一种催化乙苯等烷基苯及衍生物羟化的突变体及其应用
本发明涉及生物催化反应,具体的说是一种协同选择性催化烷基苯及衍生物羟化的突变体及其利用h2o2实现烷基苯及衍生物sp3c-h键高效区域及立体选择性羟化调控合成光学纯的(s)/(r)-苯基烷基醇的应用。
背景技术:
手性α或β位羟基烷基苯是重要的精细化学品,是香料、化妆品和药物等多个领域的重要中间体。例如,(s)-1-苯乙醇是合成抗抑郁药物氟西汀的重要前体;(r)-1-茚醇是合成长效抗抑郁药物茚达曲林的药物前体,且其r构型药效是s构型的20倍以上;(s)-1-苯基2-丙醇是合成治疗气喘和抗嗜睡药物安非他命的重要前体;(s)-四氢萘醇可用来合成jak3抑制剂,用来治疗血液系统疾病、肿瘤、类风湿性关节炎及银屑病等。
目前手性的α或β位羟基烷基苯的化学制备主要通过三种途径:1)通过动力学拆分由外消旋的羟基烷基苯获取单一构型的产物,但该途径最大的理论收率只有50%,且需要制备原料羟基烷基苯;2)以前手性羰基烷基苯为底物进行羰基的不对称还原得到手性α或β位羟基烷基苯,该途径一般均需要过渡金属铑、钌、铱等金属催化剂结合复杂的手性配基来实现不对称催化,故成本昂贵而且反应条件不温,需要额外的试剂作氢供体。3)通过对烷基苯sp3c-h键的不对称羟化来直接制备手性α或β位羟基烷基苯,此途径具有独特优势:不需要对底物进行预氧化官能团化,且原子经济性更高;此外该途径能大大提高石化资源的高附加值利用及减少挥发性芳香有机物的污染。比较典型实例为evgenii等利用手性mn-氨基吡啶复合物催化乙苯生成1-苯乙醇,其r构型的ee可以达到76%,但催化过氧化现象较严重,立体选择性有待提高。针对目前化学催化现状,生物催化因温和的反应条件及在选择性方面的先天优势,越来越受到关注。
自然界中存在能天然羟化烷基苯的微生物,但种类不多,如chen等发现假单胞菌pseudomonasmonteiliita-5可以催化乙苯、丙苯等不对称羟化生成r-α苯基醇,手性ee值可达94%;yadav等发现黄曲霉aspergillusflavusmtcc-1783和mtcc-1884羟化乙苯、丙苯的手性可达99%ee以上。由于野生菌往往受到其自身生长因素、目标酶表达量低及反应及后处理复杂等限制,均制约其实际应用。天然不对称催化烷基苯羟化的酶种类也不多,主要为苯脱氢酶、过加氧酶、细胞色素p450酶等。乙苯脱氢酶主要来源于厌氧菌,如kniemeyer等发现来源于反硝化型弧菌azoarcusstrainebn1的乙苯脱氢酶可高手性的羟化乙苯和丙苯生成s-α苯基醇,但此酶在粗酶液中还能保持活性,纯化后即失活,综合考到厌氧菌的本身特性,不适于实际催化应用。
过加氧酶因以h2o2作为末端氧化剂,催化过程简单,及高效的催化活性与选择性是目前研究的一大热点。但目前发现和研究的该类酶种类不多,如来源于caldaromycesfumago的氯过氧化物酶可催化乙苯、丙苯等不对称羟化,催化乙苯的手性可达97%(r);近期发现来源于agrocybeaegerita的过加氧酶可以高效催化烷基苯苄位不对称羟化,乙苯、丙苯手性可达99%ee以上,茚满苄位羟化的手性也在87%以上,且具有良好的催化活性。但目前仅发现一种酶,且存在一些弊端,该酶均主要选择r构型且存在过氧化现象,此外过加氧酶不易异源表达和进行分子生物学操作,目前仅在酵母、黑曲霉、米曲霉中表达成功,活性损失亦较多。
细胞色素p450酶因惰性c-h键的高效氧化能力和优异的区域及立体选择性是制备手性α位或β位羟基烷基苯研究和应用的重点。sligar等发现p450cam及其突变体可以羟化乙苯生成r-1-苯乙醇,虽手性一般。jian-hexu实验室发现p450lamo催化乙苯生成s-1-苯乙醇的手性可达99%,催化丙苯生成1-苯丙醇的手性在90%ee以上,但整体活性和区域选择性一般,也存在一定过氧化现象。zhili实验室发现p450tol对乙苯具有较好的区域选择性,其生成s-1-苯乙醇的手性亦可达97%以上,但同样存在过氧化现象。janssen等发现cyp154h1可对乙苯、丙苯等进行不对称羟化,其手性和活性较一般。zhili实验室将p450pyr进行多轮的定向进化使其具有良好的丙苯羟化能力,且羟化位点不同于大多数酶,主要选择苯β位,生成s-1-苯基-2-丙醇,手性可达98%,区域选择性在99%以上。p450bm3因其超高效的惰性c-h键催化能力一直受到人们的研究关注。qing-shanli等发现wtp450bm3氧化丙苯主要生成1-苯基-1-丙醇,而且f87单突变可改变生成α位/β位羟化比例,但立体选择性一般。wong实验室发现酶改造可改变丙苯羟化的区域选择,突变体kt5可主选择生成1-苯基-2-丙醇,选择性可达78%。之后研究又发现i401单突变可显著提高丙苯羟化活性,但未有立体选择的相关报导。除了蛋白质工程手段外,利用底物误识别策略同样可赋予和提高p450bm3选择性羟化烷基苯的能力。bell实验室利用p450bm3及其突变体搭配不同诱导pfcs分子实现了烷基苯苄位选择性羟化,区域选择性可达80%以上,但立体选择一般,手性均在80%ee以下。watanabe实验室通过对诱导分子结构的进一步优化,使wtp450bm3选择性羟化烷基苯的能力得到提升,羟化乙苯、丙苯和四氢化萘的苄位区域选择均可在94%以上,四氢化萘的手性可达96%ee,通过不同的小分子调控还可实现1-茚醇立体选择的调控,r/s构型均可实现50%ee以上。
