ALA杂合3-羟基吡啶酮衍生物及其制备方法与应用
ala杂合3
‑
羟基吡啶酮衍生物及其制备方法与应用
技术领域
1.本发明涉及有机合成和药物化学领域,具体涉及一类既具有铁离子螯合能力又具有光敏活性的3
‑
羟基吡啶酮(hpo)与5
‑
氨基乙酰丙酸(ala)杂合的ala杂合3
‑
羟基吡啶酮衍生物及其制备方法及其在光动力治疗(photodynamic therapy,pdt)中的应用。
背景技术:
2.二十世纪以来,随着工业化进程的加快,人类的生活环境污染日趋加剧,人们与致癌因素的接触日益频繁,癌症的发病率也逐年递增,在高收入国家中,癌症已经超过心血管疾病成为人类健康的头号敌人。而对于肿瘤的治疗方式主要有手术治疗、化疗和放疗三种常用手段,虽然这三种常用的治疗方式对于肿瘤的治疗有着良好的疗效,但是也会对正常机体造成不可逆的损伤。近些年,随着光动力学在抗肿瘤研究中的应用,pdt逐渐成为一种新兴的肿瘤治疗方法进入人们的视野并在某些癌症的治疗中取得了显著的疗效。
3.相比于传统的癌症治疗方法,pdt的临床应用机制有很大不同,其治疗效果依赖于靶细胞的光敏氧化。首先,光敏剂(pss)通过口服或注射进入体内,并在靶组织中积累,然后,光动力效应被适当波长的光的辐射激活,在分子氧存在的条件下,产生的单线态氧或自由基会对相关的生物分子造成损伤。通过对pdt作用机制的了解,让我们认识到ps在pdt的研究和开发中起着重要的作用,它的性能决定了pdt的治疗效果。与此同时,开发毒副作用小、光毒性强、在靶组织中有相对的选择性存留的理想光敏剂尤为重要。
4.近年来,ala作为ps原卟啉ix(ppix)的前药,它所介导的pdt成为pdt研究中最有前景的领域之一。相比于其他的ps,ala通过血红素生物合成途径代谢产生的ppix所产生的光敏效应有着较短的半衰期,减少了对正常组织的光敏持续时间和药物在体内的停留时间,从而降低了药物对正常机体组织的毒副作用。但由于ala亲水性高,细胞对ala的吸收较差,这大大降低了ala的生物利用度。
5.为了提高ala的生物利用度和提高ppix水平,科学家进行了大量的研究,主要集中在开发脂溶性较好的ala前药。通过对ala在体内的作用机制的分析,铁螯合酶可催化ppix转化为无光敏活性的血红素,导致治疗效果下降,可见铁螯合酶的活性是影响ppix在细胞中积累的关键因素,而铁螯合剂可以通过清除细胞内不稳定铁池来抑制铁螯合酶的活性。
6.研究发现在ala
‑
pdt中,由于铁螯合剂乙二胺四乙酸(edta)和去铁胺的存在,可以增加ala所诱导产生的ppix的水平,但同时也表现出一些不利影响对于已产生的ppix和较差的药代动力学性能。而具有较低的分子量和较高亲脂性的铁螯合剂hpo通过与ala杂合可以进一步提高ala在体内的生物利用度,同时螯合体内的铁离子抑制铁鳌合酶的活性,提高ppix水平。
技术实现要素:
7.为了解决现有技术中的上述问题,本发明基于合理药物设计和类药性等原则设计合成了具有铁螯合性和光敏活性的抗肿瘤的新型活性化合物。本发明的目的在于提供一系
列具有铁离子螯合能力和pdt活性的ala杂合3
‑
羟基吡啶酮(hpo)衍生物的新型活性化合物的制备方法和在抗肿瘤方面的应用。
8.为解决上述技术问题,本发明提供如下技术方案:
9.第一方面,本发明提供一种式(i)或式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物:
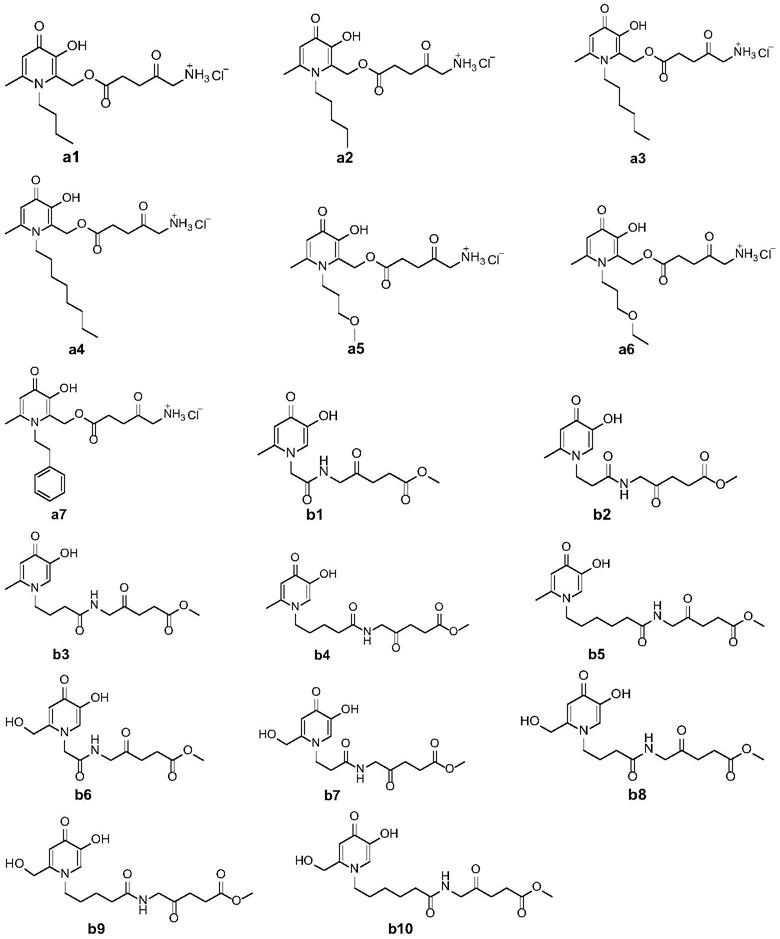
[0010][0011]
式(i)中,
[0012]
r1为c1~c10烷基、c3~c6烷氧基、c6~c10芳基,优选r1为正丁基、戊基、正己基、辛基、甲氧丙基、乙氧丙基和苯乙基;
[0013]
式(ii)中r2为
‑
h或
‑
oh,其中n1=1
‑
5。
[0014]
进一步优选地,所述的ala杂合3
‑
羟基吡啶酮衍生物为下列化合物之一:
[0015][0016]
特别优选,所述的ala杂合3
‑
羟基吡啶酮衍生物为化合物a2、a3、a4、a7或b5。
[0017]
第二方面,本发明还提供了上述式(i)所示的ala杂合3
‑
羟基吡啶酮衍生物的制备方法,所述方法为:
[0018]
(5)将化合物10、苄溴、碱性物质e1溶于溶剂e1中,回流反应0.5~2h(优选0.5~1h,特别优选1h),所得反应液e1经后处理e1,得到化合物11;所述化合物10、苄溴、碱性物质e1的物质的量之比为1:1~4:1~4,优选1:1~2:1~2,特别优选1:1.1:1.1;所述碱性物质为碳酸钾、氢氧化钾、氢氧化钠、碳酸钠、碳酸氢钠中的一种或两种以上的混合物,优选碳酸钾或氢氧化钠,特别优选碳酸钾;
[0019][0020]
(6)将步骤(5)中所述化合物11、3,4
‑
二氢
‑
2h
‑
吡喃,对甲苯磺酸溶于二氯甲烷中,
在20~60℃(优选室温25℃)下反应4~12h,所得反应液f1经后处理f1,得到化合物12;所述化合物11、3,4
‑
二氢
‑
2h
‑
吡喃、对甲苯磺酸的物质的量之比为1:2~6:0.1~0.6,优选1:2~3:0.1~0.3,特别优选1:2:0.2;
[0021][0022]
(7)将步骤(6)中所述化合物12、r1nh2溶于溶剂g1中,回流反应12~36h(优选14~30h,特别优选18h),所得反应液减压浓缩得到含化合物13的粗品;将所述含化合物13的粗品溶解在乙醇中,使用浓盐酸调节ph至1~2(优选ph为1),继续回流反应2~8h(优选2~5h),所得反应液g1经后处理g1,得到式2所示的化合物;所述化合物12与r1nh2的物质的量之比为1:4~10,优选1:4~8,特别优选1:6;
[0023][0024]
(8)保护氛围下(优选氩气或氮气,更优选氩气),将步骤(1)中所述化合物4、二环己基碳二亚胺(dcc)、4
‑
二甲氨基吡啶(dmap)溶于有机溶剂h1中,在,10~60℃(优选室温25℃)下搅拌0.5~1h(优选45分钟),缓慢滴加溶于有机溶剂y1的步骤(7)中所述的化合物2,滴完后继续反应6~24h(优选8~12h,特别优选12h),所得反应液h1经后处理h1,得到化合物5;所述化合物4、二环己基碳二亚胺、4
‑
二甲氨基吡啶与化合物2的物质的量之比为1:1~2:0.1~0.8:0.6~1.2(优选为1:1:0.17:0.83);
[0025][0026]
(9)将步骤(8)中所述化合物5溶于乙酸乙酯j1中,
‑
10~0℃(优选
‑
10℃)下滴加饱和氯化氢的无水乙酸乙酯溶液,滴毕反应1h,转移到室温(25℃)继续反应2~16h(优选4h),所得反应液j1经后处理j1,得到化合物15;所述饱和氯化氢的无水乙酸乙酯溶液的体积以所述化合物5的物质的量计为1~5ml/mmol;
[0027][0028]
(10)将步骤(9)中所述化合物15溶于有机溶剂k1中,加入钯碳催化剂(质量分数为5%),在10~60℃(优选室温25℃)、氢气气氛下搅拌反应1~16h(优选1~8h,特别优选2h),所得反应液k1经后处理k1,得到式(i)所示的ala杂合3
‑
羟基吡啶酮衍生物;所述化合物15
与钯碳催化剂的质量之比为1:0.1~0.4,优选1:0.1~0.3;
[0029][0030]
其中,化合物2、5、13、15、式(i)或r1nh2中,r1为c1~c10烷基、c3~c6烷氧基、c6~c10芳基,优选r1为正丁基、戊基、正己基、辛基、甲氧丙基、乙氧丙基和苯乙基。
[0031]
优选地,所述的式(i)所示的ala杂合3
‑
羟基吡啶酮衍生物为下列化合物之一:
[0032]
特别优选,所述的式(i)所示的ala杂合3
‑
羟基吡啶酮衍生物为化合物a2、a3、a4或a7。
[0033]
进一步,步骤(5)中所述溶剂e1为水、丙酮、乙醇、甲醇、二氯甲烷、氯仿、四氯化碳、乙腈、苯、甲苯、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺中的一种或两种以上的混合溶剂,优选体积比为1:1的水与乙醇的混合溶剂。
[0034]
步骤(5)中所述溶剂e1的体积以化合物10的物质的量计为1~6ml/mmol。
[0035]
步骤(5)中所述后处理e1为:所述反应液e1冷却至室温后浓缩,二氯甲烷溶解后用水洗涤,无水硫酸钠干燥,过滤,蒸除溶剂并干燥,得到化合物11。
