用于治疗结核病的化合物的制作方法
1.本发明涉及用于治疗结核病的化合物和组合物。
背景技术:
2.以下对背景技术的讨论旨在帮助理解本发明。但是应理解,讨论并不是承认或认可所提及材料中的任何一种在本技术的优先权日在任何司法管辖区出版、已知或为公知常识的一部分。
3.结核病(tb)是由细菌结核分枝杆菌(mycobacterium tuberculosis)引起的感染性疾病。需要新的治疗策略来应对结核病的大范围流行以及多药耐药性(mdr)和广泛耐药性(xdr)形式的tb的传播,这仍然是世界范围内的严重的公共卫生挑战。
4.贝达喹啉(bdq;)是一种属于化学类别二芳基喹啉的抗结核化合物。然而,尽管bdq在临床上成功,但是已报告了在广泛耐药性结核病(xdr-tb)患者中对bdq的临床耐药性。
5.wo 2018/151681 a2涉及用于治疗结核病的特定的嘧啶化合物以及含有它们的组合物。
6.存在对于开发另外的用于治疗结核病的化合物或组合物的持续需求。
技术实现要素:
7.在本发明的一个方面中,提供了一种具有式(ia)或(ib)的化合物在本发明的一个方面中,提供了一种具有式(ia)或(ib)的化合物其中r1是氢或甲基;r2是未取代的或取代的烷基;r3是芳基或杂芳基,其任选地被选自卤素、烷基或烷氧基的一个或多个基团取代;
并且,在式(ia)中,x是ch或n并且y是nh、s或o,或者,在式(ib)中,x是nh、s或o并且y是ch或n。
8.在本发明的优选实施例中,在式(ia)中,x是n并且y是nh,或者,在式(ib)中,x是nh并且y是n。
9.更优选地,r1是在嘧啶环的6-位处的甲基。
10.优选地,r2是乙基或-ch2cooch2ch3基团。
11.在本发明另一个优选实施例中,r3是芳基。
12.最优选地,该芳基被一个或多个卤素原子取代。
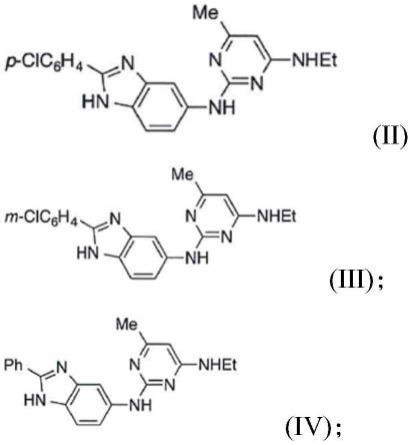
13.具体地,该化合物选自由以下组成的组合物选自由以下组成的组合物选自由以下组成的组及其互变异构体。
14.在另一个方面中,本发明涉及一种组合物,其包含根据本发明的化合物或其药学上可接受的盐和贝达喹啉(bdq)、贝达喹啉(bdq)的类似物或其混合物。
15.优选地,贝达喹啉(bdq)的类似物包含具有式(v)的化合物的外消旋体:或者具有式(vi)的化合物的外消旋体
16.在另一个方面中,本发明涉及一种根据本发明的化合物,该化合物用于在治疗中使用。
17.在另一个方面中,本发明涉及一种根据本发明的化合物,该化合物用于治疗细菌感染。
18.优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
19.在另一个方面中,本发明涉及根据本发明的化合物在制造用于治疗细菌感染的药剂中的用途。
20.优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
21.在另一个方面中,本发明涉及一种根据本发明的组合物,该组合物用于在治疗中使用。
22.在另一个方面中,本发明涉及一种根据本发明的组合物,该组合物用于治疗细菌感染。
23.优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
24.在另一个方面中,本发明涉及根据本发明的组合物在制造用于治疗细菌感染的药剂中的用途。
25.优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
26.在另一个方面中,本发明涉及一种治疗患有细菌感染的受试者的方法,该方法包括向该受试者施用治疗有效量的根据本发明的化合物或组合物的步骤。
27.优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
28.在另一个方面中,本发明涉及一种合成根据本发明的化合物的方法,其中x是nh并且y是n,或者x是n并且y是nh,该方法包括以下步骤:(a)在酸的存在下,使4-硝基苯-1,2-二胺与未取代的或用r3取代的苯甲酸反应;(b)还原硝基以获得相应的胺衍生物;以及(c)使步骤(b)的该胺衍生物与用r1和r2取代的2-氯-嘧啶-4-胺反应。
附图说明
29.图1:(a)具有式(ii)的化合物(命名为gamf1.39)对mtb h37rv的生长抑制。(b)具有式(ii)的化合物(命名为gamf1.39)对牛分枝杆菌卡介苗(bcg)细胞的生长抑制。(c)bdq
和具有式(ii)的化合物针对耻垢分枝杆菌mc
2 155的杀灭动力学。在10x mic
50
的具有式(ii)的化合物和200x mic
50
的bdq的存在下,使细菌在液体培养物(lbt)中生长。在不同时间点(从t=0天直至t=4天)收集细菌培养物的样品,并铺板至middlebrook 7h11琼脂平板上。将板在37℃下孵育3天直至出现菌落。
30.图2:(a)与bdq和具有式(vi)的化合物(命名为bdq1)相比,通过具有式(ii)的化合物(命名为gamf1.39)对牛分枝杆菌bcg细胞的细胞内atp合成的抑制。在内膜外翻的膜囊泡(imv)中通过具有式(ii)的化合物对atp合成的抑制,来自耻垢分枝杆菌具有对于耻垢分枝杆菌为90nm的ic