总结以上我们可看出,目前的生物催化底物和反应的选择性较单一,蛋白质工程手段和底物误识别策略虽已实现了烷基苯不同的选择性羟化能力,但目前的调控能力不足,整体区域选择和立体选择性能一般。此外绝大多数p450酶的催化过程高度依赖提供电子的还原辅酶nad(p)h,以及负责电子传递的还原伴侣蛋白。还原辅酶价格昂贵且其原位再生技术也不成熟,这也大大限制p450酶在手性α位或β位羟基烷基苯制备中的实际应用。为解决上述问题,人们也尝试利用底物误识别策略对极少数能利用h2o2的p450酶进行改造,watanabe实验室利用不同诱导分子实现了p450bsβ对乙苯的苄位羟化,但其手性最高仅为68%ee。
综上所述,不管是化学催化还是生物催化,都依然存在技术瓶颈,且目前不管是酶的种类数量还是催化性能都不能满足目前手性α位或β位羟基烷基苯的制备需求,也大大制约了烷基苯的高附加值利用。因此有必要开发一种新的催化策略,既能在温和条件下具有高效的催化效率,又能实现烷基苯羟化的高区域和立体选择调控,还能避免昂贵辅因子的依赖,降低成本,因此本专利具有十分重要的研究和应用价值。
技术实现要素:
鉴于现有技术的不足,本发明的目的在于提供一种协同选择性催化烷基苯及衍生物羟化的突变体及其利用h2o2实现烷基苯及衍生物sp3c-h键高效区域及立体选择性羟化调控合成光学纯的(s)/(r)-苯基烷基醇的应用。
为实现上述目的,本发明采用技术方案为:
一种催化乙苯等烷基苯及衍生物羟化的突变体,突变体为细胞色素p450bm3氧化酶第87位苯丙氨酸处和第268位苏氨酸处的两个位点突变;其中,两个位点的氨基酸可相同或不同的突变为甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、甲硫氨酸、苯丙氨酸、天冬氨酸、天冬酰胺、谷氨酸、谷氨酰胺、丝氨酸、苏氨酸、半胱氨酸或脯氨酸。
所述第87位苯丙氨酸处位点突变为丙氨酸、亮氨酸、缬氨酸或苏氨酸;所述第268位苏氨酸处位点突变为缬氨酸、异亮氨酸或脯氨酸。
所述突变体还可叠加于细胞色素p450bm3氧化酶的下述氨基酸位点中的一个或几个进行突变;下述氨基酸位点为第75位亮氨酸处、第78位缬氨酸处、第81位苯丙氨酸处、第82位丙氨酸处、第88位苏氨酸处、第168位天门冬氨酸处、第181位亮氨酸处、第184位丙氨酸处、第188位亮氨酸处、第260位苏氨酸处、第263位异亮氨酸处、第264位丙氨酸处、第267位谷氨酸处、第328位丙氨酸处、第330位丙氨酸处、第437位亮氨酸处。
所述下述氨基酸位点的氨基酸可相同或不同的突变为丙氨酸、缬氨酸、异亮氨酸、亮氨酸、甲硫氨酸、苯丙氨酸、脯氨酸、丝氨酸、苏氨酸、半胱氨酸、谷氨酰胺、天门冬酰胺、谷氨酸或天门冬氨酸。
进一步的说,利用双功能小分子化合物im-c6-phe/im-c5-phe诱导所述p450酶的氧化结构域突变体实现利用h2o2对乙苯等烷基苯及衍生物的sp3c-h键进行高效区域及立体选择性羟化生成(r)-苯基烷基醇;其中,突变体为含所述第87位和第268位进行突变的二、三或四突变体,所述第87位苯丙氨酸突变为甘氨酸或丙氨酸,第268位苏氨酸变为缬氨酸、脯氨酸或异亮氨酸;
利用双功能小分子化合物im-c5-phe诱导所述p450酶的氧化结构域突变体实现利用h2o2对乙苯等烷基苯及衍生物的sp3c-h键进行高效区域及立体选择性羟化生成(s)-苯基烷基醇;其中,突变体为含所述第87位和第268位进行突变的二、三或四突变体,所述第87位苯丙氨酸突变为缬氨酸、异亮氨酸、苏氨酸或谷氨酸,第268位苏氨酸变为缬氨酸、亮氨酸或异亮氨酸;
再进一步的说,具体的突变体为:
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为甘氨酸、丙氨酸、缬氨酸、异亮氨酸、亮氨酸、甲硫氨酸、天冬氨酸、天冬酰胺、谷氨酸、谷氨酰胺、丝氨酸、苏氨酸、半胱氨酸或脯氨酸。所得突变体依次命名为f87g、f87a、f87v、f87i、f87l、f87m、f87d、f87n、f87e、f87q、f87s、f87t、f87c、f87p。
将seqidno.1所示氨基酸序列的第268位的苯丙氨酸替换为甘氨酸、丙氨酸、缬氨酸、亮氨酸、异亮氨酸、丝氨酸、半胱氨酸或脯氨酸,所得突变体依次命名为t268g、t268a、t268v、t268l、t268i、t268s、t268c、t268p。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为甘氨酸,第268位的苯丙氨酸替换为缬氨酸,命名为f87g-t268v。
或,将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为甘氨酸,第268位的苯丙氨酸替换为异亮氨酸,命名为f87g-t268i。
或,将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为缬氨酸,命名为f87a-t268v。