[0036]
步骤(6)中所述的二氯甲烷的体积以化合物11的物质的量计为1.5~3ml/mmol。
[0037]
步骤(6)中所述后处理f1为:将所述反应液f1用质量分数为5%的碳酸钠溶液洗涤,再用水洗涤,无水硫酸钠干燥,过滤,减压蒸馏蒸除溶剂,真空干燥,得到化合物12。
[0038]
步骤(7)中所述溶剂g1为水、丙酮、乙醇、甲醇、氯仿、四氯化碳、乙腈、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选乙醇或体积比为1:1的乙醇与水混合溶剂;
[0039]
步骤(7)中乙醇的体积以化合物13的物质的量计为1~5ml/mmol;
[0040]
步骤(7)中所述后处理g1为:将所述反应液g1冷却至室温后减压浓缩,加水溶解并
用乙醚洗涤,调ph至8~10(10m氢氧化钠溶液),二氯甲烷萃取,合并有机层,无水硫酸钠干燥,过滤,浓缩,用体积比为12:1的乙醚与甲醇的混合溶液进行重结晶,得到化合物2。
[0041]
步骤(8)中所述有机溶剂h1为丙酮、乙醇、甲醇、二氯甲烷、氯仿、四氯化碳、甲苯、乙腈、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选体积比为2:1的二氯甲烷与n,n
‑
二甲基甲酰胺的混合溶剂。所述有机溶剂h1的体积以化合物4的物质的量计为1~10ml/mmol,优选3.75ml/mmol。
[0042]
步骤(8)中所述有机溶剂y1为二氯甲烷。所述有机溶剂y1的体积用量以化合物2的物质的量计为1~8ml/mmol,优选3ml/mmol。
[0043]
步骤(8)中所述后处理h1为:对所述反应液h1过滤,取滤液减压浓缩,加入二氯甲烷溶解,依次用饱和碳酸氢钠溶液和饱和氯化钠溶液洗涤,无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析,以体积比为100~20:1的二氯甲烷:甲醇混合溶液进行梯度洗脱,收集含目标产物的洗脱液,浓缩,干燥,得到化合物5。
[0044]
进一步,步骤(9)中所述化合物5的物质的量以所述乙酸乙酯j1的体积计为0.1~2ml/mmol,优选0.17ml/mmol。
[0045]
步骤(9)中所述后处理j1为:所述反应液j1经减压蒸馏除去溶剂,干燥,得到化合物15。
[0046]
步骤(10)中所述有机溶剂k1为丙酮、乙醇、甲醇、二氯甲烷、氯仿、四氯化碳、甲苯、乙腈、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选甲醇。
[0047]
步骤(10)中所述化合物15的物质的量以有机溶剂g的体积计为0.1~0.2mmol/ml。
[0048]
步骤(10)中所述后处理k1为:将所述反应液k1过滤,取滤液减压蒸馏除去溶剂,用甲醇和乙醚的混合溶剂(甲醇和乙醚的体积比为10:1)重结晶,得到式(i)所示的ala杂合3
‑
羟基吡啶酮衍生物。
[0049]
第三方面,本发明还提供上述式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物的制备方法,所述方法为:
[0050]
1)将化合物1或化合物9与溴化苄、碱性物质a2溶于有机溶剂a2中,回流反应4~18h(优选4
‑
12h),所得反应液a2经后处理a2,得到化合物16;所述化合物1或者化合物9与溴化苄、碱性物质a2的物质的量之比为1:1~4:1~4,优选1:1~3:1~3;所述碱性物质a2为碳酸钾、氢氧化钾、氢氧化钠、碳酸钠、碳酸氢钠、三乙胺中的一种或两种以上的混合物,优选碳酸钾或氢氧化钠;
[0051][0052]
2)将步骤1)中所述化合物16、nh2(ch2)
n1
cooh、碱性物质b2溶于溶剂b2中,回流反应4~24h(优选8~12h),所得反应液b2经后处理b2,得到化合物3;所述化合物16、nh2(ch2)
n1
cooh与碱性物质b2的物质的量之比为1:1~4:2~8,优选1:1~2:2~4,特别优选1:1.2:2.4;所述碱性物质b2为碳酸钾、氢氧化钾、氢氧化钠、碳酸钠、碳酸氢钠、三乙胺中的一种或两种以上的混合物,优选氢氧化钠或氢氧化钾;
[0053][0054]
3)将步骤2)中所述化合物3溶于有机溶剂c2中,依次加入化合物6、2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯(hatu),于冰水浴中缓慢滴加n
‑
甲基吗啉,搅拌反应45分钟后,体系中固体完全溶解后,再转移至20~50℃(优选室温25℃)下继续反应2~20h(优选8~16h),所得反应液c2经后处理c2,得到化合物7;所述式3化合物、2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯、化合物6与n
‑
甲基吗啉的物质的量之比为1:1~6:1~6:2~8,优选1:1~4:1~4:2~6(特别优选1:2:1.5:3);
[0055][0056]
4)将步骤3)中所述化合物7溶于有机溶剂d2中,加入钯碳催化剂(质量分数为5%),在10~50℃(优选室温25℃),氢气气氛下搅拌反应4~18h,所得反应液d2经后处理d2,得到式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物;所述式3化合物与钯碳质量之比为1:0.1~0.4;优选1:0.1~0.2,特别优选1:0.2。
[0057][0058]
化合物3、7、16中,r3为h
‑
或phcho
‑
;nh2(ch2)
n1
cooh或式(ii)中r2为
‑
h或
‑
oh,其中n1=1
‑
5。
[0059]
进一步优选地,所述的式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物为下列化合物之一:
[0060]
[0061][0062]
特别优选,所述的式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物为化合物b5。
[0063]
进一步,步骤1)中所述有机溶剂a2为丙酮、乙醇、甲醇、二氯甲烷、氯仿、四氯化碳、乙腈、甲苯、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选丙酮或四氯化碳,特别优选丙酮。
[0064]
步骤1)中所述有机溶剂a2的体积以化合物9的物质的量计为1~4ml/mmol。
[0065]
步骤1)中所述后处理a2为:将所述反应液a2冷却至室温,减压浓缩,加二氯甲烷溶解,水洗,无水硫酸钠干燥,过滤,减压浓缩,硅胶柱层析,以体积比为8~3:1的正己烷与乙酸乙酯的混合溶液进行梯度洗脱,收集含目标产物的洗脱液,浓缩,干燥,得到化合物16。
[0066]
进一步,步骤2)中所述溶剂b2为水、丙酮、乙醇、甲醇、二氯甲烷、乙腈、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选乙醇或体积比为1:1的乙醇与水的混合溶剂。
[0067]
步骤2)中所述溶剂b2的体积以化合物16的物质的量计为4~10ml/mmol;
[0068]
步骤2)中所述后处理b2为:将反应液b2冷却至室温,减压浓缩,加水溶解,二氯甲烷洗涤,然后使用2mol/l的盐酸调节ph至1~2,二氯甲烷萃取,合并有机层,无水硫酸钠干燥,过滤,滤液减压蒸除溶剂,真空干燥,所得固体用体积比为1:10的甲醇和乙醚的混合溶剂重结晶,得到化合物3。
[0069]
步骤3)中所述有机溶剂c2为丙酮、乙醇、甲醇、醋酸、二氯甲烷、乙腈、二甲基亚砜、二氧六环、哌啶、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选n,n
‑
二甲基甲酰胺。
[0070]
步骤3)中所述后处理c2为:所述反应液c2加水并用二氯甲烷萃取,合并有机层,再依次用0.1m盐酸和饱和碳酸氢钠洗涤,无水硫酸钠干燥,过滤,浓缩,进行硅胶柱层析,以体积比为80:~20:1的二氯甲烷与甲醇的混合溶液为洗脱液进行梯度洗脱,收集含有目标产物的洗脱液,浓缩,干燥,得到化合物7。
[0071]
