泰瑞拉奉结晶的制作方法
1.本发明属于药物化学领域,涉及泰瑞拉奉结晶以及泰瑞拉奉结晶的制备方法和用途。
背景技术:
2.泰瑞拉奉,化学名称:2-甲基-5-亚胺基-苯并[d][1,3]噁嗪[5-b]吡唑,结构如式i所示。泰瑞拉奉是基于先导化合物依达拉奉结构,通过分子骨架迁越药物设计方法,设计和合成的一个的全新化合物(cn101508696a)。体外试验表明泰瑞拉奉具有显著清除羟氧和超氧自由基作用。体内试验表明泰瑞拉奉能呈剂量依赖性地显著改善脑缺血再灌注动物的神经缺陷症状,缩小脑梗死面积,降低脑损伤程度,减轻脑水肿,抑制受损脑组织的脂质过氧化。
[0003][0004]
临床研究结果表明泰瑞拉奉能够显著的改善脑卒中症状,安全性和耐受性良好,没有出现严重不良事件。但泰瑞拉奉结构刚性、化学性质稳定,在水或者通常有机溶剂中溶解度都比较小,基本不溶于水,成药性较差。发明人中国专利cn 101966146 a进行了多种增溶性试验,但需要在60℃-85℃通氮气条件下,在搅拌下将泰瑞拉奉溶解在精氨酸水溶液中。
技术实现要素:
[0005]
本发明的目的在于提供具有药用价值的泰瑞拉奉结晶。
[0006]
本发明的目的可以通过以下措施达到:
[0007]
一种泰瑞拉奉结晶a,其x射线粉末衍射图谱相对强度为100%的特征峰的2θ角为8.35
±
0.2
°
。
[0008]
优选的,泰瑞拉奉结晶a,其x射线粉末衍射图谱在2θ值为8.35
±
0.2、10.55
±
0.2、13.80
±
0.2、14.60
±
0.2、15.90
±
0.2、17.95
±
0.2、19.20
±
0.2、21.65
±
0.2、27.33
±
0.2
°
处具有特征峰。
[0009]
进一步优选的,泰瑞拉奉结晶a的x射线粉末衍射图谱如图2或图3或图4所示。
[0010]
一种泰瑞拉奉结晶a的制备方法,包括:室温下,将泰瑞拉奉加入有机溶剂中,加入浓氨水溶解泰瑞拉奉,再加入有机酸,搅拌析出固体;过滤,依次用无水乙醇、纯净水、无水乙醇洗涤,60℃干燥,得到泰瑞拉奉结晶a。
[0011]
所述的有机溶剂为能与氨水互溶的有机溶剂,具体选自甲醇、乙醇或二甲基甲酰胺中的一种或多种。
[0012]
所述的有机酸为甲酸、乙酸等。
[0013]
所述的有机溶剂和泰瑞拉奉的质量比为8~10:1,所述的有机溶剂和氨水的质量比为2~2.5:1,所述的有机溶剂和有机酸的质量比为1.5~2:1。
[0014]
作为泰瑞拉奉结晶a的制备方法的进一步优选技术方案,包括:室温下,将泰瑞拉奉加入有机溶剂中,加入浓氨水溶解泰瑞拉奉,加入活性炭,搅拌、过滤,往滤液中加入有机酸,搅拌析出固体;过滤,依次用无水乙醇、纯净水、无水乙醇洗涤,60℃干燥,得到泰瑞拉奉结晶a。
[0015]
泰瑞拉奉结晶a的另一种制备方法,包括:泰瑞拉奉在二甲基甲酰胺中加热溶解,冷却至室温,析出固体;过滤,滤饼依次用二甲基甲酰胺、乙醇洗涤,再依次用纯净水、乙醇浸泡,抽干,60℃干燥,得到泰瑞拉奉结晶a。
[0016]
所述的泰瑞拉奉和二甲基甲酰胺的质量体积比为1:8~10。
[0017]
作为泰瑞拉奉结晶a的制备方法的进一步优选技术方案,包括:泰瑞拉奉在二甲基甲酰胺中加热溶解,加入活性炭,趁热过滤,冷却至室温,析出固体;过滤,滤饼依次用二甲基甲酰胺、乙醇洗涤,再依次用纯净水、乙醇浸泡,抽干,60℃干燥,得到泰瑞拉奉结晶a。
[0018]
泰瑞拉奉在二甲基甲酰胺中溶解度随温度升高而显著增加,可以作为重结晶溶剂。但二甲基甲酰胺在高温下会分解生成甲醛,甲醛和二分子泰瑞拉奉缩合生成二聚合泰瑞拉奉(式ii)。考虑到采用二甲基甲酰胺作为泰瑞拉奉重结晶溶剂会产生少量聚合泰瑞拉奉杂质。因此,二甲基甲酰胺不是泰瑞拉奉结晶a的优选重结晶溶剂。