50
(b)以及对于牛分枝杆菌bcg imv为8.7nm的ic
50
。
31.图3:300nm的具有式(ii)的化合物(命名为gamf1.39)增加了具有式(v)的化合物的外消旋体在atp合成抑制(δ)中的潜能,当将其与单独的此种外消旋体的抑制作用(
▲
)相比较时。
32.图4:具有式(ii)的化合物(命名为gamf1.39)与具有式(vi)的化合物的外消旋体(命名为bdq1)对耻垢分枝杆菌的imv中的atp合成抑制的协同作用。当将具有式(ii)的化合物以浓度依赖性方式添加到具有式(vi)的化合物的外消旋体(300nm)时,atp合成显著降低至几乎完全抑制。
33.图5:具有式(ii)的化合物(命名为gamf1.39)与具有式(vii)的化合物(命名为bdq)对耻垢分枝杆菌的imv中的atp合成抑制的协同作用。当将具有式(ii)的化合物以浓度依赖性方式添加到具有式(vii)的化合物的外消旋体(300nm)时,atp合成显著降低至几乎完全抑制。
具体实施方式
34.现在将参考附图来描述本发明的特定实施例。本文所使用的术语仅出于描述特定实施例的目的,并不旨在限制本发明的范围。此外,除非另行定义,否则本文所使用的全部技术与科学术语具有与本发明所属领域普通技术人员通常所理解的相同的含义。
35.贯穿本说明书,除非有相反指示,否则术语“包含(comprising)”、“由
……
组成(consisting of)”等应解释为非穷举性,或者换句话说,意指“包括但不限于”。
36.贯穿本说明书,除非上下文另有要求,否则词语“包含(comprise)”或变体诸如“包含(comprises)”或“包含(comprising)”将被理解为意味着包括所陈述的特征或特征的组,但不排除任何其他特征或特征的组。
37.贯穿本说明书,除非上下文另有要求,否则词语“包括(include)”或变体诸如“包括(includes)”或“包括(including)”,将被理解为意味着包括所陈述的特征或特征的组,但不排除任何其他特征或特征的组。
38.如本文所使用的,术语“约”典型地意指该陈述值的+/-5%、更典型地该陈述值的+/-4%、更典型地该陈述值的+/-3%、更典型地该陈述值+/-2%、甚至更典型地该陈述值的+/-1%、甚至更典型地该陈述值的+/-0.5%。
39.如本文所使用的,术语“药学上可接受的盐”是指由药学上可接受的无毒碱或酸制备的盐。
40.如本文所使用的,术语“治疗(treatment)”、“治疗(treat)”和“治疗(therapy)”及其同义词是指治疗性处理和预防性或防止性措施二者,其中目的为预防或减慢(减轻)tb。
需要此种处理的那些包括已经患有tb感染的那些以及容易感染tb的那些,或者待预防tb感染的那些。
41.如本文所使用的,术语化合物的“治疗有效量”是能够预防或至少减慢(减轻)tb的活性剂的量。本发明的化合物、组合物和制剂的剂量和施用可以由临床药理学或药物代谢动力学领域的普通技术人员来确定。例如,参见mordenti和rescigno,(1992)pharmaceutical research[药物研究].9:17-25;morenti等人,(1991)pharmaceutical research[药物研究].8:1351-1359;以及mordenti和chappell,"the use of interspecies scaling in toxicokinetics"in toxicokinetics and new drug development[在毒物代谢动力学和新药开发中的“种间标度在毒物代谢动力学中的用途”],yacobi等人(eds)(pergamon press[培格曼出版社]:纽约,1989),pp.42-96。待治疗使用的本发明的化合物、组合物和制剂的有效量将取决于例如治疗目的、施用途径和患者的病状。如本文说明书中所使用的,术语“患者”包括人类和动物。因此,对于治疗师将有必要根据需要确定(titer)剂量并改变施用途径以获得最佳治疗效果。典型的每日剂量可以范围从患者体重的约1μg/kg/天至约50mg/kg/天或者每天更多,约1mg/kg/天至约50mg/kg/天、约1mg/kg/天至约10mg/kg/天,优选约1μg/kg/天至约10mg/kg/天。
[0042]
如本文所使用的,术语“烷基”意指具有1至10个碳原子的支链或非支链饱和烃基团,诸如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、仲戊基、新戊基、己基、庚基、辛基、壬基、癸基。烷基可以是环状或无环的。烷基还可以是取代的或未取代的。如本文所描述的,例如,烷基可用一个或多个基团取代,该一个或多个基团包括但不限于任选地取代的烷基、环烷基、烷氧基、氨基、醚、卤素、羟基、硝基、甲硅烷基、磺氧基(sulfo-oxo)、或巯基。在优选实施例中,术语“烷基”意指为含有从1至6个碳原子的支链或非支链的烷基。在甚至更优选的实施例中,术语“烷基”意指为1至4个碳原子的支链或非支链的烷基。
[0043]