或,将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为脯氨酸,命名为f87a-t268p。
或,将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为缬氨酸,第268位的苯丙氨酸替换为异亮氨酸,命名为f87v-t268i。
或,将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为谷氨酸,第268位的苯丙氨酸替换为异亮氨酸,命名为f87q-t268i。
或,将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为苏氨酸,第268位的苯丙氨酸替换为缬氨酸,命名为f87t-t268v。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为甘氨酸,第268位的苯丙氨酸替换为异亮氨酸,第78位亮氨酸替换为甲硫氨酸,命名为f87g-t268i-v78m。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为缬氨酸,第82位亮氨酸替换为半胱氨酸,命名为f87a-t268v-a82c。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为缬氨酸,第181位亮氨酸替换为甲硫氨酸,命名为f87a-t268v-l181m。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为脯氨酸,第184位丙氨酸替换为缬氨酸,命名为f87a-t268p-a184v。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为缬氨酸,第78位缬氨酸替换为异亮氨酸,第82位丙氨酸替换为半胱氨酸,命名为f87a-t268v-v78i-a82c。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为缬氨酸,第82位丙氨酸替换为半胱氨酸,第181位亮氨酸替换为甲硫氨酸,命名为f87a-t268v-a82c-l181m。
一种表达载体,表达载体含所述任意一项突变体。
一种基因工程菌,基因工程菌含有所述的表达载体。
一种突变体的应用,所述突变体在催化烷基苯及其衍生物羟化合成光学纯的(s)/(r)-苯基烷基醇中的应用。
所述催化体系为双功能小分子化合物、所述突变体、h2o2和底物;其中,双功能小分子化合物为n-(ω-咪唑基,脂肪酰基)-l-苯丙氨酸衍生物。
一种协同选择性催化烷基苯及衍生物羟化合成光学纯的(s)/(r)-苯基烷基醇的方法,利用具有双功能的小分子n-(ω-咪唑基,脂肪酰基)-l-苯丙氨酸衍生物诱导所述p450酶的氧化结构域突变体,实现通过h2o2对烷基苯及衍生物的sp3c-h键的选择性羟化调控,进而合成光学纯的(s)/(r)-苯基烷基醇。
进一步地说,利用双功能小分子化合物im-c6-phe/im-c5-phe诱导所述p450酶的氧化结构域突变体实现利用h2o2对烷基苯及衍生物的sp3c-h键进行高效区域及立体选择性羟化生成(r)-苯基烷基醇;其中,突变体为含所述第87位和第268位进行突变的二、三或四的突变体,所述第87位苯丙氨酸突变为甘氨酸或丙氨酸,第268位苏氨酸变为缬氨酸、脯氨酸或异亮氨酸;
利用双功能小分子化合物im-c5-phe诱导所述p450酶的氧化结构域突变体实现利用h2o2对烷基苯及衍生物的sp3c-h键进行高效区域及立体选择性羟化生成(s)-苯基烷基醇;其中,突变体为含所述第87位和第268位进行突变的二、三或四的突变体,所述第87位苯丙氨酸突变为缬氨酸、异亮氨酸、苏氨酸或谷氨酸,第268位苏氨酸变为缬氨酸、亮氨酸或异亮氨酸;
所述羟化体系为将0.01μm-1mm所述突变体与0.1μm-300mm底物加入至ph5.0-9.0的缓冲溶液中,再添加1μm-300mm的所述双功能小分子化合物和1μm-600mmh2o2在0℃-45℃反应1min-24h来实现底物的催化氧化。
上述底物为烷基苯及衍生物,包括乙苯、丙苯、茚满、四氢化萘等脂肪链取代苯、苯并脂肪环取代化合物;还可为芳香邻位、对位、间位取代乙苯、丙苯等,取代基可以是卤素、硝基、腈基、三氟甲基等,其中优选乙苯、丙苯、茚满及四氢化萘。
本发明通过蛋白质工程技术与双功能小分子协同策略的双重调控能力相结合,既能实现酶催化乙苯及其类似物羟化的高催化效率及高区域、立体选择性调控,又能解除昂贵辅因子的依赖,大大降低应用成本。
具体是;以来源于巨大芽孢杆菌的细胞色素p450-bm3单加氧酶氧化酶部分为模板,利用含有87突变位点的突变引物进行pcr突变,测序获得f87单突变体;然后以单突变体基因为模板,以同样的方法pcr扩增出f87-t268组合双突变体,以此双突变p450bm3-h2o2系统对烷基苯及衍生物的羟化能力大幅提升,同时通过对f87-t268位点的氨基酸组合及与不同双功能小分子的有机结合可以实现乙苯等烷基苯及衍生物sp3c-h键的区域及立体选择性羟化调控。然后进一步通过组合突变、迭代饱和突变等其他突变方式获得含有87突变位点和268突变位点及其周围其他一个位点或多个位点相结合的多突变体,将其引入至羟化体系,大大提高此体系对乙苯及其类似物的高效选择性羟化调控性能,实现烷基苯制备光学纯的(s)/(r)-苯基烷基醇的催化应用。
