吗啡喃衍生物的晶体及其制备方法与流程
1.本发明属于药物化学领域,具体涉及吗啡喃衍生物的晶体及其制备方法。
背景技术:
2.通常,一个具有多晶型的化合物有时在不同晶体之间具有不同物理性质。特别是在医药领域,在溶解度、溶解速度、稳定性或吸收度等方面有所不同。因此,即便使用相同化合物,其效价亦随其晶型而有所不同。所以,当具有多晶型的化合物用作药物时,必须稳定地提供具有均一晶型的化合物以确保其均一的质量和稳定的效力。
3.17-(环丙基甲基)-3,14β-二羟基-4,5α-环氧基-6β-[n-甲基-反-3-(3-呋喃基)丙烯酰胺基]吗啡喃盐酸盐(如式i所示)已被证实其可做为镇痛、利尿及止痒药剂的有效活性成分。现有技术cn101155814b公开了该吗啡喃衍生物具有a型、b型、及c型三种晶型。
[0004][0005]
为优化该吗啡喃衍生物的物性,本发明提供新型吗啡喃衍生物的结晶形式及其制备方法。
技术实现要素:
[0006]
一方面,本发明涉及式i化合物的晶体结构:
[0007][0008]
在一些实施方案中,提供了式i化合物的晶型i,使用cu-ka辐射,在2θ角度表示的x射线粉末衍射图中在2θ值为7.28
±
0.2
°
、11.88
±
0.2
°
、15.57
±
0.2
°
、17.21
±
0.2
°
和21.11
±
0.2
°
处有特征峰。
[0009]
在一些实施方案中,所述晶型i,在以2θ角度表示的x射线粉末衍射图中在2θ值为7.28
±
0.2
°
、10.60
±
0.2
°
、11.88
±
0.2
°
、14.57
±
0.2
°
、15.57
±
0.2
°
、17.21
±
0.2
°
、18.21
±
0.2
°
、21.11
±
0.2
°
和22.23
±
0.2
°
处有特征峰。
[0010]
在一些实施方案中,所述晶型i,在以2θ角度表示的x射线粉末衍射图中在2θ值为7.28
±
0.2
°
、9.71
±
0.2
°
、10.32
±
0.2
°
、10.60
±
0.2
°
、11.12
±
0.2
°
、11.67
±
0.2
°
、11.88
±
0.2
°
、14.57
±
0.2
°
、14.98
±
0.2
°
、15.57
±
0.2
°
、17.21
±
0.2
°
、18.21
±
0.2
°
、21.11
±
0.2
°
和22.23
±
0.2
°
处有特征峰。
[0011]
在一些实施方案中,所述晶型i,在以2θ角度表示的x射线粉末衍射图中在2θ值为7.28
±
0.2
°
、9.11
±
0.2
°
、9.71
±
0.2
°
、10.32
±
0.2
°
、10.60
±
0.2
°
、11.12
±
0.2
°
、11.67
±
0.2
°
、11.88
±
0.2
°
、14.18
±
0.2
°
、14.57
±
0.2
°
、14.98
±
0.2
°
、15.57
±
0.2
°
、17.21
±
0.2
°
、18.21
±
0.2
°
、19.68
±
0.2
°
、20.39
±
0.2
°
、20.72
±
0.2
°
、21.11
±
0.2
°
、21.46
±
0.2
°
、21.90
±
0.2
°
、22.23
±
0.2
°
、23.44
±
0.2
°
、23.79
±
0.2
°
、25.59
±
0.2
°
和29.40
±
0.2
°
处有特征峰。
[0012]
在一些实施方案中,所述式i化合物的晶型i,其x射线粉末衍射图谱解析数据如表1所示:
[0013]
表1式i化合物的晶型i的x射线粉末衍射图谱解析数据
[0014]
编号衍射角2θ相对强度编号衍射角2θ相对强度17.28
°
100.00%2724.15
°
6.20%29.11
°
7.50%2825.05
°
9.70%39.71
°
15.40%2925.59
°
12.10%410.32
°
16.50%3025.90
°
8.60%510.60
°
29.20%3126.31
°
3.30%611.12
°
20.20%3226.72
°
3.00%711.67
°
21.40%3327.15
°
3.00%811.88
°
32.10%3428.09
°
4.70%913.32