[0019][0020]
一种泰瑞拉奉结晶b,其x射线粉末衍射图谱相对强度为100%的特征峰的2θ角为12.88
±
0.2
°
。
[0021]
优选的,泰瑞拉奉结晶b,其x射线粉末衍射图谱在2θ值为7.22
±
0.2、10.64
±
0.2、12.88
±
0.2、14.42
±
0.2、17.79
±
0.2、21.42
±
0.2、25.54
±
0.2、26.62
±
0.2、27.06
±
0.2、31.46
±
0.2、34.26
±
0.2
°
处具有特征峰。
[0022]
进一步优选的,泰瑞拉奉结晶b的x射线粉末衍射图谱如图6所示。
[0023]
泰瑞拉奉结晶b的制备方法,包括:以小分子羧酸或小分子羧酸水溶液为重结晶溶剂,将泰瑞拉奉加热溶解在重结晶溶剂中,冷却至室温,搅拌结晶,过滤,依次用无水乙醇、纯净水、无水乙醇洗涤,60℃干燥,得到泰瑞拉奉结晶b;其中,所述的小分子羧酸为甲酸、乙酸等与水互溶的小分子羧酸;所述的小分子羧酸水溶液中小分子羧酸的浓度为50%(v/v);所述的重结晶溶剂和泰瑞拉奉体积质量之比≥50:1ml/g或l/kg。
[0024]
作为泰瑞拉奉结晶b的制备方法的进一步优选技术方案,包括:将泰瑞拉奉加热溶解在重结晶溶剂中,加入活性炭,趁热过滤,冷却至室温,搅拌结晶,过滤,依次用无水乙醇、纯净水、无水乙醇洗涤,60℃干燥,得到泰瑞拉奉结晶b。
[0025]
在小分子羧酸(如甲酸、乙酸等)或小分子羧酸水溶液中,泰瑞拉奉溶解度随温度变化增加不显著,且需要采用大量重结晶溶剂(如50%乙酸水溶液和泰瑞拉奉体积质量之比≥50:1ml/g或l/kg)才能加热溶解泰瑞拉奉。用该方法制备泰瑞拉奉结晶b,生产效率较低,因此,泰瑞拉奉结晶b不是泰瑞拉奉优选的药用晶型。
[0026]
一种泰瑞拉奉结晶c,其x射线粉末衍射图谱相对强度为100%的吸收峰的2θ角为10.61
±
0.2
°
。
[0027]
优选的,泰瑞拉奉结晶c,其x射线粉末衍射图谱在2θ值为7.16
±
0.2、10.61
±
0.2、12.81
±
0.2、14.37
±
0.2、17.77
±
0.2、18.66
±
0.2、21.35
±
0.2、25.52
±
0.2、26.53
±
0.2、27.09
±
0.2、32.22
±
0.2
°
处具有特征峰。
[0028]
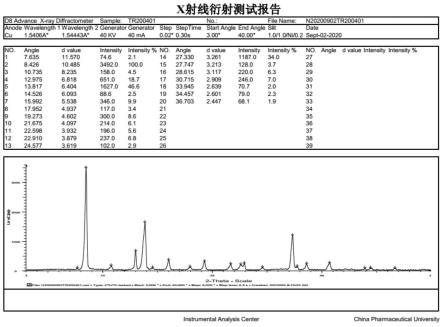
进一步优选的,泰瑞拉奉结晶c的x射线粉末衍射图谱如图8所示。
[0029]
泰瑞拉奉结晶c的制备方法:以乙醇、浓盐酸和水的混合溶剂为重结晶溶剂,先加热回流使泰瑞拉奉溶解,冷却结晶,过滤,滤饼依次用重结晶溶剂和水洗涤,再分散于水中,用40%的氢氧化钠溶液中和至ph7~8,过滤,滤饼依次用水、无水乙醇洗涤,60℃左右干燥,得到泰瑞拉奉结晶c。其中,所述的乙醇、浓盐酸和水的质量比为8~10:1~1.5:1;所述的重结晶溶剂和泰瑞拉奉的质量比为15~20:1。
[0030]
泰瑞拉奉的亚胺结构具有酸性,使泰瑞拉奉在氨水中溶解,再利用有机酸中和氨水得到泰瑞拉奉结晶。该方法制得的泰瑞拉奉结晶a性状稳定,便于贮存和使用,并且溶解条件要求相对较低,溶解速度更快,溶解度能满足制剂的需要。因此,泰瑞拉奉结晶a是泰瑞拉奉优选药用晶型。
[0031]
表1.泰瑞拉奉结晶a在精氨酸水溶液中的溶解度(25℃)
[0032][0033]
表2.泰瑞拉奉结晶在5%精氨酸水溶液(ph9)中的溶解情况