与现有技术相比,本发明具有以下有益效果:
本发明首次通过蛋白质工程手段与双功能小分子结构优化的双重调控相结合,实现乙苯等烷基苯及衍生物sp3c-h键的高效化学、区域及立体选择性羟化调控。即烷基链sp3c-h及苯环sp2c-h键的不同区域选择羟化调控,而是创新地实现烷基苯烷基侧链中具有多个相似性质的sp3c-h键的高效区域选择及立体选择调控。而且相对于以往野生酶及利用蛋白质工程手段或化学干预策略研究中对立体选择调控能力的不足,目前最优调控为watanabe实验室利用化学干预策略对茚满羟化生成1-茚醇立体选择调控,最优r/s构型却仅为50%ee以上。而本发明实现了90%ee左右的1-茚醇立体选择调控,这充分体现了该体系在烷基苯选择性羟化利用方面的巨大能力。本发明首次实现了利用同一个酶进行乙苯等多种烷基苯及衍生物sp3c-h键高效区域及立体选择性羟化调控合成光学纯的(s)/(r)-苯基烷基醇,而且利用特定的双功能小分子诱导的p450-h2o2系统羟化解除了p450酶对辅因子和电子传递链的依赖,并且显著提高野生酶对乙苯及其类似物的羟化能力,使工业应用成本大大降低,这都大大提高了利用此体系进行乙苯等烷基苯及衍生物的高附加值利用,具有很强的工业应用潜力。
附图说明
图1为本发明实施例提供的im-c6-phe/im-c5-phe协同p450bm3典型突变体利用h2o2调控乙苯苄位的选择性羟化的示意图。
图2为本发明实施例提供的im-c5-phe协同p450bm3典型突变体利用h2o2调控正丙苯不同sp3c-h的区域及立体选择性羟化的示意图。
图3为本发明实施例提供的im-c5-phe协同p450bm3典型突变体利用h2o2调控四氢化萘苄位羟化的立体选择性的示意图。
具体实施方式
以下对本发明的原理和特征做进一步的描述,所列举实施例只用于解释本发明,并非用于限定本发明的保护范围。另外,下述实施例中所使用的实验方法如无特殊说明,均为本领域的常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
本发明首先以双功能小分子协同p450过加氧酶策略为基础,t268位和f87位的组合及小分子结构优化的辅助调控是实现烷基苯及衍生物sp3c-h键高效区域及立体羟化的关键位点。选取实现不同区域及立体选择r/s构型的父本体系,以细胞色素p450-bm3单加氧酶的结构为基础,选取对底物结合构象有明显影响的其他位点,进行半理性设计的氨基酸突变,通过分析获得的每个位置的突变指纹图谱,进而通过组合迭代获得显著增强乙苯等烷基苯及其衍生物sp3c-h区域及立体选择性羟化区域的体系。
同时下述实施例的小分子化合物可商业购买或根据现有技术合成获得。
下述实施例中涉及的工程菌构建、酶的诱导表达与纯化、催化活性测定均采用下列方法,具体如下:
1.野生型p450bm3氧化酶部分(bmp)基因的构建
设计p450bm3氧化酶部分的上下游引物,以野生型p450bm3(seqidno.1,其ncbi登录号为nz_cp009920.1)为模板用pcr技术来扩增获得目的基因。
上游引物bmp-f:catgccatgggcatgacaattaaagaaatgcc
下游引物bmp-r:
ccggaattcttagtggtggtggtggtggtgaagcggaatttttttcgattttgcttttacc
pcr反应体系如下。
反应条件:94℃15s→(94℃15s→60℃5s→72℃80s)×2→(94℃15s→58℃5s→72℃80s)×2→(94℃15s→55℃5s→72℃80s)×26→72℃10min→16℃∞
pcr扩增产物经琼脂糖凝胶电泳检测后,在紫外条件下,切下目的条带按照胶回收试剂盒的操作说明回收目的片段。
建立双酶切体系
用限制性内切酶ncoi、ecori分别对载体pet-28a(+)质粒和bmp目的片段进行双酶切。质粒和bmp目的片段酶切体系分别如下。
37℃酶切2h后通过琼脂糖凝胶电泳确认酶切产物大小,电泳完后在紫外条件下,分别切下目的条带,然后按照胶回收试剂盒的操作说明回收目的片段。
建立连接体系
将用相同限制性内切酶处理过的bmp、pet-28a(+)质粒进行连接反应,构建重组质粒pet-28a(+)-bmp。连接体系如下。
16℃过夜连接。
采用热激法将连接产物转入大肠杆菌bl21(de3)感受态细胞中,测序正确的单菌落,即为构建成功的pet-28a(+)-bmp。
2、突变体的构建
利用pcr诱变技术对野生型细胞色素p450-bm3单加氧酶的基因(基因序列如seqidno.1所示)进行定点诱变。
以连接有p450bm3氧化酶部分(bmp)基因的重组质粒pet-28a(+)-bmp为模板,设计一对包含突变位点的正反向引物,进行pcr,得到重组基因。
其中,pcr的反应体系为:
pcr程序:
94℃5min→(94℃15s→63℃15s→72℃4min)×1→(94℃15s→61℃15s→72℃4min)×1→(94℃15s→59℃15s→72℃4min)×1→(94℃15s→57℃15s→72℃4min)×2→(94℃15s→55℃15s→72℃4min)×23→72℃10min→16℃∞