°
2.90%3528.52
°
1.00%1014.18
°
6.20%3629.02
°
3.60%1114.57
°
29.90%3729.40
°
8.10%1214.99
°
19.50%3829.80
°
2.10%1315.57
°
48.60%3930.22
°
2.90%1417.21
°
75.90%4030.51
°
1.80%1518.21
°
29.00%4130.87
°
2.80%1618.69
°
1.30%4231.35
°
3.90%1719.50
°
3.80%4332.19
°
1.60%1819.68
°
6.70%4432.62
°
3.60%1920.39
°
10.20%4533.75
°
1.20%2020.72
°
6.10%4634.40
°
1.90%2121.11
°
42.50%4734.90
°
1.20%2221.46
°
7.10%4835.68
°
1.30%2321.90
°
13.40%4936.15
°
1.90%2422.23
°
24.90%5037.37
°
1.60%2523.45
°
12.30%5138.47
°
4.00%2623.79
°
15.30%5239.62
°
0.50%
[0015]
。
[0016]
在一些实施方案中,所述晶型i以2θ角度表示的x射线粉末衍射具有如图1所示的图谱,即具有如图1所示的xrpd图谱所代表的特征。
[0017]
在一些实施方案中,所述晶型i在差示扫描量热法测得的热分析图中,在峰温为68.4
±
5℃、94.9
±
5℃和215.5
±
5℃处存在3个吸热峰;在一些典型的实施方案中,所述晶型i在差示扫描量热法测得的热分析图中,在峰温为68.4
±
3℃、94.9
±
3℃和215.5
±
3℃处存在3个吸热峰。
[0018]
在一些典型的实施方案中,所述晶型i具有如图2所示的dsc图谱,即具有如图2所示的dsc图谱所代表的特征。
[0019]
在一些实施方案中,所述晶型i的热重分析曲线在175
±
3℃处失重为4.1
±
0.2%。
[0020]
在一些典型的实施方案中,所述晶型i具有如图3所示的tga图谱,即具有如图3所示的tga图谱所代表的特征。
[0021]
在一些实施方案中,提供了式i化合物的晶型iii,使用cu-ka辐射,在2θ角度表示的x射线粉末衍射图中在2θ值为9.75
±
0.2
°
、11.54
±
0.2
°
、14.93
±
0.2
°
、15.14
±
0.2
°
和20.97
±
0.2
°
处有特征峰。
[0022]
在一些实施方案中,所述晶型iii,在2θ角度表示的x射线粉末衍射图中在2θ值为7.54
±
0.2
°
、9.40
±
0.2
°
、9.75
±
0.2
°
、11.54
±
0.2
°
、14.93
±
0.2
°
、15.14
±
0.2
°
、18.39
±
0.2
°
、20.97
±
0.2
°
和26.27
±
0.2
°
处有特征峰。
[0023]
在一些实施方案中,所述晶型iii,在2θ角度表示的x射线粉末衍射图中在2θ值为7.54
±
0.2
°
、9.40
±
0.2
°
、9.75
±
0.2
°
、11.54
±
0.2
°
、12.06
±
0.2
°
、14.93
±
0.2
°
、15.14
±
0.2
°
、18.39
±
0.2
°
、18.68
±
0.2
°
、19.81
±
0.2
°
、20.97
±
0.2
°
、22.11
±
0.2
°
、22.59
±
0.2
°
和26.27
±
0.2
°
处有特征峰。
[0024]
在一些实施方案中,所述晶型iii,在2θ角度表示的x射线粉末衍射图中在2θ值为2θ值为7.54
±
0.2
°
、9.40
±
0.2
°
、9.75
±
0.2
°
、11.05
±
0.2
°
、11.54
±
0.2
°
、12.06
±
0.2
°
、14.93
±
0.2
°
、15.14
±
0.2
°
、15.70
±
0.2
°
、18.39
±
0.2
°
、18.68
±
0.2
°
、19.81
±
0.2
°
、20.97
±
0.2
°
、21.31
±
0.2
°
、22.11