步骤4)中所述式3化合物的物质的量以有机溶剂d2的体积计为0.01~0.12mmol/ml。
[0072]
步骤4)中所述有机溶剂d2为丙酮、乙醇、甲醇、二氯甲烷、氯仿、四氯化碳、甲苯、乙腈、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的
混合溶剂,优选甲醇或者乙醇。
[0073]
步骤4)中所述后处理d2为:反应结束后,反应液过滤,滤液减压浓缩,用甲醇/乙醚重结晶,得到式(ii)所示化合物;将所述反应液d2过滤,取滤液减压蒸馏除去溶剂,用甲醇和乙醚的混合溶剂(甲醇和乙醚的体积比为1:10)重结晶,得到式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物。
[0074]
本发明还推荐上述化合物9、10按照以下方法制备:
[0075]
(1)将化合物14、二碳酸二叔丁酯(boc2o)、碱性物质a1溶于有机溶剂a1中,于20~60℃(优选25~50℃,特别优选室温)下反应18~36h(优选20~25h),所得反应液a1经后处理a1后,得到化合物4(boc保护的ala);所述化合物14、二碳酸二叔丁酯与碱性物质a1的物质的量之比为1:1~1.5:4~12,优选1:1.1:6;所述碱性物质a1为碳酸钾、氢氧化钾、氢氧化钠、碳酸钠、碳酸氢钠、碳酸氢钾或三乙胺中的一种或两种以上的混合物(优选碳酸氢钠或者碳酸氢钾);
[0076][0077]
进一步,步骤(1)中所述有机溶剂a1为丙酮、乙醇、甲醇、二氯甲烷、氯仿、四氯化碳、甲苯、乙腈、二甲基亚砜、二氧六环、n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺中的一种或两种以上的混合溶剂,优选甲醇。
[0078]
优选地,步骤(1)中所述有机溶剂a1的体积以化合物14的物质的量计为2~4ml/mmol。
[0079]
进一步,步骤(1)中所述后处理a1为:将所述反应液a1过滤,取滤液减压蒸馏除去溶剂,加水溶解,用质量分数为10%硫酸氢钾溶液酸化至ph=1~2,乙酸乙酯萃取,合并有机层,用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压蒸除溶剂并干燥,即得化合物4。
[0080]
(2)化合物1与氯化亚砜在室温下反应3~9h(优选为4~6h),所得反应液b1经后处理b1,得到化合物8;氯化亚砜的体积以化合物1的物质的量计为0.5
‑
1ml/mmol(优选0.6ml/mmol);此时氯化亚砜既做溶剂也做反应物。
[0081][0082]
步骤(2)中所述后处理b1为:向反应液b1中加水淬灭,过滤得到含化合物8的粗品(淡黄色的物质),再用正己烷对所述含化合物8的粗品进行洗涤,干燥,得到化合物8。
[0083]
(3)将步骤(2)中所述化合物8溶于水c1中,在40~60℃(优选40~55℃)搅拌5分钟,随后加入1~6物质的量当量(优选为2~3eq)的锌粉,然后缓慢滴加浓盐酸,控制温度在70~80℃。待浓盐酸滴加完毕后,维持温度在75℃,继续反应6~8h,所得反应液c1经后处理c1得到化合物9;所述的化合物8与浓盐酸中hcl的物质的量之比为1:0.8~2,优选为1:0.8~1.5,特别优选1:1.17;
[0084][0085]
步骤(3)中所述水c1的体积以化合物8的物质的量计为0.5~1ml/mmol;
[0086]
步骤(3)中所述后处理c1为:将反应液c1趁热过滤,取滤液用二氯甲烷萃取,合并有机层,无水硫酸钠干燥,最后经减压蒸馏除去溶剂,异丙醇重结晶后干燥,得到化合物9;
[0087]
(4)将步骤(3)中所述化合物9用1.1mol/l的氢氧化钠水溶液溶解,室温条件下搅拌40分钟,然后缓慢滴加质量分数为37%的甲醛水溶液,反应18~24h,所得反应液d1经后处理d1,得到化合物10;所述的化合物9与所述甲醛水溶液中甲醛的物质的量之比为1:1~2,优选为1:1.1;
[0088][0089]
进一步,步骤(4)中所述的1.1mol/l的氢氧化钠水溶液的体积以化合物8的物质的量计为1~3ml/mmol。
[0090]
步骤(4)中所述后处理d1为:用浓盐酸将反应液d1的ph调至1,冰浴条件下冷却析出结晶,过滤,干燥滤饼,得到化合物10。
[0091]
第四方面,本发明还提供所述式(i)或式(ii)所示的ala杂合3
‑
羟基吡啶酮衍生物在制备预防或治疗肿瘤的光动力学治疗药物中的应用。
[0092]
进一步,所述肿瘤为鳞状细胞癌、皮肤癌、基底细胞癌或皮肤癌。
[0093]
优选地,所述肿瘤细胞为hela细胞、mcf
‑
7细胞或者a375细胞。
[0094]
进一步优选地,所述的ala杂合3
‑
羟基吡啶酮衍生物为化合物a2、a3、a4、a7或b5。
[0095]
更进一步,所述光为蓝光。
[0096]
与现有技术相比,本发明的有益效果在于:本发明合成了一系列新型的具有pdt活性的化合物,在抗肿瘤活性方面具有显著优势。
附图说明
[0097]
图1:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物a1
‑
a7(浓度100~400μm)在hela细胞系中的光毒性,蓝光照射(2.5j
·
cm
‑
2),化合物与细胞共孵育4h。
[0098]
图2:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物a1
‑
a7(浓度100~400μm)在mcf
‑
7细胞系中的光毒性,蓝光照射(2.5j
·
cm
‑
2),化合物与细胞共孵育4h。
[0099]
图3:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物a1
‑
a7(浓度100~400μm)在a375细胞系中的光毒性,蓝光照射(2.5j
·
cm
‑
2),化合物与细胞共孵育4h。
[0100]
图4:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物a1
‑
a7(浓度100μm)在a375细胞系中的暗毒性,(a)化合物与hela细胞共孵育4h,(b)化合物与mcf
‑
7细胞共孵育4h,(c)化合物与a375细胞共孵育4h。
[0101]
图5:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物b1
‑
b10(浓度100~400μm)在hela细胞系中的光毒性,蓝光照射(5j
·
cm
‑
2),(a)化合物与细胞共孵育4h,(b)化合物与细胞共孵育24h.
[0102]
图6:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物b1
‑
b10(浓度100~400μm)在mcf
‑
7细胞系中的光毒性,蓝光照射(5j
·
cm
‑
2),(a)化合物与细胞共孵育4h,(b)化合物与细胞共孵育24h.
[0103]
图7:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物b1
‑
b10(浓度100~400μm)在a375细胞系中的光毒性,蓝光照射(5j
·
cm
‑
2),(a)化合物与细胞共孵育4h,(b)化合物与细胞共孵育24h.
[0104]
图8:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物b1
‑
b10(浓度100~400μm)在hela细胞系中的暗毒性,(a)化合物与细胞共孵育4h,(b)化合物与细胞共孵育24h.
[0105]
图9:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物b1
‑
b10(浓度100~400μm)在mcf
‑
7细胞系中的暗毒性,(a)化合物与细胞共孵育4h,(b)化合物与细胞共孵育24h.
[0106]
图10:用mtt法评估ala、ala+cp20和ala
‑
hpo杂合物b1
‑
b10(浓度100~400μm)在a375细胞系中的暗毒性,(a)化合物与细胞共孵育4h,(b)化合物与细胞共孵育24h.