pcr后进行琼脂糖凝胶电泳鉴定大小,电泳完后在紫外条件下分别切下目的条带然后利用胶回收试剂盒回收目的片段。
将pcr扩增后的不同突变体的序列末端进行磷酸化处理,磷酸化体系如下。
37℃孵育1h。
将磷酸化后的突变体的序列进行连接,连接体系如下。
16℃过夜连接。
采用热激法将纯化产物转入大肠杆菌bl21(de3)感受态细胞中,测序正确的单菌落,即为构建成功的突变子。
3、突变体的诱导表达及纯化
挑取突变成功的单菌落,随机挑取单菌落至含kana的10mllb培养基中,活化一段时间后,转接500μl菌液于50ml的lb液体培养基(kana),活化一段时间后,转接10ml菌液于1l的lb液体培养基(kana),待od值为0.6-0.8时,加入0.5mm的fecl3,5’-ala,30℃,200rpm培养20min后,加入1mm的iptg,30℃,200rpm过夜诱导。
将表达好的菌液于4200rpm,4℃的条件下离心15min,弃去上清,菌体用bindingbuffer(100mmk2hpo4-kh2po4,500mmnacl,ph7.4)重悬洗涤两次,得静息细胞。将静息细胞用30ml的bindingbuffer重悬,并加入1mm的pmsf(蛋白酶抑制剂),混匀后经超声波破碎,将破碎后的产物于13000rpm,4℃的条件下离心30min去除细胞碎片,得突变体的粗酶液。
粗酶液的纯化采用ni柱亲和层析。将粗酶液经0.22μm滤膜过滤。采用
4、烷基苯及衍生物sp3c-h键催化活性、区域选择性及立体选择性测定
反应体系如下:
将15mm烷基苯及衍生物(2%dmso助溶),0.5μm酶,0.5mm的双功能小分子,60mmh2o2加入到终体积为1ml的ph8.0的pbs(100mm)中,25℃水浴反应30min后,用1ml的乙酸乙酯对反应体系进行涡旋萃取2min,取有机相过0.22μm膜,用na2s2o4干燥,添加1mm内标,跑gc测定。
gc条件:
活性测定:db-5柱;进样口温度280℃;fid检测器温度280℃;程序升温条件为60℃保留3min,10℃/min升至140℃保留5min,60℃/min升至280℃保留2min;总分析时间为20.33min,内标为二苯甲酮。
立体选择性测定:cp-chirasildexcb柱;进样口温度220℃;fid检测器温度220℃;程序升温条件为80℃保留1min,10℃/min升至170℃保留5min,60℃/min升至190℃保留5min;总分析时间为20.33min,内标为二苯甲酮。
实施例1
f87单突变体制备
以连接有p450bm3氧化酶部分(bmp)基因的重组质粒pet-28a(+)-bmp为模板,分别与含不同突变位点的正反向引物(见表1)作为引物对,进行pcr,得到f87单突变体f87a、f87g、f87v、f87i、f87l的重组基因。扩增体系以及pcr条件见通用方法。
表1
实施例2
f87-t268双突变的制备
以连接有p450bmp-f87单突变的重组质粒pet-28a(+)-f87a为模板,分别与包含不同突变位点的正反向引物作为引物对(见表2),进行pcr,分别得到不同f87-t268双突变体的重组基因。扩增体系以及pcr条件见通用方法。
表2
实施例3
在f87-t268双突变的基础上,选取活性口袋周围的氨基酸位点进行较系统的研究:l75,v78,f81,a82,t88,d168,l181,a184,t260,i263,a264,e267,a328,a330,l437。
1)将氨基酸突变为更大的疏水氨基酸或某些带极性基团的氨基酸,改变活性中心的底物口袋以更好的结合乙苯及其类似物底物;
2)合理地引入带极性基团的氨基酸,增加h2o2的结合和利用效率或改变底物在活性中心的构象。
根据f87-t268双突变搭配不同的双功能小分子对烷基苯及其衍生物sp3c-h羟化的活性、区域及立体选择性分析,选取活性和选择性较优的f87g-t268i,f87a-t268v等,以上述获得的各突变体的重组质粒为模板,分别与包含不同突变位点的正反向引物作为引物对(见表3),进行pcr,得到重组基因。扩增体系以及pcr条件见通用方法。
表3
每个位点取一对引物为例,突变成其他氨基酸时只需将下划线处密码子替换成对应氨基酸的密码子即可。
进而可以获得不同的突变体,举例如下:
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为甘氨酸,第268位的苯丙氨酸替换为异亮氨酸,第78位丙氨酸替换为甲硫氨酸,命名为f87g-t268i-v78m。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为缬氨酸,第82位丙氨酸替换为半胱氨酸,命名为f87a-t268v-a82c。
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苯丙氨酸替换为脯氨酸,第184位丙氨酸替换为丙氨酸,命名为f87a-t268p-a184v。
四突变的获得是上述构建好的三突变体的重组质粒为模板,然后再利用选择突变位点的正反向引物,进行pcr,得到重组基因。