[0034][0035]
本发明的另一个目的是提供一种药物组合物,该药物组合物含有药理学上有效量的泰瑞拉奉结晶a、结晶b或结晶c,以及药学上可接受的辅料。
[0036]
优选的,所述的药物组合物为可注射用的药物制剂。
[0037]
本发明的另一个目的是提供所述的药物组合物在制备治疗或预防心脑血管疾病药物方面的用途。
[0038]
发明人在中国专利cn 101508696a公开了泰瑞拉奉的合成方法:在碱性条件下,2-氰基苯肼和乙酰乙酸甲酯在40~80℃高温下缩合生成2-(3-甲基-5-氧代-4,5-二氢-1h-吡唑-1-基)苯腈,再由氯化氢催化环合生成泰瑞拉奉。该反应条件温度高,副产物多,产品不容易纯化,收率在40%左右。
[0039][0040]
本发明的另一个目的是提供一种改进的泰瑞拉奉的合成工艺,包括:以饱和氯化氢乙醇溶液为反应溶剂,2-氰基苯肼和乙酰乙酸甲酯在饱和氯化氢乙醇溶液中缩合、环合生成泰瑞拉奉。
[0041][0042]
所述的2-氰基苯肼和乙酰乙酸甲酯的摩尔比为0.8~1:1。
[0043]
反应温度为室温。
[0044]
反应结束后,反应液降温至5℃左右,加入反应溶剂2倍质量的冰水,搅拌析出产
物,过滤,依次采用33%乙醇和无水乙醇洗涤,得到泰瑞拉奉纯品。
[0045]
本发明采用一锅法制备泰瑞拉奉,反应条件温和,在室温下即可进行,避免高温副产,反应收率高,至少达到75%,且产品易于纯化。本发明泰瑞拉奉的合成工艺更适宜工业化生产。
附图说明
[0046]
图1为泰瑞拉奉结晶a(实施例2,氨水/乙酸溶液)的显微照片。
[0047]
图2为泰瑞拉奉结晶a(实施例2,氨水/乙酸溶液,0月)的x-射线粉末衍射图。
[0048]
图3为泰瑞拉奉结晶a(实施例2,氨水/乙酸溶液,40℃考察6个月)的x-射线粉末衍射图。
[0049]
图4为泰瑞拉奉结晶a(实施例3,dmf溶液)的x-射线粉末衍射图。
[0050]
图5为泰瑞拉奉结晶b的显微照片。
[0051]
图6为泰瑞拉奉结晶b(实施例4,50%乙酸溶液)的x-射线粉末衍射图。
[0052]
图7为泰瑞拉奉结晶c的显微照片。
[0053]
图8为泰瑞拉奉结晶c(实施例5,乙醇盐酸水溶液)的x-射线粉末衍射图。
具体实施方式
[0054]
下面结合具体实施方式对本发明的技术方案做进一步说明。
[0055]
本发明使用的浓氨水(市售)的浓度为25%~28%,浓盐酸(市售)浓度为36%~38%。
[0056]
实施例1
[0057]
泰瑞拉奉制备
[0058]
50l反应釜中,搅拌下加入饱和氯化氢乙醇溶液10.5kg、2-氰基苯肼1.5kg和乙酰乙酸甲酯1.57kg,室温搅拌反应过夜;反应液降温至5℃左右,加入冰水21kg,并在5℃下搅拌1~1.5小时,抽滤,用33%乙醇溶液洗涤滤饼2~3次,再用无水乙醇洗涤滤饼1次,抽干后在60℃左右干燥,得泰瑞拉奉1.91kg。
[0059]
实施例2
[0060]
50l反应釜中,加入泰瑞拉奉(按照实施例1方法制得)1.97kg和无水乙醇17.57kg,室温搅拌下加入浓氨水7.64kg,待全部溶解后,加活性炭0.1kg,保持25~35℃搅拌30分钟,抽滤,滤液转移到干净的反应釜中,搅拌下加入冰醋酸8.83kg,自然冷却至室温,搅拌结晶过夜;过滤,用无水乙醇洗涤,再用纯净水分4次浸泡洗涤,最后用无水乙醇洗涤,充分抽干,60℃干燥,得到泰瑞拉奉结晶a1.58kg。
[0061]
泰瑞拉奉结晶a的显微照片见图1。
[0062]