贯穿本说明书,通常使用“烷基”指未取代的烷基和取代的烷基二者;在适当情况下,取代的烷基在本文中还通过鉴别烷基上的一个或多个特定取代基来特别提及。例如,术语“卤代烷基”特别地是指用一个或多个卤素原子(例如氟、氯、溴或碘)取代的烷基。术语“烷氧基烷基”特别地是指用一个或多个烷氧基取代的烷基。术语“烷基氨基”特别地是指用一个或多个氨基取代的烷基。术语“氮杂烷基”特别地是指其中至少一个碳被氮替代的烷基。术语“氧杂烷基”特别地是指其中至少一个碳被氧替代的烷基。
[0044]
如本文所使用的,术语“烷氧基(alkoxy)”和“烷氧基(alkoxyl)”是指通过醚键键合的烷基或环烷基;也就是说,“烷氧基”基团可以定义为—oa1,其中a1是如以上定义的烷基或环烷基。“烷氧基”还包括如刚刚所描述的烷氧基的低聚物;也就是说,烷氧基可以是聚醚,诸如-oa1-oa2或-oa1—(oa2)a—oa3,其中“a”是从1至200的整数,并且a1、a2和a3是烷基和/或环烷基。
[0045]
如本文所使用的,术语“衍生物”或“类似物”是指具有与该使用的术语有关的化合物相似或相关结构的化合物。
[0046]
如本文所使用的,环结构上的取代基的键不指向其特定位置而是指向该环结构的中心,意指该取代基可以结合至该环结构的任何可能位置。作为实例,式(ia)或式(ib)的r1可以结合至嘧啶环上可能位置中的任何一个,即,至其5-或6-位。
[0047]
贯穿本披露内容,可以以范围格式披露某些实施例。应理解的是,以范围格式的描述仅是为了方便和简洁,而不应被解释为对所披露范围的范围的限制。因此,范围的描述应被认为已经特别披露了所有可能的子范围以及该范围内的各个数值。例如,对诸如从1至6的范围的描述应被认为已特别披露了子范围诸如从1至3、从1至4、从1至5、从2至4、从2至6、从3至6等,以及在该范围内的各个数字,例如,1、2、3、4、5和6。范围不限于整数,并且可以包括小数测量值。无论范围的广度如何,这都适用。
[0048]
对于本领域的普通技术人员来说,在结合附图阅读完本发明的具体实施例的以下描述之后,本发明的其他方面将变得清楚。
[0049]
在本发明的一个方面中,提供了一种具有式(ia)或(ib)的化合物在本发明的一个方面中,提供了一种具有式(ia)或(ib)的化合物其中r1是氢或甲基;r2是未取代的或取代的烷基;r3是芳基或杂芳基,其任选地被选自卤素、烷基或烷氧基的一个或多个基团取代;并且,在式(ia)中,x是ch或n并且y是nh、s或o,或者,在式(ib)中,x是nh、s或o并且y是ch或n。
[0050]
有利地,本发明的化合物靶向f-atp合酶。f1f
0 atp合酶(f-atp合酶)是满足分枝杆菌生命周期增殖性需氧以及低氧休眠阶段二者的能量需求的必需的酶之一。该酶由九个化学计量亚单位α3:β3:γ:δ:ε:a:b:b’:c9构成,并且分为膜嵌入的f0域(a:b:b’:c9)和水溶性f1部分(α3:β3:γ:δ:ε)。该f1域含有形成α3:β3六聚物的三个催化性αβ-对,在其中发生atp合成或atp水解。该催化性α3:β3-顶部通过两个中心柄亚单元γ、ε和外围柄与离子泵送f0部分相连。该f0域含有亚单元a、b和b’以及由9个c亚单元组成的环结构。建议以c-环的旋转运动来触发中心亚单元γ和ε旋转,从而致使核苷酸结合亚单元α和β发生连续构象变化,随后将adp+pi合成为atp。
[0051]
已经示出f-atp合酶对于耻垢分枝杆菌和结核分枝杆菌(mtb)(后者导致tb)的最佳生长非常重要。这在其他原核生物和真核生物(即人类)中是不同的,其中该酶对于在可发酵碳源上的生长不是必要的,并且其中增加的糖酵解通量可以补偿氧化磷酸化的损失。
该差异归因于合成分枝杆菌细胞所需的atp的量非常高。该分枝杆菌f-atp合酶的独特性还在于,其没有质子转位能力,以及其在快速或缓慢生长形式下的atp酶活性分别为低活性或潜在活性。
[0052]
bdq(f1f
0 atp合酶的抑制剂)的临床上的成功确认该酶复合物作为用于抗结核病药物开发的要害靶标有效。
[0053]
此外,f-atp合酶属于形成电子传递链(etc)的酶的组合(orchestra),其中细胞色素c氧化酶(cyt-bc1-aa3)和细菌特异性细胞色素bd型甲基萘醌氧化酶(cyt-bd)归属于该酶的组合,并且f-atp合酶有助于atp的生成。
[0054]
更有利地,本发明的化合物靶向耐药性mdr和xdr-tb中分枝杆菌f1f
0-atp合酶的可溶性f1部分。该概念的依据在于诸位发明人对将能量固定在分枝杆菌内部的自然范例的新颖见解、关键催化剂内部负责atp合成的新药物靶标、以及新化合物的开发。此外,本发明的化合物被发现有助于在多种药物组合中与新bdq类似物的协同功效,从而解决了mdr和xdr-tb的挑战。
[0055]
本发明的化合物是wo 2018/151681 a2中描述的化合物的苯甲酰胺类似物,其在以前尚未被描述过。
[0056]
在各个实施例中,在式(ia)中,x是n并且y是nh,或者,在式(ib)中,x是nh并且y是n。
[0057]
在各个实施例中,r1是在嘧啶环的6-位置处的甲基。
[0058]
在各个实施例中,r2是乙基或-ch2cooch2ch3基团。
[0059]
在各个实施例中,r3是芳基。
[0060]
在各个实施例中,该芳基被一个或多个卤素原子取代。