±
0.2
°
、22.59
±
0.2
°
、24.29
±
0.2
°
、24.70
±
0.2
°
和26.27
±
0.2
°
处有特征峰。
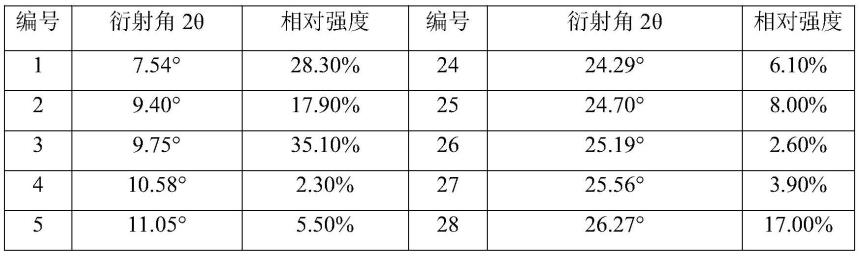
[0025]
在一些实施方案中,所述式i化合物的晶型iii,其x射线粉末衍射图谱解析数据如表2所示:
[0026]
表2式i化合物的晶型iii的x射线粉末衍射图谱解析数据
[0027]
[0028]
。
[0029]
在一些实施方案中,所述晶型iii以2θ角度表示的x射线粉末衍射具有如图4所示的图谱,即具有如图4所示的xrpd图谱所代表的特征。
[0030]
在一些实施方案中,所述晶型iii在差示扫描量热法测得的热分析图中,在起始温度为231.95
±
5℃处有一个放热峰;在一些典型的实施方案中,所述晶型iii在差示扫描量热法测得的热分析图中,在起始温度为231.95
±
3℃处有一个放热峰。
[0031]
在一些典型的实施方案中,所述晶型iii具有如图5所示的dsc图谱,即具有如图5所示的dsc图谱所代表的特征。
[0032]
在一些典型的实施方案中,所述晶型iii具有如图6所示的tga图谱,即具有如图6所示的tga图谱所代表的特征。
[0033]
另一方面,本发明提供了所述晶型i或晶型iii在用于制备止痛、利尿和止痒药物中的用途。
[0034]
再一方面,本发明还提供了所述晶型iii的制备方法,其包括以下步骤:
[0035]
(1)将式i化合物无定型加入a类溶剂中,其中a类溶剂选自甲醇、乙醇、乙酸乙酯、四氢呋喃、甲苯或二氯甲烷中的一种或两种的组合,优选为甲醇、乙醇或两者的混合物;
[0036]
(2)升温打浆;
[0037]
(3)缓慢降温搅拌析晶;
[0038]
(4)抽滤、干燥得到晶型iii。
[0039]
在一些实施方案中,a类溶剂与式i化合物的体积质量比为10~15ml/g;优选为11~14ml/g。
[0040]
在一些实施方案中,步骤(2)中升温打浆的温度为50-80℃;在一些典型的实施方案中,步骤(2)中升温打浆的温度为50-70℃;在一些更为典型的实施方案中,步骤(2)中升温打浆的温度为55-60℃。
[0041]
在一些实施方案中,步骤(2)中打浆的时间为1~5h;在一些典型的实施方案中,步骤(2)中打浆的时间为1~2h;在一些更为典型的实施方案中,步骤(2)中打浆的时间为1h。
[0042]
在一些实施方案中,步骤(3)中的缓慢降温搅拌析晶是温度降至-5~30℃;在一些典型的实施方案中,步骤(3)中的缓慢降温搅拌析晶是温度降至0~25℃;在一些更为典型的实施方案中,步骤(3)中的缓慢降温搅拌析晶是温度降至0~10℃。
[0043]
在一些实施方案中,步骤(3)中的缓慢降温搅拌析晶的析晶时间为1~10h;在一些典型的实施方案中,步骤(3)中的缓慢降温搅拌析晶的析晶时间为1~6h;在一些更为典型的实施方案中,步骤(3)中的缓慢降温搅拌析晶的析晶时间为1~2h。
[0044]
再一方面,本发明还提供了所述晶型iii的制备方法,其包括以下步骤:
[0045]
(a)将式i化合物无定型加入c类溶剂中,其中c类溶剂选自甲醇、乙醇、乙酸乙酯、四氢呋喃、甲苯或二氯甲烷一种或两种的组合;优选为甲醇、乙醇或两者的混合物;更优选为甲醇;
[0046]
(b)升温溶解、过滤得到澄清溶液;
[0047]
(c)降至室温后边搅拌边加入d类溶剂,滴加完毕后保温搅拌,其中d类溶剂选自正丙醇、异丙醇或正丁醇中的一种或两种;优选为异丙醇;
[0048]
(d)抽滤、干燥得到晶型iii。
[0049]
在一些实施方案中,c类溶剂与式i化合物的体积质量比为5~10ml/g;优选为5~6ml/g。
[0050]
在一些实施方案中,c类溶剂与d类溶剂的体积比为1:1~5,优选为1:1~3;进一步优选为1:3。
[0051]
在一些实施方案中,步骤(c)中滴加完毕后保温搅拌的时间为12~36h;在一些典型的实施方案中,步骤(c)中滴加完毕后保温搅拌的时间为12~24h;在一些更为典型的实施方案中,步骤(c)中滴加完毕后保温搅拌的时间为24h。
[0052]
本发明的有益效果:
[0053]