泰瑞拉奉结晶a的x-射线粉末衍射图(0月)见图2,泰瑞拉奉结晶a进行稳定性考察(40℃加速试验6月),其x-射线粉末衍射图见图3,说明泰瑞拉奉结晶a性状稳定,便于贮存和使用。
[0063]
表3.泰瑞拉奉结晶a的x-射线粉末衍射数据
[0064][0065]
实施例3
[0066]
1000ml反应瓶中,加入泰瑞拉奉(按照实施例1方法制得)43g、二甲基甲酰胺(dmf)400ml,加热至微沸,全部溶解,稍冷,加入活性碳1g,再加热20min;热过滤,冷却至室温结晶,过滤;滤饼,依次用二甲基甲酰胺50ml、乙醇50ml洗涤,再用纯净水50ml浸泡20min,最后用乙醇50ml浸泡20min,充分抽干,60℃干燥,得到泰瑞拉奉结晶a36g。
[0067]
泰瑞拉奉结晶a的x-射线粉末衍射图见图4。
[0068]
表4.泰瑞拉奉结晶a的x-射线粉末衍射数据
[0069]
2θd值相对强度/%8.38810.53210010.4318.4743.113.7726.42543.414.7675.99415.016.0495.51821.617.9074.9494.519.2364.6116.721.6514.10130.627.2843.26643.7
[0070]
实施例4
[0071]
1000ml反应瓶中,加入泰瑞拉奉10g、乙酸250ml、水250ml,加热回流溶解,待全部溶解后,稍冷加活性炭0.5g,再加热至回流,趁热过滤,滤液自然冷却至室温,搅拌结晶过夜。过滤,用无水乙醇洗涤,再用纯净水分4次浸泡洗涤,最后用无水乙醇洗涤,充分抽干。60℃干燥得到泰瑞拉奉结晶b 6.58g。
[0072]
泰瑞拉奉结晶b的显微照片见图5。
[0073]
泰瑞拉奉晶型b的x射线粉末衍射图谱见图6。
[0074]
表5.泰瑞拉奉结晶a的x-射线粉末衍射数据
[0075]
2θd值相对强度/%
7.22812.2201.510.6398.30918.612.8766.870100.014.4166.13912.517.7974.9803.721.4224.1458.425.5493.4842.826.6213.3464.627.0573.2939.231.4692.8412.534.2612.6153.5
[0076]
实施例5
[0077]
50l反应釜中,加入泰瑞拉奉(按照实施例1方法制得)1.91kg、纯净水26kg、浓盐酸3.91kg和无水乙醇2.6kg,加热至回流,保持回流20~30分钟使物料溶解,停止加热,温度降至85~90℃,加入活性炭后继续加热回流30~40分钟;趁热抽滤,滤液冷却至室温结晶;抽滤,滤饼用重结晶溶剂(纯净水、浓盐酸和无水乙醇按照质量比26:3.91:2.6kg配制)和水依次洗涤,再加入到反应釜中,加入水16kg,室温搅拌下滴加40%的氢氧化钠溶液中和至ph7~8,抽滤,滤饼用水洗涤固体3~4次、无水乙醇洗涤1次,60℃左右干燥,得到泰瑞拉奉结晶c 0.86kg。
[0078]
泰瑞拉奉结晶c的显微照片见图7。
[0079]
泰瑞拉奉晶型c的x射线粉末衍射图谱见图8。
[0080]
表6.泰瑞拉奉结晶c的x-射线粉末衍射数据
[0081]
2θd值相对强度/%7.16512.3272.510.6078.334100.012.8106.90569.214.3716.15827.117.7754.9869.218.6624.7518.221.3554.15710.825.5293.48622.126.5343.35714.927.0903.28915.532.2192.7765.6
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1