例如:
将seqidno.1所示氨基酸序列的第87位的苯丙氨酸替换为丙氨酸,第268位的苏替换为缬氨酸,第78位缬氨酸替换为亮氨酸,第82位丙氨酸替换为半胱氨酸,基是以f87a-t268v-a82c质粒为模板,以v78l的引物进行pcr得到f87a-t268v-a82c-v78l的重组质粒。
实施例4
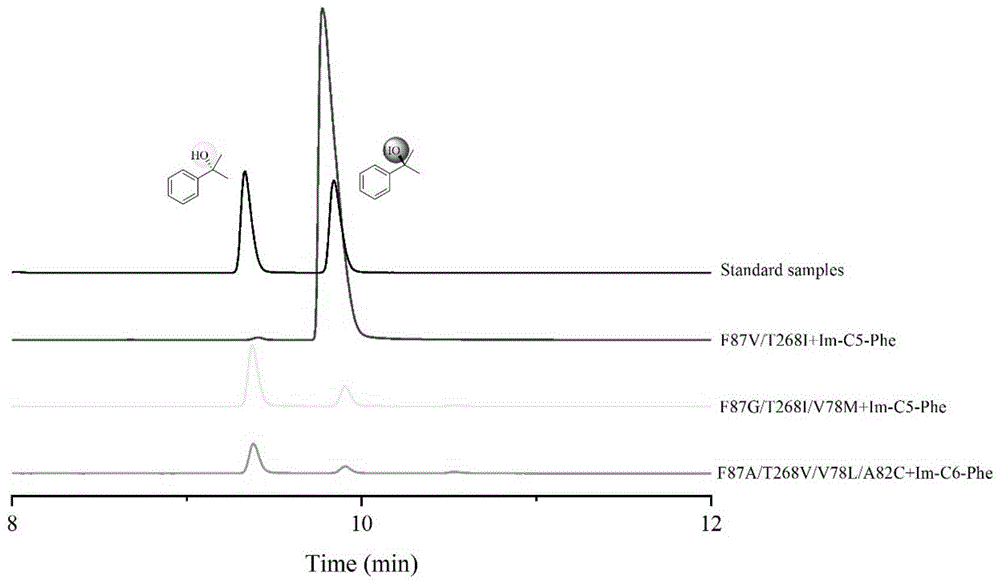
以上述获得的突变体中的f87v-t268i利用
将15mm乙苯(2%dmso助溶),0.5μmf87v-t268i,0.5mm的小分子
双突变f87v-t268i在im-c5-phe诱导分子作用下利用h2o2催化乙苯羟化30minton可达6000以上,且>97%选择生成1-苯乙醇,另其立体选择性亦非常优异,>99%ee选择s-1-苯乙醇。
实施例5
以上述获得的突变体中的f87a-t268v利用
将15mm乙苯(2%dmso助溶),0.5μmf87a-t268v,0.5mm的小分子
双突变f87a-t268v在不同协同分子im-c6-phe/im-c5-phe作用下利用h2o2催化乙苯羟化的区域及立体选择性均有明显差异,其中在im-c6-phe下,85%区域选择及18%ee选择r-1-苯乙醇,而在im-c5-phe下,94%区域选择及23%ee选择s-1-苯乙醇。
实施例6
以上述获得的突变体中的f87g-t268i与f87a-t268i利用
将15mm乙苯(2%dmso助溶),0.5μmf87g-t268i/f87a-t268i,0.5mm的小分子
同样在im-c5-phe诱导分子作用下,双突变f87g-t268i与f87a-t268i利用h2o2催化乙苯羟化生成1-苯乙醇的立体选择差异异常明显,其中f87g-t268i21%ee选择r-1-苯乙醇,而f87a-t268i91%ee选择s-1-苯乙醇。
实施例7
以上述获得的突变体中的f87g-t268i-v78m/f87g-t268i-v78c利用
将15mm乙苯(2%dmso助溶),0.5μmf87g-t268i-v78m/c,0.5mm的小分子
v78m突变位点的引入,使双突变f87g-t268i在im-c5-phe诱导分子作用下利用h2o2催化乙苯羟化生成r-1-苯乙醇的活性和立体选择性进一步提升,50%ee选择r-1-苯乙醇,而v78c的引入却使双突变f87g-t268i催化乙苯羟化生成r构型选择性明显下降。
实施例8
以上述获得的突变体中的f87a-t268v-a82c/f87a-t268v-a82l利用
将15mm乙苯(2%dmso助溶),0.5μmf87a-t268v-a82c/f87a-t268v-a82l,0.5mm的小分子
a82c突变位点的引入,使双突变f87a-t268v在im-c6-phe诱导分子作用下利用h2o2催化乙苯羟化生成r-1-苯乙醇的区域和立体选择性进一步提升,89%选择生成1-苯乙醇,40%ee选择r-1-苯乙醇,而a82l却使立体选择性明显下降。不添加双功能分子的对照实验均无明显活性。
实施例9
以上述获得的突变体中的f87a-t268v-a82c-v78l/f87a-t268v-a82c-v78i利用
将15mm乙苯(2%dmso助溶),0.5μmf87a-t268v-a82c-v78l/f87a-t268v-a82c-v78i,0.5mm的小分子
v78l突变位点的进一步引入,使im-c6-phe诱导分子作用下利用h2o2催化乙苯羟化生成r-1-苯乙醇的区域和立体选择性进一步提升,92%选择生成1-苯乙醇,62%ee选择r-1-苯乙醇,v78i的提升效果不明显。
实施例10
以上述获得的突变体中的f87v-t268i/f87v-t268v利用
将15mm正丙苯(2%dmso助溶),0.5μmf87v-t268i/f87v-t268v,0.5mm的小分子
双突变f87v-t268i在im-c5-phe诱导分子作用下利用h2o2催化正丙苯羟化生成1-苯基-1-丙醇的30minton可达2900以上,且>87%选择生成1-苯基-1-丙醇,其立体选择性亦非常优异,>95%ee选择s-1-苯基-1-丙醇,而f87v-t268v的立体选择性仅有51%ee。
实施例11
以上述获得的突变体中的f87a-t268p/f87a-t268i利用
将15mm正丙苯(2%dmso助溶),0.5μmf87a-t268p/f87a-t268i,0.5mm的小分子
双突变f87a-t268p在im-c5-phe诱导分子作用下利用h2o2催化正丙苯羟化生成1-苯基-2-丙醇的30minton为511,且89%选择生成1-苯基-2-丙醇,其立体选择性亦非常优异,>88%ee选择r-1-苯基-2-丙醇,而f87a-t268i却选择生成1-苯基-1-丙醇的比例较多。不添加双功能分子的对照实验同样均无明显活性。
实施例12
以上述获得的突变体中的f87a-t268v/f87a-t268i利用
将15mm茚满(2%dmso助溶),0.5μmf87a-t268v/f87a-t268i,0.5mm的小分子
双突变f87a-t268v在im-c5-phe诱导分子作用下利用h2o2催化茚满羟化生成1-茚醇的30minton可达3000以上,选择性高达99%以上,且>78%ee选择r-1-茚醇,而f87a-t268i的立体选择性不高。
