0.05%βββ-异构体(面积)。
[0144]
mp.:153.5-154.3℃
[0145]
ei hrms:m=478.19866;δ=0.06ppm;c
28h30
o7[0146]1h nmr(499.9mhz,cdcl3)δ=5.41(1h,dd,j=8.4hz,j=6.6hz,h-16),5.18(1h,dd,j=10.5hz,j=8.4hz,h-15),5.03(1h,d,j=6.6hz,h-17),(3h,s,17-乙酰基),2.10(3h,s,15-乙酰基),2.07(3h,s,16-乙酰基),1.77(1h,t,j=11.1hz,h-14),0.95(3h,s,h-18)
[0147]
13
c nmr(125.7mhz,cdcl3)δ=170.9(17-乙酰基co),170.1(15-乙酰基co),169.8(16-乙酰基co),84.1(c-17),73.5(c-16),70.8(c-15),52.4(c-14),40.2(c-13),20.9(17-乙酰基-ch3),20.7(15-乙酰基-ch3),20.6(16-乙酰基-ch3),14.5(c-18)
[0148]
实施例4
[0149]
(15α,16α,17β)-3-羟基雌-1,3,5(10)-三烯-15,16,17-三基三甲酸酯
[0150]
在20-25℃下,在n2气氛下,将5.0g的(15α,16α,17β)-3-(苄氧基)雌-1,3,5(10)-三烯-15,16,17-三甲酸酯(实施例3)溶于150ml的乙酸乙酯中。将380mg的10%pd/c催化剂悬浮于5ml的深冷乙酸乙酯中,并加入到溶液中。将n2气氛改变为h2气氛,在大气压下,在20-25℃下,搅拌反应混合物4小时。
[0151]
后处理:过滤出催化剂,将反应混合物在减压下浓缩至四分之一(38ml),然后加入52ml的正庚烷。在0-5℃下搅拌1小时之后,将其过滤,在过滤器上用16ml的正庚烷洗涤两次,干燥至恒重,从而得到3.51g(94%)的白色结晶产物。
[0152]
纯度(hplc):99.42%ααβ-异构体,0.04%βββ-异构体(面积)。
[0153]
mp.:234-235℃
[0154]
ms:m-h=387(esi)
[0155]1h nmr(499.9mhz,dmso-d6)δ=8.17(1h,s,17-甲酰基-h),8.09(1h,s,15-甲酰基-h),8.04(1h,s,16-甲酰基-h),5.52(1h,t,j=7.4hz,h-16),5.24(1h,dd,j=10.1hz,j=8.6hz,h-15),5.11(1h,d,j=6.5hz,h-17),0.99(3h,s,h-18)
[0156]
13
c nmr(125.7mhz,dmso-d6)δ=159.5(17-甲酰基-c),159.3(15-甲酰基-c),158.8(16-甲酰基-c),82.4(c-17),71.7(c-16),69.2(c-15),51.3(c-14),39.6(c-13),13.5(c-18)
[0157]
实施例5
[0158]
雌四醇((15α,16α,17β)-雌-1,3,5(10)-三烯-3,15,16,17-四醇)
[0159]
方法a
[0160]
在20-25℃下,在n2气氛下,将19.88g(46.18mmol)的(15α,16α,17β)-3-羟基雌-1,3,5(10)-三烯-15,16,17-三基三乙酸酯(实施例2)悬浮于596ml的甲醇中,然后分批加入19.88g的碳酸钾,并搅拌3小时。通过tlc(正庚烷:丙酮1:1)监测反应。
[0161]
后处理:将14.91ml的浓乙酸加入到反应混合物中,并搅拌30分钟,在加入298ml的水之后,通过蒸馏除去甲醇,然后将沉淀的晶体保持在0-5℃下1小时,过滤并在过滤器上用水洗涤。然后,将其在40℃下真空干燥至恒重。因此,得到13.66g(97.22%)的白色结晶产物。
[0162]
纯度(hplc):99.67%ααβ-异构体,0.04%βββ异构体(面积),所有杂质《0.10%
[0163]
方法b
[0164]
在20-25℃下,在n2气氛下,将5g(12.87mmol)的(15α,16α,17β)-3-羟基雌-1,3,5(10)-三烯-15,16,17-三基三甲酸酯(实施例4)悬浮于150ml的甲醇中,然后分批加入5.34g(38.6mmol)的碳酸钾,并搅拌3小时。通过tlc(正庚烷:丙酮1:1)监测反应。
[0165]
后处理:将4ml的乙酸加入到反应混合物中,并搅拌30分钟,在加入75ml的水之后,通过蒸馏从该混合物中除去甲醇,将沉淀的晶体保持在0-5℃下1小时,然后过滤并在过滤器上用5ml的0-5℃的水洗涤。然后,将其在40℃下真空干燥至恒重。因此,得到3.80g(97%)的白色结晶产物。
[0166]
纯度(hplc):99.67%ααβ-异构体,0.04%βββ异构体(面积),所有杂质《0.10%
[0167]
mp.:240-243℃
[0168]
ei-hrms:c
18h24
o4[m
+
]的理论值:304.16691;实测值:304.16716;δ=0.82ppm.
[0169]1h nmr(499.9mhz,dmso-d6)δ=4.86(1h,d,j=4.8hz,oh(17)),4.61(1h,br s,oh(16)),4.26(1h,br d,j=3.3hz,oh(15)),3.55-3.78(2h,m,h-16,15),3.25(1h,dd,j=5.7,4.7hz,h-17),1.05(1h,dd,j=10.9hz,j=9.4hz,h-14),0.67(3h,s,h-18)
[0170]
13
c nmr(125.7mhz,dmso-d6)δ=86.3(c-17),75.0(c-16),69.2(c-15),55.5(c-14),39.5(c-13),14.0(c-18)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1