本发明公开了式i化合物的晶型i和晶型iii,具有优异的稳定性和良好的溶解性,且溶剂残留量低,能达到合格范围。
[0054]
相关定义
[0055]
除非有特定说明,下列用在说明书和权利要求书中的术语具有下述含义:
[0056]
本发明中,“xrpd”是指x射线粉末衍射;
[0057]
本发明中,“dsc”是指差示扫描量热法;
[0058]
本发明中,“tga”是指热重分析;
[0059]
本发明中,“h”是指小时,“min”是指分钟,“ml”指毫升,“μl”指微升;
[0060]
本发明中,“室温”是指25℃;
[0061]
本发明中,“抽滤”是指减压过滤。
[0062]
本发明中的“x-射线粉末衍射图谱”为使用cu-kα辐射测量得到。
[0063]
本发明中的“2θ”或“2θ角度”是指衍射角,θ为布拉格角,单位为
°
或度;每个特征峰
2θ的误差范围为
±
0.20
°
。
[0064]
需要说明的是,在x射线粉末衍射光谱(xrpd)中,由结晶化合物得到的衍射谱图对于特定的结晶往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的结晶并非是特征性的。判断是否与已知的结晶相同时,更应该注意的是峰的相对位置而不是它们的相对强度。此外,对任何给定的结晶而言,峰的位置可能存在轻微误差,这在结晶学领域中也是公知的。例如,由于分析样品时温度的变化、样品移动、或仪器的标定等,峰的位置可以移动,2θ值的测定误差有时约为
±
0.2
°
。因此,在确定每种结晶结构时,应该将此误差考虑在内。在xrpd图谱中通常用2θ角或晶面距d表示峰位置,两者之间具有简单的换算关系:d=λ/2sinθ,其中d代表晶面距,λ代表入射x射线的波长,θ为衍射角。对于同种化合物的同种结晶,其xrpd谱的峰位置在整体上具有相似性,相对强度误差可能较大。还应指出的是,在混合物的鉴定中,由于含量下降等因素会造成部分衍射线的缺失,此时,无需依赖高纯试样中观察到的全部谱带,甚至一条谱带也可能对给定的结晶是特征性的。
[0065]
差示扫描量热法(dsc)测定当晶体由于其晶体结构发生变化或晶体熔融而吸收或释放热时的转变温度。对于同种化合物的同种晶型,在连续的分析中,热转变温度和熔点误差典型的在约5℃之内,通常在约3℃之内。当描述某个化合物具有某一给定的dsc峰或熔点时,指的是该dsc峰或熔点
±
5℃。dsc提供了一种辨别不同晶型的辅助方法。不同的晶体形态可根据其不同的转变温度特征而加以识别。需要指出的是对于混合物而言,其dsc峰或熔点可能会在更大的范围内波动。此外,由于在物质熔化的过程中伴有分解,因此熔化温度与升温速率相关。
[0066]
热重分析(tga)指的是在程序控制温度下测量待测样品的质量与温度变化之间关系的一种热分析技术。当被测物质在加热过程中有升华或汽化现象时,其分解出了气体或失去了结晶水时,引起被测物质量发生变化。这时,热重曲线就不是直线而是有所下降。通过分析热重曲线,即可知道被测物质在什么温度下产生变化,并且根据所失重量,可计算失去了多少物质量。
[0067]
在提到例如xrpd图谱、dsc图谱或tga图谱时,术语“如
……
所示”包括与本文描绘的那些不一定相同,但在被本领域技术人员考虑时落入实验误差的限度内的图谱。
附图说明
[0068]
图1式i化合物晶型i的xrpd图谱
[0069]
图2式i化合物晶型i的dsc曲线
[0070]
图3式i化合物晶型i的tga曲线
[0071]
图4式i化合物晶型iii的xrpd图谱
[0072]
图5式i化合物晶型iii的dsc曲线
[0073]
图6式i化合物晶型iii的tga曲线
[0074]
图7式i化合物晶型a的xrpd图谱
具体实施方式
[0075]
下面通过实施例更详细地描述本发明。但这些具体描述仅用于说明本发明的技术
方案,不对本发明构成任何限制。
[0076]
各仪器测试条件如下:
[0077]
本发明中,x-射线衍射(又称“x-射线粉末衍射”,x-raypowder diffractometer,xrpd)的测定方法如下:
[0078]
仪器型号:d8 advance
[0079]
测试条件:详细的xrpd参数如下:
[0080]
x-ray发生器:cu,kα,
[0081]
管电压:40kv,管电流:40ma.