实施例13
以上述获得的突变体中的f87v-t268i/f87v-t268v利用
将15mm茚满(2%dmso助溶),0.5μmf87v-t268i/f87v-t268v,0.5mm的小分子
双突变f87v-t268i在im-c5-phe诱导分子作用下利用h2o2催化茚满羟化生成1-茚醇的30minton可达1900以上,选择性高达99%以上,且>93%ee选择s-1-茚醇,而f87v-t268v基本无立体选择性。
实施例14
以上述获得的突变体中的f87a-t268p/f87a-t268i利用
将15mm四氢化萘(2%dmso助溶),0.5μmf87a-t268p/f87a-t268i,0.5mm的小分子
双突变f87a-t268p在im-c5-phe诱导分子作用下利用h2o2催化四氢化萘羟化生成1-四氢萘酚的30minton可达1800以上,选择性高达92%以上,且>85%ee选择r-1-四氢萘酚,而f87a-t268i活性降低明显,且选择s构型多。
实施例15
以上述获得的突变体中的f87v-t268i/f87v-t268v利用
将15mm四氢化萘(2%dmso助溶),0.5μmf87v-t268i/f87v-t268v,0.5mm的小分子
双突变f87v-t268i在im-c5-phe诱导分子作用下利用h2o2催化四氢化萘羟化生成1-四氢萘酚的30minton为445,选择性可达88%以上,且>82%ee选择s-1-四氢萘酚,而f87v-t268v选择性大大降低。
由上述各实施例可见,不同f87-t268双突变体及其基础上的叠加突变体与不同诱导小分子的组合对乙苯等烷基苯及其衍生物烷基侧链sp3c-h羟化的区域及立体选择性具有重要影响。本研究首次实现利用同一酶体系进行乙苯等烷基苯及其衍生物不同sp3c-h键的高效区域及立体选择性羟化调控。这大大提高了目前乙苯等烷基苯及其衍生物高附加值利用的效率,具有很强的工业应用潜力。
seqidno1
序列表
<110>中国科学院青岛生物能源与过程研究所
<120>一种催化乙苯等烷基苯及衍生物羟化的突变体及其应用
<160>1
<170>siposequencelisting1.0
<210>1
<211>3150
<212>dna
<213>人工序列(artificialsequence)
<400>1
atgacaattaaagaaatgcctcagccaaaaacgtttggagagcttaaaaatttaccgtta60
ttaaacacagataaaccggttcaagctttgatgaaaattgcggatgaattaggagaaatc120
tttaaattcgaggcgcctggtcgtgtaacgcgctacttatcaagtcagcgtctaattaaa180
gaagcatgcgatgaatcacgctttgataaaaacttaagtcaagcgcttaaatttgtacgt240
gattttgcaggagacgggttatttacaagctggacgcatgaaaaaaattggaaaaaagcg300
cataatatcttacttccaagcttcagtcagcaggcaatgaaaggctatcatgcgatgatg360
gtcgatatcgccgtgcagcttgttcaaaagtgggagcgtctaaatgcagatgagcatatt420
gaagtaccggaagacatgacacgtttaacgcttgatacaattggtctttgcggctttaac480
tatcgctttaacagcttttaccgagatcagcctcatccatttattacaagtatggtccgt540
gcactggatgaagcaatgaacaagctgcagcgagcaaatccagacgacccagcttatgat600
gaaaacaagcgccagtttcaagaagatatcaaggtgatgaacgacctagtagataaaatt660
attgcagatcgcaaagcaagcggtgaacaaagcgatgatttattaacgcatatgctaaac720
ggaaaagatccagaaacgggtgagccgcttgatgacgagaacattcgctatcaaattatt780
acattcttaattgcgggacacgaaacaacaagtggtcttttatcatttgcgctgtatttc840
ttagtgaaaaatccacatgtattacaaaaagcagcagaagaagcagcacgagttctagta900
gatcctgttccaagctacaaacaagtcaaacagcttaaatatgtcggcatggtcttaaac960
gaagcgctgcgcttatggccaactgctcctgcgttttccctatatgcaaaagaagatacg1020
gtgcttggaggagaatatcctttagaaaaaggcgacgaactaatggttctgattcctcag1080
cttcaccgtgataaaacaatttggggagacgatgtggaagagttccgtccagagcgtttt1140