[0061]
在各个具体实施例中,该化合物选自由以下组成的组
[0062][0063][0064]
及其互变异构体。
[0065]
对于化合物(ii)、(iii)和(iv)的测试结果示出于表1中。表1:苯甲酰胺类似物化合物的测试结果
[0066]
非常出人意料地,全部这些类似物显示出良好的ic
50
和mic
50
值,其中这些化合物中的一种(具有式(ii)的化合物)甚至明显优于wo 2018/151681 a2的主要化合物(在那里命名为“cpd6”)。
[0067]
具有式(ii)的化合物展现出有关于最小抑制浓度(mic
50
)的10倍的改进,在mtb h37rv中为3μm(见以上表1;图1a),以及在牛分枝杆菌卡介苗(bcg)中为6.8μm(图1b)。具有式(ii)的化合物在20倍其mic
50
下对耻垢分枝杆菌mc2 155具有杀细菌作用,其通过如图1c中示出的观察到的抑制的细胞生长表明。相反,bdq已经延迟了杀细菌活性,如以前所报告的(a.koul,l.vranckx,n.dhar,h.w.gohlmann,e.ozdemir,j.m.neefs,m.schulz,p.lu,e.mortz,j.d.mckinney,k.andries,d.bald,nat commun[自然通讯]2014,5,3369.)。
[0068]
为确定是否抗分枝杆菌活性归因于氧化磷酸化抑制,进行了对牛分枝杆菌bcg的细胞内atp合成测定(图2a)。具有式(ii)的化合物在3.3μm的ic
50
下对于atp水平具有作用,表明它对于在细胞内抑制atp合成的能力,并且具有式(vi)的化合物(命名为bdq-1)揭示了对于细胞内atp合成抑制的3.4nm的ic
50
,当将其与bdq(ic
50
=11.5nm;图2a)相比较时略有改进。发现作为对照的ic
50 bdq为11.5nm,其与报告的ic
50
相似(preiss l,langer jd,yildizeckhardt-strelau l,guillemont jeg,koul a&meier t(2015)structure of the mycobacterial atp synthase f
o rotor ring in complex with anti-tb drug bedaquiline[具有抗tb药物贝达喹啉的复合物中分枝杆菌atp合酶fo转子环的结构].sci.adv.[科学进展]1:e1500106.)。有趣的是,具有式(ii)的化合物提供了在atp合成抑制方面的18倍的增强,对于耻垢分枝杆菌约90nm(图2b)以及对于牛分枝杆菌bcg imv甚至为8.7nm(图2c)。
[0069]
具有式(ii)的化合物(具有6.51的clogp值)与bdq(clogp=7.25)相比亲脂性较低,并且在小鼠肝脏微粒体中具有良好的代谢稳定性(t
1/2
为29.6min、cl
hep
为60.5ml/min/kg、并且cl
int
为46.8ml/min/mg蛋白质)。
[0070]
为探索具有式(ii)的化合物的抗tb潜能,已由具有与结核分枝杆菌相似基因组的
牛分枝杆菌bcg来感染thp1细胞(单核细胞)。
[0071]
在第二个方面中,本发明涉及一种组合物,其包含根据本发明的化合物或其药学上可接受的盐和贝达喹啉(bdq)、贝达喹啉(bdq)的类似物或其混合物。
[0072]
在各个实施例中,该贝达喹啉(bdq)包含具有式(vii)的化合物:
[0073]
在耻垢分枝杆菌imv上测试了具有式(vii)的化合物(bdq)的外消旋体与不同浓度的具有式(ii)的化合物的协同作用。如图5中所示出的,3x ic
50
(270nm)的具有式(ii)的化合物的添加完全抑制了atp合成。
[0074]
在各个实施例中,贝达喹啉(bdq)的类似物包含具有式(v)的化合物的外消旋体:或者具有式(vi)的化合物的外消旋体:其中式(v)和(vi)中的星号标明手性碳原子。
[0075]
最近,已对具有在结核分枝杆菌菌株以及药理特性方面的改进的潜能的bdq类似物进行了描述(tong ast,choi pj,blaser a,sutherland hs,tsang sky,guillemont j,motte m,cooper cb,andries k,van den broeck w,franzblau sg,upton am,denny wa,
palmer bd,conole d.2017.6-cyano analogues of bedaquiline as less lipophilic and potentially safer diarylquinolines for tuberculosis[贝达喹啉的6-氰基类似物作为较少亲脂性和潜在地更安全的二芳基喹啉用于结核病].acs med chem lett[美国化学会医药化学通讯]8:1019-1024;choi pj,sutherland hs,tong ast,blaser a,franzblau sg,cooper cb,lotlikar mu,upton am,guillemont j,motte m,queguiner l,andries