具体实施方式
[0107]
下面结合具体实例对本发明作进一步阐述,但本发明不局限于这些实施例。
[0108]
下述使用的浓盐酸质量分数为37%。
[0109]
下述实施例中收率的计算公式(不考虑纯度)为:
[0110]
y=(m
产量
/m
产物
)/n
原料
[0111]
m
产量
为保护杂质的产物的质量,m
产物
为目标产物的相对分子质量,n
原料
为物质的量较小的反应物的物质的量。
[0112]
实施例1
[0113]
(1)在250ml的圆底烧瓶中加入ala盐酸盐(4.20g,25.0mmol)、nahco3(12.60g,150mmol)和boc2o(6.00g,27.5mmol),并加入75ml无水甲醇为溶剂,氩气保护下室温搅拌,反应期间用tlc检测反应进程,在碘缸中显色。24h后,停止反应,此时反应液呈米黄色,将未反应的nahco3固体使用布氏漏斗过滤除去,并用甲醇进行冲洗,滤液经减压蒸馏后并用30ml水进行溶解,然后用质量分数为10%硫酸氢钾溶液酸化至ph=1~2,乙酸乙酯萃取3次,合并有机层并用饱和食盐水洗涤(3
×
25ml),无水硫酸钠干燥12h,过滤除去固体,减压蒸馏除去溶剂,真空干燥,得到n
‑
boc
‑
ala(5.05g,87%)淡黄色固体,,hplc分析含量≥98%。
[0114]
(2)在500ml的圆底烧瓶中加入曲酸(23.86g,168mmol)和101ml的氯化亚砜,室温下反应5h后加水淬灭,并使用氢氧化钠溶液对尾气进行吸收,得到黄色固体悬浮物,布氏漏斗过滤后,滤饼用正己烷洗涤,真空干燥,得到2
‑
氯甲基
‑5‑
羟基吡喃
‑4‑
酮白色固体化合物22.80g,产率为95%,hplc分析含量≥96%。
[0115]
(3)在500ml的圆底烧瓶中加入2
‑
氯甲基
‑5‑
羟基吡喃
‑4‑
酮(25.69g,160.0mmol)和112ml的水,油浴50℃下充分搅拌后加入锌粉(20.92g,320.0mmol)继续搅拌5分钟,将96ml浓盐酸缓慢滴加到反应体系中,维持体系温度在70~80℃之间,滴加完毕浓盐酸后维
持温度在75℃,tlc检测反应情况,反应8h后,趁热过滤,二氯甲烷萃取(6
×
60ml),合并有机相,无水硫酸钠干燥,减压蒸馏除去有机溶剂,得到褐色固体,使用异丙醇重结晶后自然风干得到2
‑
甲基
‑5‑
羟基吡喃
‑4‑
酮白色固体化合物13.10g,产率为65%,hplc分析含量≥97%。
[0116]
(4)在500ml的圆底烧瓶中加入2
‑
甲基
‑5‑
羟基
‑
吡喃
‑4‑
酮(13.10g,104.0mmol)和1.1mol/l的氢氧化钠溶液112ml,室温条件下搅拌40分钟,缓慢滴加质量分数为37%的甲醛水溶液(3.43g,114.4mmol),反应24h,期间使用tlc监测反应情况,反应结束后使用浓盐酸调至ph=1,冰浴冷却后析出固体,过滤,真空干燥,得到3
‑
羟基
‑2‑
羟甲基
‑6‑
甲基
‑
吡喃
‑4‑
酮白色固体化合物12.17g,产率为75%,hplc分析含量≥97%。
[0117]
(5)在250ml的圆底烧瓶中依次加入3
‑
羟基
‑2‑
羟甲基
‑6‑
甲基吡喃
‑4‑
酮(12.17g,78mmol)、碳酸钾(12.92g,93.6mmol)、乙醇和水(1:1)140ml、溴化苄(16g,93.6mmol),回流反应1h,期间使用tlc检测反应进程,反应结束后,冷却至室温,真空浓缩溶剂,二氯甲烷溶解后用水洗涤3次,无水硫酸钠干燥过夜,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体3
‑
苄氧基
‑2‑
羟甲基
‑6‑
甲基吡喃
‑4‑
酮18.23g,产率为95%,hplc分析含量≥96%。
[0118]
(6)在250ml的圆底烧瓶中依次加入3
‑
苄氧基
‑2‑
羟甲基
‑6‑
甲基吡喃
‑4‑
酮(18.23g,74.1mmol)、对甲苯磺酸(2.55g,14.8mmol)、120ml二氯甲烷、3,4
‑
二氢
‑
2h
‑
吡喃(12.47g,148.2mmol),室温下反应6h,tlc监测反应进程,反应结束后依次用5%的碳酸钠溶液和水各洗涤3遍,无水硫酸钠干燥过夜,过滤,减压蒸馏除去溶剂,真空干燥后得到黄色油状产物23.71g,产率为97%,hplc分析含量≥95%。
[0119]
(7)在100ml的圆底烧瓶中加入上述得到的黄色油状物(3.30g,10.0mmol)和正丁胺(4.39g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑1‑
丁基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮白色固体化合物0.96g,产率为32%,hplc分析含量≥96%。
[0120]
(8)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑1‑
丁基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮(0.602g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和饱和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=100~20:1体积比,梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.504g,产率为49%,hplc分析含量≥98%。
[0121]
(9)在25ml圆底烧瓶中加入上述黄色油状化合物(0.257g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压
蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.205g,收率为91%,hplc分析含量≥97%。
[0122]
(10)称取0.205g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.041g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去pd/c,滤液经减压蒸馏除去溶剂,用乙醚/甲醇(v:v=10:1)重结晶,风干得白色固体化合物a1 0.079g,产率48%,hplc分析含量≥99%。
[0123]
m.p.=149.1
‑
151.9℃.1h nmr(400mhz,dmso
‑
d6)δ8.32(s,3h),7.27(s,1h),5.37(s,2h),4.23(t,j=5.2hz,2h),3.96(m,2h),2.85(t,j=4.4hz,2h),2.63(s,3h),2.61(t,j=4.4hz,2h),1.73(m,2h),1.24(m,2h),0.94(t,j=4.8hz,3h),
13
c nmr(400mhz,dmso
‑
d6)δ203.0,172.0,160.9,148.9,144.7,135.5,114.3,56.6,51.4,46.9,34.7,31.5,27.4,20.4,19.8,13.9;esi
‑
hrms:m/z calcd for c
16
h
25
n2o5[m+h]
+
:325.1758;found:325.1762.
[0124]
实施例2
[0125]
(1)在100ml的圆底烧瓶中加入实施例1(6)得到的黄色油状物(3.30g,10.0mmol)和戊胺(5.23g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑1‑
戊基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮白色固体化合物0.946g,产率为30%,hplc分析含量≥96%。
[0126]
(2)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和实施例1(1)制备的n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑1‑
戊基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮(0.63g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=100:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.539g,产率为51%,hplc分析含量≥95%。
[0127]
(3)在25ml圆底烧瓶中加入上述黄色油状化合物(0.264g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.214g,收率为92%,hplc分析含量≥96%。
[0128]
(4)称取0.214g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.043g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去钯碳,滤液经减压蒸馏除去溶剂,用甲醇/乙醚(v:v=10:1)重结晶得白色固体化合物a2 0.079g,产率46%,hplc
分析含量≥99%。
[0129]
m.p.=149.2
‑
151.4℃.1h nmr(400mhz,dmso
‑
d6)δ8.40(s,3h),7.37(s,1h),5.37(s,2h),4.24(t,j=7.6hz,2h),3.94(m,2h),2.85(t,j=6.0hz,2h),2.64(s,3h),2.61(t,j=6.0hz,2h),1.74(m,2h),1.35(m,4h),0.90(t,j=6.4hz,3h),
13
c nmr(400mhz,dmso
‑
d6)δ203.0,172.0,161.1,148.9,144.8,135.5,114.3,56.6,51.6,47.0,34.7,29.4,28.5,27.4,22.1,20.4,14.3;esi
‑
hrms:m/z calcd.for c
17
h
27
n2o5[m+h]
+
:339.1914;found:339.1909.
[0130]
实施例3
[0131]
(1)在100ml的圆底烧瓶中加入实施例1(6)得到的黄色油状物(3.30g,10.0mmol)和正己胺(6.07g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑1‑
己基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮白色固体化合物1.22g,产率为37%,hplc分析含量≥98%。
[0132]
(2)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和实施例1(1)制备的n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑1‑
己基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮(0.66g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=100:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.575g,产率为53%,hplc分析含量≥97%。
[0133]
(3)在25ml圆底烧瓶中加入上述黄色油状化合物(0.271g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.213g,收率为89%,hplc分析含量≥98%。
[0134]
(4)称取0.213g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.043g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去钯碳,滤液经减压蒸馏除去溶剂,用甲醇/乙醚(v:v=10:1)重结晶得白色固体化合物a3 0.086g,产率50%,hplc分析含量≥99%。
[0135]
m.p.=139.3
‑
141.0℃.1h nmr(400mhz,dmso
‑
d6)δ8.40(s,3h),7.35(s,1h),5.36(s,2h),4.24(t,j=8.0hz,2h),3.94(m,2h),2.85(t,j=6.4hz,2h),2.64(s,3h),2.60(t,j=6.4hz,2h),1.72(m,2h),1.38(m,2h),1.30(m,4h),0.88(t,j=6.8hz,3h),
13
c nmr(400mhz,dmso
‑
d6)δ202.9,172.0,161.1,148.9,144.8,135.4,114.3,56.6,51.6,47.0,34.7,31.1,29.6,27.4,26.1,22.5,20.4,14.4;esi
‑
hrms:m/z calcd.for c
18
h
29
n2o5[m+h
]
+
:353.2071;found:353.2074.