gaaaatccaagtgcgattccgcagcatgcgtttaaaccgtttggaaacggtcagcgtgcg1200
tgtatcggtcagcagttcgctcttcatgaagcaacgctggtacttggtatgatgctaaaa1260
cactttgactttgaagatcatacaaactacgagctggatattaaagaaactttaacgtta1320
aaacctgaaggctttgtggtaaaagcaaaatcgaaaaaaattccgcttggcggtattcct1380
tcacctagcactgaacagtctgctaaaaaagtacgcaaaaaggcagaaaacgctcataat1440
acgccgctgcttgtgctatacggttcaaatatgggaacagctgaaggaacggcgcgtgat1500
ttagcagatattgcaatgagcaaaggatttgcaccgcaggtcgcaacgcttgattcacac1560
gccggaaatcttccgcgcgaaggagctgtattaattgtaacggcgtcttataacggtcat1620
ccgcctgataacgcaaagcaatttgtcgactggttagaccaagcgtctgctgatgaagta1680
aaaggcgttcgctactccgtatttggatgcggcgataaaaactgggctactacgtatcaa1740
aaagtgcctgcttttatcgatgaaacgcttgccgctaaaggggcagaaaacatcgctgac1800
cgcggtgaagcagatgcaagcgacgactttgaaggcacatatgaagaatggcgtgaacat1860
atgtggagtgacgtagcagcctactttaacctcgacattgaaaacagtgaagataataaa1920
tctactctttcacttcaatttgtcgacagcgccgcggatatgccgcttgcgaaaatgcac1980
ggtgcgttttcaacgaacgtcgtagcaagcaaagaacttcaacagccaggcagtgcacga2040
agcacgcgacatcttgaaattgaacttccaaaagaagcttcttatcaagaaggagatcat2100
ttaggtgttattcctcgcaactatgaaggaatagtaaaccgtgtaacagcaaggttcggc2160
ctagatgcatcacagcaaatccgtctggaagcagaagaagaaaaattagctcatttgcca2220
ctcgctaaaacagtatccgtagaagagcttctgcaatacgtggagcttcaagatcctgtt2280
acgcgcacgcagcttcgcgcaatggctgctaaaacggtctgcccgccgcataaagtagag2340
cttgaagccttgcttgaaaagcaagcctacaaagaacaagtgctggcaaaacgtttaaca2400
atgcttgaactgcttgaaaaatacccggcgtgtgaaatgaaattcagcgaatttatcgcc2460
cttctgccaagcatacgcccgcgctattactcgatttcttcatcacctcgtgtcgatgaa2520
aaacaagcaagcatcacggtcagcgttgtctcaggagaagcgtggagcggatatggagaa2580
tataaaggaattgcgtcgaactatcttgccgagctgcaagaaggagatacgattacgtgc2640
tttatttccacaccgcagtcagaatttacgctgccaaaagaccctgaaacgccgcttatc2700
atggtcggaccgggaacaggcgtcgcgccgtttagaggctttgtgcaggcgcgcaaacag2760
ctaaaagaacaaggacagtcacttggagaagcacatttatacttcggctgccgttcacct2820
catgaagactatctgtatcaagaagagcttgaaaacgcccaaagcgaaggcatcattacg2880
cttcataccgctttttctcgcatgccaaatcagccgaaaacatacgttcagcacgtaatg2940
gaacaagacggcaagaaattgattgaacttcttgatcaaggagcgcacttctatatttgc3000
ggagacggaagccaaatggcacctgccgttgaagcaacgcttatgaaaagctatgctgac3060
gttcaccaagtgagtgaagcagacgctcgcttatggctgcagcagctagaagaaaaaggc3120
cgatacgcaaaagacgtgtgggctgggtaa3150
- 还没有人留言评论。精彩留言会获得点赞!