k,van den broeck w,denny wa,palmer bd.2017.synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic b-ring units[含有杂环b-环单元的结核病药物贝达喹啉的类似物的合成和评估].bioorg med chem[生物有机化学与医药化学通讯]27:5190-5196;sutherland hs,tong ast,choi pj,conole d,blaser a,franzblau sg,cooper cb,upton am,lotlikar mu,denny wa,palmer bd.2018.structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles[对于具有被二环杂环替代的萘单元的结核病药物贝达喹啉类似物的结构-活性关系].bioorg med chem[生物有机化学与医药化学]26:1797-1809;sarathy,j.,ragunathan,p.,joon,s.,cooper,c.,upton,a.,gr
ü
ber,g.,和dick,t.(2019)tbaj-876 retains bedaquiline’s activity against subunit c and ε of mycobacterium tuberculosis f-atp synthase[tbaj-876保留了贝达喹啉对结核分枝杆菌f-atp合酶的亚单元c和ε的活性].antimicrob.agents chemother[抗菌物和化学疗法].63(10).pii:e01191-19),包括了具有式(v)的化合物的外消旋体,其似乎与分枝杆菌f-atp合酶的亚单元c-环和亚单元ε相互作用。使用改变的合成方案,合成具有式(v)的化合物的外消旋体(以下实例部分)。
[0076]
图3示出了300nm的具有式(ii)的化合物增加了具有式(v)的化合物的外消旋体在耻垢分枝杆菌imv的atp合成抑制(δ)中的潜能,当将其与单独的此种外消旋体的抑制作用(
▲
)相比较时。
[0077]
具有式(vi)的化合物的外消旋体(命名为bdq1)与不同浓度的具有式(ii)的化合物的协同作用在耻垢分枝杆菌imv上测试。如图4中所示出的,3x ic
50
(270nm)的具有式(ii)的化合物的添加完全抑制了atp合成。
[0078]
另外,使用了人类胚胎干细胞(hesc)系(e3)以检验潜在的药物诱导的遗传毒性和导致hesc分化的对hesc转录程序的扰动。测定揭示了测试的具有式(vi)的化合物的外消旋体(100nm)与300nm的具有式(ii)的化合物组合既没有示出实质性的遗传毒性作用,亦没有在整体转录程序中诱导重大改变。
[0079]
在另一个方面中,本发明涉及一种根据本发明的化合物,该化合物用于在治疗中使用。
[0080]
在另一个方面中,本发明涉及一种根据本发明的化合物,该化合物用于治疗细菌感染,优选地用于治疗结核病,特别地用于治疗多药耐药性或广泛耐药性结核病。
[0081]
在另一个方面中,本发明涉及根据本发明的化合物在制造用于治疗细菌感染的药剂中的用途。
[0082]
优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
[0083]
在另一个方面中,本发明涉及一种根据本发明的组合物,该组合物用于在治疗中
使用。
[0084]
在另一个方面中,本发明涉及一种根据本发明的组合物,该组合物用于治疗细菌感染,优选用于治疗结核病,特别地用于治疗多药耐药性或广泛耐药性结核病。
[0085]
在另一个方面中,本发明涉及根据本发明的组合物在制造用于治疗细菌感染的药剂中的用途。
[0086]
优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
[0087]
在另一个方面中,本发明涉及一种治疗患有细菌感染的受试者的方法,该方法包括向该受试者施用治疗有效量的根据本发明的化合物或组合物的步骤。
[0088]
优选地,该细菌感染是结核病,特别地是多药耐药性或广泛耐药性结核病。
[0089]
在另一个方面中,本发明涉及一种合成根据本发明的化合物的方法,其中x是nh并且y是n,或者x是n并且y是nh,该方法包括以下步骤:(a)在酸的存在下,使4-硝基苯-1,2-二胺与未取代的或用r3取代的苯甲酸反应;(b)还原硝基以获得相应的胺衍生物;以及(c)使步骤(b)的该胺衍生物与用r1和r2取代的2-氯-嘧啶-4-胺反应。实例材料和方法从耻垢分枝杆菌的倒膜囊泡的制备
[0090]
选择耻垢分枝杆菌作为结核分枝杆菌的替代模型,这是因为使用耻垢分枝杆菌有多个优点。耻垢分枝杆菌是腐生性的,并且与结核分枝杆菌不同,它不是致病性的,且可以在生物安全等级2(bsl2)的条件下安全地处理,而无生物安全等级3(bsl3)的要求。此外,与结核分枝杆菌的生长(生成时间:大约24小时)相比,耻垢分枝杆菌的生长快得多(生成时间:大约3小时)。而且,对于结核分枝杆菌需要几乎3至4周来在琼脂平板上产生菌落,而对于耻垢分枝杆菌在琼脂平板上产生菌落仅需要2至3天,从而减少了实验持续时间。重要的是,耻垢分枝杆菌倒膜囊泡(imv)示出了可检测的atp水解活性,其对于待使用或进行的酶测定至关重要。