[0136]
实施例4
[0137]
(1)在100ml的圆底烧瓶中加入实施例1(6)得到的黄色油状物(3.30g,10.0mmol)和辛胺(7.75g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑1‑
辛基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮白色固体化合物1.25g,产率为35%,hplc分析含量≥97%。
[0138]
(2)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和实施例1(1)制备的n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑1‑
辛基
‑2‑
羟甲基
‑6‑
甲基吡啶
‑4‑
酮(0.71g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=100:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.548g,产率为48%,hplc分析含量≥96%。
[0139]
(3)在25ml圆底烧瓶中加入上述黄色油状化合物(0.285g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.215g,收率为85%,hplc分析含量≥95%。
[0140]
(4)称取0.215g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.043g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去钯碳,滤液经减压蒸馏除去溶剂,用甲醇/乙醚(v:v=10:1)重结晶得白色固体化合物a4 0.081g,产率46%,hplc分析含量≥99%。
[0141]
m.p.=80.2
‑
82.1℃.1h nmr(400mhz,dmso
‑
d6)δ8.35(s,3h),7.35(s,1h),5.36(s,2h),4.23(t,j=8.0hz,2h),3.94(m,2h),2.84(t,j=6.4hz,3h),2.64(s,3h),2.61(t,j=6.4hz,2h),1.72(m,2h),1.29(m,10h),0.85(t,j=6.8hz,3h),
13
c nmr(400mhz,dmso
‑
d6)δ202.9,172.0,161.1,148.8,144.8,135.4,114.3,56.6,51.5,47.0,34.7,31.7,29.7,29.0,28.9,27.4,26.4,22.5,20.4,14.4;esi
‑
hrms:m/z calcd.for c
20
h
33
n2o5[m+h]
+
:381.2384;found:381.2384.
[0142]
实施例5
[0143]
(1)在100ml的圆底烧瓶中加入实施例1(6)得到的黄色油状物(3.30g,10.0mmol)和3
‑
甲氧基丙胺(5.35g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇
溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑2‑
羟甲基
‑1‑
(3
‑
甲氧基丙基)
‑6‑
甲基吡啶
‑4‑
酮白色固体化合物1.33g,产率为42%,hplc分析含量≥96%。
[0144]
(2)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和实施例1(1)制备的n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑2‑
羟甲基
‑1‑
(3
‑
甲氧基丙基)
‑6‑
甲基吡啶
‑4‑
酮(0.63g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=100:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.594g,产率为56%,hplc分析含量≥97%。
[0145]
(3)在25ml圆底烧瓶中加入上述黄色油状化合物(0.265g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.219g,收率为94%,hplc分析含量≥96%。
[0146]
(4)称取0.219g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.044g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去钯碳,滤液经减压蒸馏除去溶剂,用甲醇/乙醚(v:v=10:1)重结晶得白色固体化合物a5 0.095g,产率54%,hplc分析含量≥99%。
[0147]
m.p.=138.1
‑
140.3℃.1h nmr(400mhz,dmso
‑
d6)δ8.42(s,3h),7.37(s,1h),5.37(s,2h),4.34(t,j=7.8hz,2h),3.94(m,2h),3.42(t,j=5.6hz,2h),3.26(s,3h),2.84(t,j=6.4hz,3h),2.64(s,3h),2.61(t,j=6.4hz,2h),2.00(m,2h),
13
c nmr(400mhz,dmso
‑
d6)δ202.9,171.9,161.2,149.1,144.7,135.6,114.3,68.9,58.6,56.6,49.2,47.0,34.7,29.7,27.4,20.4;esi
‑
hrms:m/z calcd.for c
16
h
25
n2o6[m+h]
+
:341.1707;found:341.1713.
[0148]
实施例6
[0149]
(1)在100ml的圆底烧瓶中加入实施例1(6)得到的黄色油状物(3.30g,10.0mmol)和3
‑
乙氧基丙胺(6.19g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑2‑
羟甲基
‑1‑
(3
‑
乙氧基丙基)
‑6‑
甲基吡啶
‑4‑
酮白色固体化合物1.56g,产率为47%,hplc分析含量≥98%。
[0150]
(2)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和实施例1(1)制
备的n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑2‑
羟甲基
‑1‑
(3
‑
乙氧基丙基)
‑6‑
甲基吡啶
‑4‑
酮(0.66g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=100:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.62g,产率为57%,hplc分析含量≥97%。
[0151]
(3)在25ml圆底烧瓶中加入上述黄色油状化合物(0.272g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.209g,收率为87%,hplc分析含量≥97%。
[0152]
(4)称取0.209g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.042g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去钯碳,滤液经减压蒸馏除去溶剂,用甲醇/乙醚(v:v=12:1)重结晶得白色固体化合物a6 0.083g,产率49%,hplc分析含量≥99%。
[0153]
m.p.=140.1
‑
142.3℃.1h nmr(400mhz,dmso
‑
d6)δ8.39(s,3h),7.37(s,1h),5.37(s,2h),4.35(t,j=7.2hz,2h),3.95(m,2h),3.44(m,4h),2.84(t,j=6.4hz,3h),2.64(s,3h),2.60(t,j=6.4hz,2h),2.00(m,2h),1.10(t,j=6.8hz,3h),
13
c nmr(400mhz,dmso
‑
d6)δ202.9,171.9,161.2,149.1,144.7,135.6,114.3,66.7,66.0,56.7,49.2,47.0,34.7,29.8,27.4,20.4,15.5;esi
‑
hrms:m/z calcd.for c
17
h
27
n2o6[m+h]
+
:355.1864;found:355.1863.
[0154]
实施例7
[0155]
(1)在100ml的圆底烧瓶中加入实施例1(6)得到的黄色油状物(3.30g,10.0mmol)和3
‑
乙氧基丙胺(6.19g,60.0mmol),乙醇和水(v:v=1:1)20ml作为溶剂,回流条件下反应18h,期间使用tlc监测反应进程,反应结束后,减压蒸馏除去溶剂后得到褐色油状物,乙醇溶解并用浓盐酸调至ph=1,继续回流4h,tlc监测反应进程,反应结束后,冷却至室温,减压蒸馏除去溶剂,残余物用水溶解并用乙醚洗涤两次,随后用10mol/l的氢氧化钠溶液调至ph=9,二氯甲烷萃取(3
×
20ml),合并有机层,无水硫酸钠干燥,过滤,减压蒸馏除去溶剂,真空干燥,得到淡黄色固体,乙醚/甲醇(v:v=12:1)重结晶得到3
‑
苄氧基
‑2‑
羟甲基
‑6‑
甲基
‑1‑
苯乙基吡啶
‑4‑
酮白色固体化合物2.06g,产率为59%,hplc分析含量≥96%。
[0156]
(2)氩气保护下,将dcc(0.50g,2.4mmol)、dmap(0.049g,0.4mmol)和n
‑
boc
‑
ala(0.555g,2.4mmol)溶解在二氯甲烷(6ml)和dmf(3ml)中,室温下搅拌45分钟。将溶解于二氯甲烷(6ml)中的3
‑
苄氧基
‑2‑
羟甲基
‑6‑
甲基
‑1‑
苯乙基吡啶
‑4‑
酮(0.70g,2mmol)置于恒压滴液漏斗中,在40分钟内逐滴加入,反应液在室温下反应12h,期间用tlc监测反应进程,反应结束后,滤出沉淀物,滤液经减压蒸馏除去溶剂,残余物溶解在二氯甲烷中,依次用饱和碳酸氢钠溶液和氯化钠溶液对其进行洗涤,无水硫酸钠干燥,过滤,减压浓缩有机层,经硅
胶柱层析纯化(二氯甲烷:甲醇=100:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.58g,产率为52%,hplc分析含量≥97%。
[0157]
(3)在25ml圆底烧瓶中加入上述黄色油状化合物(0.281g,0.5mmol),溶解于3ml乙酸乙酯溶液中,将饱和的氯化氢的乙酸乙酯溶液(3ml)置于恒压滴液漏斗中,
‑
10℃缓慢滴加,滴毕,继续反应1h,随后移至室温继续反应4h,通过tlc监测反应进程,反应结束后,减压蒸馏除去有机溶剂,真空干燥,得到的中间体化合物为黄色油状物0.222g,收率为89%,hplc分析含量≥98%。
[0158]
(4)称取0.222g的上述化合物溶于4ml甲醇并置于25ml圆底烧瓶中,用0.044g的5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)催化氢化,保持30psi氢气压力,室温下搅拌反应2h,期间使用tlc监测反应进程,反应结束后,过滤除去钯碳,滤液经减压蒸馏除去溶剂,用甲醇/乙醚(v:v=10:1)重结晶得白色固体化合物a7 0.112g,产率62%,hplc分析含量≥99%。
[0159]
m.p.=147.2
‑
148.9℃.1h nmr(400mhz,dmso
‑
d6)δ8.40(t,j=5.2hz,3h),7.25
‑
7.37(m,6h),5.37(s,2h),4.51(t,j=7.6hz,2h),3.94(m,2h),3.12(t,j=7.6hz,2h),2.84(t,j=6.4hz,2h),2.61(t,j=6.4hz,2h),2.59(s,3h),
13
c nmr(400mhz,dmso
‑
d6)δ203.0,172.0,161.2,149.3,144.8,137.2,135.7,129.4,129.2,127.6,114.3,56.6,52.4,46.9,35.3,34.7,27.4,20.6;esi
‑
hrms:m/z calcd.for c
20
h
25
n2o5[m+h]
+
:373.1758;found:373.1759.