[0091]
为了纯化用于atp合成和水解测定的耻垢分枝杆菌的imv,使细胞在37℃下在用10%adc、0.5%甘油和0.05%tween80补充的7h9中生长过夜,直至它们达到0.4的od600值。将培养物在200ml的补充的7h9中扩增,并在滚瓶(2rpm)生长直至od
600
=0.4。使用该培养物以接种5l培养物,使其在滚瓶中生长过夜,直至od600=0.4。将约5g(湿重)的耻垢分枝杆菌重悬浮于20ml膜制备缓冲液(50mm mops,2mm mgcl
2 ph 7.5)中,该缓冲液含有不含edta的蛋白酶抑制剂混合物(20ml缓冲液中1片,美国罗氏公司(roche-usa))和1.2mg/ml溶菌酶。将悬浮液在室温下搅拌45分钟,并另外用300μl 1m mgcl2和50μl dna酶i(赛默飞世尔公司(thermo fischer),美国)补充,并在室温下继续再搅拌15分钟。所有后续步骤均在冰上进行。通过冰预冷的m-110l型微流化处理器(m-110l)在18,000psi下通过三个通道对细胞进行破碎。将含有裂解细胞的悬浮液在4℃下在4,200x g下离心20min。使含有膜组分的上清液在4℃下在45,000x g下进一步经受超速离心1h。弃去上清液,并且将沉淀的膜组分重悬浮在含有15%甘油的膜制备缓冲液中,将其等分、速冻并在-80℃下贮存。通过bca法确定囊泡中蛋白质的浓度。将倒膜囊泡在-80℃下贮存。
atp合成测定
[0092]
在平底白色微量滴定96孔板(美国康宁公司(corning usa))中测量atp合成。在含有10μm adp、250μm pi和1mm nadh的测定缓冲液(50mm mops,ph 7.5,10mm mgcl2)中制得反应混合物。通过添加溶解在测定缓冲液中的100mm kh2po4盐来调节pi的浓度。通过添加耻垢分枝杆菌的倒囊泡至最终蛋白浓度为5μg/ml来开始atp合成。将反应混合物在室温下孵育30min,然后添加50μlcelltiter-glow试剂,并将混合物在室温下于黑暗中再孵育10min。使用下列参数,通过帝肯板读数器infinite 200pro(美国帝肯公司(tecan usa))测量所产生的与合成的atp相关的发光:发光、积分时间500ms、无衰减。抗分枝杆菌活性
[0093]
针对耻垢分枝杆菌mc2 155、结核分枝杆菌h37rv和牛分枝杆菌bcg筛选测试化合物和对照药物。在90%dmso中将测试化合物的初始储备溶液制成10mm的浓度。使用环丙沙星作为阳性对照,并且使用媒介物dmso作为阴性对照。在第一种方法中,在固定浓度为50μm的微生物培养物上测试化合物。将上述菌株中的每一种在用0.2%甘油和10%adc(白蛋白葡萄糖过氧化氢酶(albumin dextrose catalase))补充的middlebrook 7h9液体培养基中在37℃下培养,直至实现对数生长(od
600 0.4-0.6)。通过在试管中将悬浮液相对于od
600 0.1稀释到最终体积为1ml来获得测试接种物,并将其在37℃下孵育24小时(耻垢分枝杆菌)和7天(mtb)。选择与阳性和阴性对照相比未示出可见杆菌生长的测试化合物作为命中物。用于具有式(ia)或(ib)的化合物的合成的通用操作
[0094]
根据以下方案(方案s1)获得具有式(ia)或(ib)的化合物,其通过在多聚磷酸的存在下加热相应的芳基或杂芳基酸(诸如,例如未取代的或取代的苯甲酸)与4-硝基苯-1,2-二胺。使用fe粉末在氯化铵的存在下的硝基还原提供了苯并咪唑胺衍生物,将其在微波加热下与对应的取代的2-氯嘧啶-4-胺(在方案中命名为s2)反应以给出最终产物。方案s1.具有式(ii)、(iii)和(iv)的化合物的合成
[0095]
2-(4-氯苯基)-5-硝基-1h-苯并[d]咪唑s6a
[0096]
将4-氯苯甲酸(292mg,1.87mmol)和4-硝基苯-1,2-二胺(300mg,1.96mmol,1.05eq)在多聚磷酸(4ml)中的混合物在140℃下搅拌4小时。通过倾倒入水(5ml)中使反应淬灭并用10n naoh溶液调节至ph 7。将沉淀物过滤并在真空中干燥以给出黑色残余物。通过硅胶快速色谱法(0%-40%etoac/己烷)使粗产物纯化以提供作为淡绿色固体的化合物
s6a(142mg,28%产率);1h nmr(400mhz,dmso-d6)δ13.7(br.s,1h),8.49(s,1h),8.23(d,j=6.8hz,2h),8.14(dd,j=8.8,2.4hz,1h),7.78(d,j=8.4hz,1h),7.69(d,j=8.4hz,2h);ms(esi)m/z 274.0[c
13
h8cln3o2+h]
+
。
[0097]
2-(3-氯苯基)-5-硝基-1h-苯并[d]咪唑s6b
[0098]