[0160]
实施例8
[0161]
(1)在500ml的圆底烧瓶中加入曲酸(34.08g,240mmol)和144ml的氯化亚砜,室温下反应5h后加水淬灭,并使用氢氧化钠溶液对尾气进行吸收,得到黄色固体悬浮物,布氏漏斗过滤后,滤饼用正己烷洗涤,真空干燥,得到2
‑
氯甲基
‑5‑
羟基吡喃
‑4‑
酮白色固体化合物36.71g,产率为95%,hplc分析含量≥96%。
[0162]
(2)在500ml的圆底烧瓶中加入2
‑
氯甲基
‑5‑
羟基吡喃
‑4‑
酮(36.71g,228mmol)和160ml的水,油浴50℃下充分搅拌后加入锌粉(29.82g,456.0mmol)继续搅拌5分钟,将137ml浓盐酸缓慢滴加到反应体系中,维持体系温度在70~80℃之间,滴加完毕浓盐酸后维持温度在75℃,tlc检测反应情况,反应8h后,趁热过滤,二氯甲烷萃取(6
×
60ml),合并有机相,无水硫酸钠干燥,减压蒸馏除去有机溶剂,得到褐色固体,使用异丙醇重结晶后得到2
‑
甲基
‑5‑
羟基吡喃
‑4‑
酮白色固体化合物18.67g,产率为65%,hplc分析含量≥97%。
[0163]
(3)在500ml圆底烧瓶中加入原料2
‑
甲基
‑5‑
羟基吡喃
‑4‑
酮(18.67g,148.2mmol),无水k2co3(24.54g,177.8mmol),溴化苄(30.4g,177.8mmol)和丙酮(288ml),反应体系加热至回流,tlc监测反应进程,反应完成后,混合物冷却至室温,减压蒸馏除去溶剂,残余物用二氯甲烷(120ml)溶解,水洗(3
×
100ml),无水硫酸钠干燥,过滤,减压浓缩有机层,经硅胶柱层析纯化(正己烷:乙酸乙酯=8:1~3:1梯度洗脱),收集正己烷:乙酸乙酯=5:1~3:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物29.77g,收率为93%,hplc分析含量≥95%。
[0164]
(4)将甘氨酸(0.90g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入上述的黄色油状吡喃酮(2.16g,10.0mmol),然
后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水30ml溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.48g,收率为54%,hplc分析含量≥99%。
[0165]
(5)在25ml圆底烧瓶中加入上述白色固体中间体(0.41g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为60%,hplc分析含量≥98%。
[0166]
(6)在25ml圆底烧瓶中加入上述黄色油状中间体(0.2g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.04g)和6ml的甲醇,在30psi氢气气氛下搅拌反应4h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b10.14g,收率为88%,hplc分析含量≥99%。
[0167]
m.p.=183.5
‑
184.9℃.1h nmr(400mhz,dmso
‑
d6)δ8.48(t,j=4.8hz,1h),7.34(s,1h),6.08(s,1h),4.65(s,2h),4.05(d,j=5.2hz,2h),3.57(s,3h),2.72(t,j=6.4hz,2h),2.52(t,j=6.8hz,2h),2.17(s,3h);
13
c nmr(400mhz,dmso
‑
d6)δ205.2,173.1,171.4,167.4,146.6,145.9,124.7,113.9,55.0,51.9,48.8,34.5,27.6,18.8;esi
‑
hrms:m/z calcd for c
14
h
19
n2o6[m+h]
+
:311.1243;found:311.1224.
[0168]
实施例9
[0169]
(1)将丙氨酸(1.07g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例8(3)制备的黄色油状吡喃酮(2.16g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.35g,收率为47%,hplc分析含量≥96%。
[0170]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.43g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为63%,hplc分析含量≥97%。
[0171]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.21g,0.5mmol)、5%pd/c(萨恩化学技术(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.041g)和6ml的甲醇,在30psi氢气气氛下搅拌反应4h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b2 0.13g,收率为83%,hplc分析含量≥99%。
[0172]
m.p.=192.0
‑
194.2℃.
[0173]1h nmr(400mhz,dmso
‑
d6)δ8.30(t,j=4.8hz,1h),7.39(s,1h),6.20(s,1h),4.41(s,2h),4.11(t,j=6.8hz,2h),3.96(d,j=5.2hz,2h),2.64(m,4h),2.48(t,j=6.0hz,2h),2.27(s,3h);
13
c nmr(400mhz,dmso
‑
d6)δ205.6,173.1,171.0,169.9,147.1,145.1,122.9,114.0,51.9,51.9,48.8,36.1,34.3,27.6,18.8;esi
‑
hrms:m/z calcd for c
15
h
21
n2o6[m+h]
+
:325.1400;found 325.1390.
[0174]
实施例10
[0175]
(1)将4
‑
氨基丁酸(1.24g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例8(3)制备的黄色油状吡喃酮(2.16g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水(30ml)溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.36g,收率为45%,hplc分析含量≥97%。
[0176]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.45g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为57%,hplc分析含量≥96%。
[0177]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.21g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.041g)和6ml的甲醇,在30psi氢气气氛下搅拌反应4h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b30.15g,收率为88%,hplc分析含量≥99%。
[0178]
m.p.=117.0
‑
117.9℃.1h nmr(400mhz,dmso
‑
d6)δ8.23(t,j=5.6hz,1h),7.37(s,1h),6.06(s,1h),3.96(d,j=5.6hz,2h),3.83(t,j=7.6hz,2h),3.57(s,3h),2.70(t,j=6.4hz,2h),2.49(t,j=6.4hz,2h),2.25(s,3h),2.21(t,j=7.2hz,2h),1.86(m,2h);
13
c nmr(400mhz,dmso
‑
d6)δ205.9,173.1,172.0,170.9,147.2,145.0,122.9,114.0,51.9,51.8,48.8,34.4,31.8,27.6,26.4,18.7;esi
‑
hrms:m/z calcd for c
16
h
23
n2o6[m+h]
+
:339.1556;found 339.1542.
[0179]
实施例11
[0180]
(1)将5
‑
氨基颉草酸(1.41g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例8(3)制备的黄色油状吡喃酮(2.16g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水30ml溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.36g,收率为43%,hplc分析含量≥96%。
[0181]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.47g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为51%,hplc分析含量≥97%。
[0182]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.22g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.044g)和6ml的甲醇,在30psi氢气气氛下搅拌反应4h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b40.15g,收率为86%,hplc分析含量≥99%。
[0183]
m.p.=84.2
‑
85.2℃.1h nmr(400mhz,dmso
‑
d6)δ8.17(t,j=4.4hz,1h),7.39(s,1h),6.06(s,1h),3.94(d,j=5.2hz,2h),3.83(t,j=7.2hz,2h),3.57(s,3h),2.69(t,j=6.4hz,2h),2.48(t,j=6.4hz,2h),2.25(s,3h),2.18(t,j=6.8hz,2h),1.62(m,2h),1.52(m,2h);
13
c nmr(400mhz,dmso
‑
d6)δ205.9,173.1,172.6,170.9,147.1,144.8,123.2,114.1,52.4,51.9,48.7,34.9,34.4,29.9,27.6,22.5,18.7;esi
‑
hrms:m/z calcd for c
17
h
25
n2o6[m+h]
+
:353.1713;found 353.1708.
[0184]
实施例12
[0185]
(1)将6
‑
氨基己酸(1.57g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例8(3)制备的黄色油状吡喃酮(2.16g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水30ml溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.88g,收率为57%,hplc分析含量≥96%。
[0186]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.49g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,
无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为48%,hplc分析含量≥97%。
[0187]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.23g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.046g)和6ml的甲醇,在30psi氢气气氛下搅拌反应4h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b50.16g,收率为86%,hplc分析含量≥99%。
[0188]
m.p.=65.2
‑
67.2℃.1h nmr(400mhz,dmso
‑
d6)δ8.13(t,j=5.6hz,1h),7.40(s,1h),6.07(s,1h),3.92(d,j=5.6hz,2h),3.81(t,j=7.6hz,2h),3.57(s,3h),2.68(t,j=6.4hz,2h),2.48(t,j=6.4hz,2h),2.26(s,3h),2.15(t,j=7.2hz,2h),1.63(m,2h),1.53(m,2h),1.27(m,2h);
13
c nmr(400mhz,dmso
‑
d6)δ206.0,173.2,172.8,170.7,147.1,144.8,123.3,114.0,52.5,51.9,48.7,35.2,34.3,30.1,27.6,25.8,25.2,18.8;esi
‑
hrms:m/z calcd for c
18
h
27
n2o6[m+h]
+
:367.1869;found 367.1864.
[0189]
实施例13
[0190]
(1)在500ml圆底烧瓶中加入原料曲酸(14.2g,100mmol),氢氧化钠(8.4g,210mmol),溴化苄(35.9g,210mmol)和丙酮(200ml),反应体系加热至回流,tlc监测反应进程,反应完成后,混合物冷却至室温,减压蒸馏除去溶剂,残余物用二氯甲烷(160ml)溶解,水洗涤(3
×
100ml),无水硫酸钠干燥,过滤,减压浓缩,经硅胶柱层析纯化(正己烷:甲乙酸乙酯=8:1~3:1梯度洗脱),收集正己烷:乙酸乙酯=5:1~3:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物28.04g,收率为87%,hplc分析含量≥98%。
[0191]
(2)将甘氨酸(0.90g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入上述的黄色油状吡喃酮(3.22g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水(30ml)溶解残余物,二氯甲烷洗洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.86g,收率为49%,hplc分析含量≥96%。
[0192]
(3)在25ml圆底烧瓶中加入上述白色固体中间体(0.57g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为51%,hplc分析含量≥97%。
[0193]
(4)在25ml圆底烧瓶中加入上述黄色油状中间体(0.2g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.051g)和6ml的甲醇,在30psi氢气气氛下搅拌反应16h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用
甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b60.11g,收率为67%,hplc分析含量≥99%。
[0194]
m.p.=186.2
‑
188.4℃.1h nmr(400mhz,dmso
‑
d6)δ8.48(t,j=6.0hz,1h),7.33(s,1h),6.22(s,1h),4.73(s,2h),4.27(s,2h),4.04(d,j=5.2hz,2h),3.57(s,3h),2.71(t,j=6.4hz,2h),2.50(t,j=6.8hz,2h);
13
c nmr(400mhz,dmso
‑
d6)δ205.2,173.1,171.6,167.6,148.2,146.8,125.3,112.5,59.8,53.9,51.9,48.9,34.5,27.6;esi
‑
hrms:m/z calcd for c
14
h
18
n2nao7[m+na]
+
:349.1012;found 349.1006.
[0195]
实施例14
[0196]
(1)将丙氨酸(1.07g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例13(1)制备的黄色油状吡喃酮(3.22g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水(30ml)溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.69g,收率为43%,hplc分析含量≥96%。
[0197]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.59g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为49%,hplc分析含量≥96%。
[0198]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.26g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.052g)和6ml的甲醇,在30psi氢气气氛下搅拌反应16h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b70.092g,收率为54%,hplc分析含量≥99%。
[0199]
m.p.=176.7
‑
177.4℃.1h nmr(400mhz,dmso
‑
d6)δ8.30(t,j=4.8hz,1h),7.39(s,1h),6.20(s,1h),4.41(s,2h),4.11(t,j=6.8hz,2h),3.96(d,j=5.2hz,2h),3.57(s,3h),2.66(m,4h),2.48(t,j=8.0hz,2h);
13
c nmr(400mhz,dmso
‑
d6)δ205.7,173.1,171.2,170.1,147.7,147.4,123.4,113.0,60.0,51.9,48.9,48.1,36.5,34.4,27.7;esi
‑
hrms:m/z calcd for c
15
h
20
n2nao7[m+na]
+
:363.1168;found 363.1180.
[0200]
实施例15
[0201]
(1)将4
‑
氨基丁酸(1.24g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例13(1)制备的黄色油状吡喃酮(3.22g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水(30ml)溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去
溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.71g,收率为42%,hplc分析含量≥97%。
[0202]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.61g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为51%,hplc分析含量≥98%。
[0203]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.27g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.053g)和6ml的甲醇,在30psi氢气气氛下搅拌反应16h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b80.105g,收率为59%,hplc分析含量≥99%。
[0204]
m.p.=157.6
‑
158.2℃.1h nmr(400mhz,dmso
‑
d6)δ8.21(t,j=5.2hz,1h),7.40(s,1h),6.23(s,1h),4.39(s,2h),3.96(d,j=5.6hz,2h),3.87(t,j=7.2hz,2h),3.58(s,3h),2.70(t,j=6.4hz,2h),2.50(t,j=7.6hz,2h),2.21(t,j=7.2hz,2h),1.90(m,2h);
13
c nmr(400mhz,dmso
‑
d6)δ205.9,173.1,172.1,171.1,147.8,147.4,123.4,112.7,59.7,51.9,51.2,48.8,34.4,32.0,27.6,26.9;esi
‑
hrms:m/z calcd for c
16
h
22
n2nao7[m+na]
+
:377.1325;found 377.1332.
[0205]
实施例16
[0206]
(1)将5
‑
氨基颉草酸(1.41g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例13(1)制备的黄色油状吡喃酮(3.22g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水(30ml)溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体2.19g,收率为52%,hplc分析含量≥96%。
[0207]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.63g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为55%,hplc分析含量≥96%。
[0208]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.27g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.053g)和6ml的甲醇,在30psi氢气气氛下搅拌
反应16h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b90.103g,收率为56%,hplc分析含量≥99%。
[0209]
m.p.=169.0
‑
170.2℃.1h nmr(400mhz,dmso
‑
d6)δ8.15(t,j=5.2hz,1h),7.41(s,1h),6.22(s,1h),4.38(s,2h),3.94(d,j=5.6hz,2h),3.87(t,j=7.2hz,2h),3.57(s,3h),2.68(t,j=6.4hz,2h),2.48(t,j=6.4hz,2h),2.18(t,j=7.2hz,2h),1.68(m,2h),1.51(m,2h);
13
c nmr(400mhz,dmso
‑
d6)δ205.9,173.1,172.6,171.0,147.6,147.4,123.5,112.7,59.8,51.9,51.6,48.7,34.9,34.4,30.5,27.6,22.6;esi
‑
hrms:m/z calcd for c
17
h
25
n2o7[m+h]
+
:369.1662;found 369.1640.
[0210]
实施例17
[0211]
(1)将6
‑
氨基己酸(1.57g,12.0mmol)、naoh(0.96g,24.0mmol)溶于h2o:etoh(v:v=1:1)混合溶液60ml中,待固体完全溶解后,加入实施例13(1)制备的黄色油状吡喃酮(3.22g,10.0mmol),然后将混合物回流反应9h,期间使用tlc检测反应进程。反应结束后冷却至室温,减压蒸馏除去乙醇,加水(30ml)溶解残余物,二氯甲烷洗涤(3
×
40ml),稀盐酸调ph=2,二氯甲烷萃取(5
×
40ml),合并有机层经无水硫酸钠干燥,过滤,滤液减压蒸馏除去溶剂,真空干燥,得淡黄色固体,并用甲醇/乙醚(v:v=1:10)重结晶得白色固体1.79g,收率为41%,hplc分析含量≥97%。
[0212]
(2)在25ml圆底烧瓶中加入上述白色固体中间体(0.65g,1.5mmol)并溶解于n,n
‑
二甲基甲酰胺(6ml)中,再加入hatu(1.14g,3mmol)和ala甲酯盐酸盐(0.33g,1.8mmol),于冰水浴中缓慢滴加n
‑
甲基吗啉(0.45g,4.5mmol),搅拌反应45分钟,待体系中固体完全溶解后,再转移至室温继续反应12h,期间用tlc监测反应进程。反应结束后,加水并用二氯甲烷萃取(3
×
20ml),合并有机层,再依次用0.1mol/l稀盐酸和饱和碳酸氢钠溶液洗涤有机层,无水硫酸钠干燥,过滤,真空浓缩溶剂,减压浓缩有机层,经硅胶柱层析纯化(二氯甲烷:甲醇=80:1~20:1梯度洗脱),收集二氯甲烷:甲醇=40:1~20:1时的洗脱液,经减压浓缩,干燥,得到黄色油状产物0.36g,收率为44%,hplc分析含量≥98%。
[0213]
(3)在25ml圆底烧瓶中加入上述黄色油状中间体(0.28g,0.5mmol)、5%pd/c(萨恩化学技术(上海)有限公司,e060062
‑
25g)(0.056g)和6ml的甲醇,在30psi氢气气氛下搅拌反应16h,用tlc检测反应进程,反应结束后,过滤除去不溶性杂质,滤液经减压蒸馏浓缩,用甲醇/乙醚(v:v=1:10)进行重结晶,得到白色固体化合物b100.094g,收率为49%,hplc分析含量≥99%。
[0214]
m.p.=151.2
‑
152.4℃.1h nmr(400mhz,dmso
‑
d6)δ8.11(t,j=5.2hz,1h),7.42(s,1h),6.22(s,1h),4.37(s,2h),3.92(d,j=5.6hz,2h),3.85(t,j=7.6hz,2h),3.57(s,3h),2.69(t,j=6.4hz,2h),2.50(t,j=9.2hz,2h),2.14(t,j=7.2hz,2h),1.67(m,2h),1.53(m,2h),1.28(m,2h);
13
c nmr(400mhz,dmso
‑
d6)δ206.0,173.1,172.8,170.9,147.6,147.4,123.6,112.7,59.9,51.9,51.8,48.7,35.3,34.4,30.6,27.6,25.9,25.2;esi
‑
hrms:m/z calcd for c
18
h
26
n2nao7[m+na]
+
:405.1638;found 405.1644.