将3-氯苯甲酸(292mg,1.87mmol)和4-硝基苯-1,2-二胺(300mg,1.96mmol,1.05eq)在多聚磷酸(4ml)中的混合物在140℃下搅拌2小时。通过倾倒入水(5ml)中使反应淬灭并用10n naoh溶液调节至ph 7。将沉淀物过滤并在真空中干燥以给出黑色残余物。通过硅胶快速色谱法(0%-40%etoac/己烷)使粗产物纯化以提供作为橙色固体的化合物s6b(82.6mg,16%产率);1h nmr(400mhz,dmso-d6)δ13.7(br.s,1h),8.51(s,1h),8.26(s,1h),8.22-8.14(m,2h),7.80(d,j=8.4hz,1h),7.65(s,2h);ms(esi)m/z 274.0[c
13
h8cln3o2+h]
+
。
[0099]
2-(4-氯苯基)-1h-苯并[d]咪唑-5-胺s7a
[0100]
将s6a(132mg、0.482mmol)、铁粉末(269mg,4.82mmol,10eq.)和nh4cl(258mg,4.82mmol,10eq.)在4:1etoh/水(5ml)中的混合物在80℃下加热5小时。将混合物通过硅藻土垫过滤,用meoh洗涤。将滤液浓缩并将残余物吸收在水(10ml)中并且用etoac(3x 5ml)萃取。将合并的有机物用盐水洗涤,经na2so4干燥,过滤并在真空中浓缩,以给出作为褐色油的s7a(87.6mg,75%产率),将其不经进一步纯化而在下一个步骤中使用。1h nmr(400mhz,cdcl3)δ7.91(d,j=8.4hz,2h),7.48-7.42(m,3h),6.84(s,1h),6.69(dd,j=8.4,2.0hz,1h);ms(esi)m/z 244.0[c
13h10
cln3+h]
+
。
[0101]
2-(3-氯苯基)-1h-苯并[d]咪唑-5-胺s7b
[0102]
将s6b(82.6mg、0.302mmol)、铁粉末(169mg,3.02mmol,10eq.)和nh4cl(161mg,3.02mmol,10eq.)在4:1etoh/水(5ml)中的混合物在80℃下加热3小时。将混合物通过硅藻土垫过滤,用meoh洗涤。将滤液浓缩并将残余物吸收在水(10ml)中并且用etoac(3x 5ml)萃取。将合并的有机层用盐水洗涤,经na2so4干燥,过滤并在真空中浓缩,以给出作为褐色油的s7b(72.4mg,98%产率),将其不经进一步纯化而在下一个步骤中使用。1h nmr(400mhz,cdcl3)δ7.99(s,1h),7.88-7.85(m,1h),7.47(d,j=8.4hz,1h),7.36(d,j=3.2hz,2h),6.82(br.s,1h),6.68(dd,j=8.4,2.0hz,1h);ms(esi)m/z 244.0[c
13h10
cln3+h]
+
。
[0103]n2-(2-(4-氯苯基)-1h-苯并[d]咪唑-5-基)-n
4-乙基-6-甲基嘧啶-2,4-二胺(式(ii))
[0104]
将2-氯-n-乙基-6-甲基嘧啶-4-二胺s2(35.0mg,0.204mmol)和苯胺s7a(74.5mg,0.306mmol,1.5eq.)在n-buoh(1ml)中的溶液在微波反应器中在180℃下加热2小时。使反应混合物与甲苯共沸以给出白色残余物,将其通过制备型hplc(20%-50%mecn/h2o;0.1%甲酸)纯化以提供具有式(ii)的化合物(甲酸盐),其经冷冻干燥为灰白色固体(12.0mg,15.5%产率);1h nmr(400mhz,dmso-d6)δ12.6(br.s,1h),9.00(s,1h),8.28(s,1h),8.14(d,j=8.8hz,2h),7.59(d,j=8.8hz,2h),7.45(q,j=8.4hz,2h),6.98(br.s,1h),5.78(s,1h),3.37(br.s,2h),2.14(s,3h),1.18(t,j=7.2hz,3h);13c nmr(100mhz,dmso-d6)δ163.2,163.0,159.6,133.8,129.3,129.1,128.9,127.8,127.7,34.9,23.3,14.7;ms(esi)m/z 379.1[c
20h19
cln6+h]
+
;[c
20h19
cln6+h]
+
的hrms(esi)m/z计算值为379.1433,实测379.1428;熔点=142℃-143℃。
[0105]n2-(2-(3-氯苯基)-1h-苯并[d]咪唑-5-基)-n
4-乙基-6-甲基嘧啶-2,4-二胺(式(iii))
[0106]
将2-氯-n-乙基-6-甲基嘧啶-4-二胺s2(34.0mg,0.198mmol)和苯胺s7b(72.4mg,0.297mmol,1.5eq.)在n-buoh(1ml)中的溶液在微波反应器中在180℃下加热2小时。使反应混合物与甲苯共沸以给出白色残余物,将其通过制备型hplc(20%-60%mecn/h2o;0.1%甲酸)纯化以提供具有式(iii)的化合物(甲酸盐),其经冷冻干燥为白色固体(38.4mg,51.2%产率);1h nmr(400mhz,dmso-d6)δ12.7(br.s,1h),9.14(s,1h),8.30(s,1h),8.18(s,1h),8.10(d,j=7.6hz,1h),7.57-7.44(m,4h),7.04(br.s,1h),5.79(s,1h),3.37(br.s,2h),2.15(s,3h),1.18(t,j=7.2hz,3h);13c nmr(100mhz,dmso-d6)δ163.5,163.0,159.3,148.7,137.2,133.7,132.5,130.8,128.9,125.7,125.6,124.6,115.5,35.0,23.1,14.7;ms(esi)m/z 379.1[c