[0215]
实施例18
[0216]
下面是本发明中式(i)所示的ala
‑
hpo杂合衍生物的药理实验数据:
[0217]
1、光毒性实验
[0218]
实验方法:将不同的肿瘤细胞以约1.0
×
104/孔的密度接种与96孔板中,培养48h后,用磷酸缓冲液洗涤细胞。100μl含有不同浓度(20~100μm)的化合物溶液加入指定的孔中,其中阳性药分别是ala以及ala与3
‑
羟基
‑
1,2
‑
二甲基吡啶
‑4‑
酮(cp20)的混合给药,培养4h后,使用2.5j
·
cm
‑2的蓝光照射5分钟,随后吸掉含有药物的培养基,使用不含有血清的培养基继续培养18h。采用mtt测定细胞毒性。mtt检测方法为:除去孔板中的所有培养基,每孔加入100μl含0.5%mtt的培养基,加盖放入培养箱中培养4h。再次除去板中所有培养基,每孔加入100μl二甲基亚砜,振摇5分钟,测定520nm处吸光度,计算每个前药在每种浓度下的平均细胞存活率,并以对照的百分比表示。
[0219]
将细胞存活率和浓度的数值制成表格并以折线图的形式呈现。
[0220]
平均细胞存活率=吸光度(每种前药的平均值)/吸光度(对照组的平均值)
×
100%。
[0221]
从表1中可以得出以下结论:在hela细胞系中,光毒性最强的偶联物是a2,其ld
50
值为46.11μm;在mcf
‑
7细胞系中偶联物a2和a3都表现出较强的细胞光毒性,ld
50
值分别为52.74μm和45.42μm;而在a375细胞系中偶联物a3、a4、a7表现出很强的细胞光毒性,其ld
50
值分别为33.22μm、21.32μm、34.89μm。进一步说明酯键类化合物可以快速的进入细胞并在细胞内肽酶的作用下破坏ppix前药与hpo分子之间的键,游离出ala,并在细胞内通过代谢产生高浓度的光敏活性物质ppix。
[0222]
表1:ala、ala与cp20混合物以及式(i)所示的ala
‑
hpo杂合衍生物在在不同肿瘤细胞系中的ld
50
值。
[0223][0224]
2、暗毒性试验
[0225]
为了测定化合物的“暗”毒性,重复光毒性实验步骤,但不要用光照射。
[0226]
将细胞存活率和浓度的数值制成表格并以柱状图的形式呈现。
[0227]
平均细胞存活率=吸光度(每种前药的平均值)/吸光度(对照组的平均值)
×
100%。
[0228]
从图4来看可以得出以下结论:式(i)所示的ala
‑
hpo杂合衍生物在100μm浓度下与hela、mcf
‑
7、a375肿瘤细胞系共孵育4h,发现该系列化合物未表现出细胞暗毒性。说明在没有光照的条件下,该系列化合物对细胞是安全的。
[0229]
实施例19
[0230]
下面是本发明中式(ii)所示的ala
‑
hpo杂合衍生物的药理实验数据:
[0231]
1、光毒性实验
[0232]
实验方法:将不同的肿瘤细胞以约1.0
×
104/孔的密度接种与96孔板中,培养48h后,用磷酸缓冲液洗涤细胞。100μl含有不同浓度(100~400μm)的化合物溶液加入指定的孔中,其中阳性药分别是ala、3
‑
羟基
‑
1,2
‑
二甲基吡啶
‑4‑
酮(cp20)、ala与3
‑
羟基
‑
1,2
‑
二甲基吡啶
‑4‑
酮(cp20)的混合给药,培养4h或24h后,使用5j
·
cm
‑2的蓝光照射5分钟,随后吸掉含有药物的培养基,使用不含有血清的培养基继续培养18h。采用mtt测定细胞毒性。mtt检测方法为:除去孔板中的所有培养基,每孔加入100μl含0.5%mtt的培养基,加盖放入培养箱中培养4h。再次除去板中所有培养基,每孔加入100μl二甲基亚砜,振摇5分钟,测定520nm处吸光度,计算每个前药在每种浓度下的平均细胞存活率,并以对照的百分比表示。
[0233]
将细胞存活率和浓度的数值制成表格并以柱状图的形式呈现。
[0234]
平均细胞存活率=吸光度(每种前药的平均值)/吸光度(对照组的平均值)
×
100%。
[0235]
从图5(a)、图6(a)以及图7(a)来看可以得出以下结论:式(ii)所示的ala
‑
hpo杂合衍生物与hela、mcf
‑
7、a375肿瘤细胞系共孵育4h,使用5j
·
cm
‑2的蓝光照射后,发现化合物b1、b2、b3、b4、b6、b7、b8、b9、b10浓度在100~400μm对肿瘤细胞都没有明显的细胞杀伤作用,而化合物b5在三种不同的肿瘤细胞系则表现出较为明显的细胞杀伤作用,其细胞杀伤作用高于ala,或者cp20,但是明显低于ala与cp20混合给药。其中ala、化合物b5在100μm、200μm、400μm浓度下处理的hela细胞系的存活百分比分别是94.5%、82.1%、70.5%、70.4%、64.9%、48.6%;ala、化合物b5在100μm、200μm、400μm浓度下处理的mcf
‑
7细胞系的存活百分比分别是98.4%、57.8%、28.0%、40.2%、32.1%、28.9%;ala、化合物b5在100μm、200μm、400μm浓度下处理的a375细胞系的存活百分比分别是73.0%、52.7%、43.6%、47.0%、36.7%、28.3%。
[0236]
此外,还研究了式(ii)所示的系列化合物与hela、mcf
‑
7、a375肿瘤细胞系共孵育24h后的细胞光毒性,所使用的蓝光光剂量也为5j
·
cm
‑2。从图5(b)、图6(b)以及图7(b)发现化合物b1、b2、b3、b4、b6、b7、b8、b9、b10浓度在100~200μm对三种不同的肿瘤细胞未表现出明显的细胞杀伤作用,而在400μm浓度时表现出一定的细胞毒性,但是其细胞杀伤作用低于相同浓度下的ala。其中ala、化合物b5在100~400μm浓度下处理的hela细胞系的存活百分比分别是65.5%、64.1%、48.6%、40.0%、32.8%、25.0%;ala、化合物b5在100~400μm浓度下处理的mcf
‑
7细胞系的存活百分比分别是92.5%、61.7%、13.1%、32.6%、24.6%、18.3%;ala、化合物b5在100~400μm浓度下处理的a375细胞系的存活百分比分别是72.2%、57.3%、27.2%、32.6%、24.4%、19.8%;
[0237]
以上结果表明,化合物b5可以更好的进入细胞并在细胞内肽酶的作用下破坏ppix前药与hpo分子之间的键,游离出ala,并能通过代谢产生光敏活性物质ppix。
[0238]
2、暗毒性试验
[0239]
为了测定化合物的“暗”毒性,重复光毒性实验步骤,但不要用光照射。
[0240]
将细胞存活率和浓度的数值制成表格并以柱状图的形式呈现。
[0241]
平均细胞存活率=吸光度(每种前药的平均值)/吸光度(对照组的平均值)
×
100%。
[0242]
从图8(a)、图9(a)以及图10(a)来看可以得出以下结论:式(ii)所示的ala
‑
hpo杂
合衍生物与hela、mcf
‑
7、a375肿瘤细胞系共孵育4h,发现该系列化合物在100~400μm浓度下对肿瘤细胞未表现出明显的细胞暗毒性。当孵育时间延长至24h时,从图8(b)、图9(b)以及图10(b)来看该系列化合物在100~400μm浓度下对hela细胞未表现出明显的暗毒性;而对于在mcf
‑
7细胞系中,b2、b3、b5、b6、b7、b8、b9、b10在400μm浓度下孵育24h则表现出较明显的细胞暗毒性;当该系列化合物与a375细胞孵育24h后,仅化合物b8和b10在400μm浓度下才表现出明显的暗毒性。
[0243]
最后,需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1