20h19
cln6+h]
+
;[c
20h19
cln6+h]
+
的hrms(esi)m/z计算值为379.1433,实测379.1426;熔点=142℃-143℃。
[0107]n4-乙基-6-甲基-n
2-(2-苯基-1h-苯并[d]咪唑-5-基)嘧啶-2,4-二胺(式(iv))
[0108]
将2-氯-n-乙基-6-甲基嘧啶-4-二胺s2(30.0mg,0.175mmol)和可商购的苯胺s7c(54.8mg,0.262mmol,1.5eq.)在n-buoh(1ml)中的溶液在微波反应器中在180℃下加热2小时。使反应混合物与甲苯共沸以给出白色残余物,将其通过制备型hplc(5%-60%mecn/h2o;0.1%甲酸)纯化以提供具有式(iv)的化合物(甲酸盐),其经冷冻干燥为白色固体(46.2mg,76.7%产率);1h nmr(400mhz,dmso-d6)δ12.6(br.s,1h),9.28(s,1h),8.25(s,1h),8.13(d,j=7.2hz,2h),7.52(t,j=7.2hz,2h),7.48-7.43(m,3h),7.13(br.s,1h),5.79(s,1h),3.37(br.s,2h),2.15(s,3h),1.18(t,j=7.2hz,3h);13c nmr(100mhz,dmso-d6)δ163.5,163.0,159.0,150.4,136.6,130.4,129.3,128.8,126.1,115.3,35.0,22.8,14.6;ms(esi)m/z 345.2[c
20h20
n6+h]
+
;[c
20h20
n6+h]
+
的hrms(esi)m/z计算值为345.1822,实测345.1818;熔点=149℃-150℃。用于具有式(v)的化合物的外消旋体的合成的通用操作试剂和条件:(i)litmp,thf,-78℃,1.5h然后适当的醛,-78℃,4h(4,37%),(5,55%);(ii)incl3,ph2sihcl,dce,80℃,12h,44%;(iii)et3sih,tfa,ch2cl2,50℃,60%;(iv)二异丙基氨基锂(lda),thf,-78℃,1.5h然后8,然后hoac,(5307(从11与8的反应形成),9%),(5366,32%),(5316,33%);(v)pd(pph3)4,cs2co3,phme/dmf,110℃,5h,55%。
[0109]
具有式(v)的化合物的外消旋体的合成如以下进行:
[0110]
四甲基哌啶锂(litmp)介导的甲氧基喹啉(1)和5-异丙氧基-2-甲氧基烟碱醛(2)和2,3-二氢苯并[b][1,4]二噁英-5-甲醛(3)的添加分别提供了中间体苄基醇(4)和(5)。随后使用incl3、ph2sihcl、dce和et3sih/tfa在酸性条件下进行脱氧,分别成为对应的二氢加成物(6)和(7)。不幸的是,由于吡啶部分的碱性抑制反应,使用任一脱氧条件均未形成6-溴-3-((2,3-二甲氧基吡啶-4-基)甲基)-2-甲氧基喹啉(11)。可替代地,采用了更长的合成途径,其使用在甲氧基喹啉硼酸(9)与4-(溴甲基)-2,3-二甲氧基吡啶(10)之间的铃木反应(suzuki reaction)以提供6-溴-3-((2,3-二甲氧基吡啶-4-基)甲基)-2-甲氧基喹啉(11)。
[0111]
然后通过lda介导的适当的苄基喹啉(11)和1-(2,6-二甲氧基吡啶-4-基)-3-(二
甲基氨基)丙烷-1-酮(8)的添加来合成具有式(v)的化合物的外消旋体,其由sutherland等人报告(sutherland hs,tong ast,choi pj,conole d,blaser a,franzblau sg,cooper cb,upton am,lotlikar mu,denny wa,palmer bd.2018.structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles[对于具有被二环杂环替代的萘单元的结核病药物贝达喹啉类似物的结构-活性关系].bioorg med chem[生物有机化学与医药化学]26:1797-1809)。使得到的二芳基喹啉形成为两种非对映异构体的外消旋混合物,将其通过柱色谱法将其分离。通过x-射线晶体学进一步确定期望的非对映异构体的结构并且在atp合成和细胞生长测定中表征。
[0112]
用于具有式(vi)的化合物的合成的通用操作
[0113]
如以下方案中所示出的合成具有式(vi)的化合物:
[0114]
本领域的技术人员应进一步认识到,以上描述的特征的变化和组合并不是替代或取代,而是可以组合以形成落在本发明预期范围内的其他实施例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1