[0082]
散射狭缝:0.6mm
[0083]
探测器狭缝:5mm
[0084]
反散射狭缝:0.6mm
[0085]
扫描范围:3-40deg
[0086]
步长:0.02deg
[0087]
速率:0.3s
[0088]
样品盘转速:0rpm(未设)
[0089]
本发明中差热分析(又称“差示扫描量热法”,differential scanning calorimeter,dsc)测试方法如下:
[0090]
仪器型号:dsc 25
[0091]
测试方法:取样品(1-10mg)置于dsc铝锅内进行测试,方法为:室温~目标温度,升温速率为10℃/min。
[0092]
本发明热重分析(thermal gravimetric analyzer,tga)测试方法如下:
[0093]
仪器型号:tga550
[0094]
测试条件:取样品(1-10mg)置于tga铂金锅内进行测试,方法为:30~目标温度,升温速率为10℃/min。
[0095]
参考实施例1 17-(环丙基甲基)-3,14β-二羟基-4,5α-环氧基-6β-[n-甲基-反-3-(3-呋喃基)丙烯酰胺基]吗啡喃盐酸盐的制备方法
[0096]
[0097]
i)式b化合物的制备
[0098]
依次向反应瓶中加入二氯甲烷、n,n-二异丙基乙胺和式a化合物,搅拌均匀;氮气置换三次,降温至0℃~10℃缓慢滴加草酰氯;滴毕,升温至20℃~30℃反应至终点(式a化合物的斑点基本消失);将反应液真空浓缩,浓缩物中加入二氯甲烷,再次浓缩,备用。
[0099]
ii)式d化合物的制备
[0100]
依次向反应瓶中加入二氯甲烷、三乙胺和式c化合物(参照文献方法制备),搅拌均匀;氮气置换三次,控制温度-5℃~5℃滴加式b化合物的二氯甲烷溶液;滴毕,控制温度-5℃~5℃反应30分钟,tlc监控反应至终点(式c化合物斑点基本消失);反应完全后,控制温度-5℃~5℃加入碳酸氢钠水溶液搅拌10分钟~20分钟,分液,舍弃水相(上层),有机相用碳酸氢钠水洗3次,舍弃水相(上层),有机相进行干燥浓缩,柱层析纯化得式d化合物。
[0101]
iii)式i化合物的制备
[0102]
向反应瓶里加入甲醇,开启搅拌,加入式d化合物,搅拌均匀;氮气置换三次,控制温度为20℃~30℃滴加2m氯化氢乙酸乙酯溶液;滴毕,控制温度20℃~30℃搅拌1小时;过滤,滤饼用甲醇淋洗;将滤饼转移至反应瓶中,加入甲醇,减压蒸除溶剂,重复2次;再向反应瓶中加入无水乙醇,减压蒸除溶剂;将浓缩物转移至真空干燥箱中干燥得式i化合物无定型。
[0103]
参考实施例2式i化合物的晶型a的制备
[0104]
参照专利cn101155814b中公开的纳呋拉啡晶型a的制备方法,具体操作如下:
[0105]
称取1g参考实施例1中制备得到的式i化合物无定型加入到样品瓶中,加入4.5ml甲醇,升温至40℃-50℃搅拌溶清,向体系滴加13.5ml异丙醇,滴加结束后,降至室温加入1%晶种,室温析晶8h,过滤,用2ml异丙醇淋洗滤饼,45℃-55℃真空干燥4h。该晶体由xrpd进行固体形态的表征(图7),为晶型a。
[0106]
参考实施例3式i化合物的晶型a的制备
[0107]
称取1g参考实施例1中制备得到的式i化合物无定型加入到样品瓶中,加入20ml甲醇溶解,减压浓缩至干,并用20ml乙醇带蒸,加入24ml乙醇,升温至回流1h,降至室温,室温搅拌过夜,过滤,60℃真空干燥5h。经xrpd表征,该晶体为晶型a。
[0108]
参考实施例4式i化合物的晶型a的制备
[0109]
将5g式i化合物无定型用50ml甲醇溶解,减压浓缩至干,加入15ml甲醇和35ml乙酸乙酯,升温至回流,加入晶型a晶种,回流1h,降至室温,保温析晶过夜,60℃真空干燥3h。经xrpd表征,该晶体为晶型a。
[0110]
实施例1式i化合物的晶型i的制备
[0111]
称取1g参考实施例1中制备得到的式i化合物无定型加入到样品瓶中,加入20ml无水乙醇,升温至75℃保温搅拌0.5h,缓慢降温至25℃,保温搅拌1-2h,抽滤、干燥。该晶体经xrpd、dsc和tga进行固体形态的表征,分别得到如图1、2、3所示的图谱。
[0112]
实施例2a式i化合物的晶型iii的制备
[0113]
称取1g参考实施例1中制备得到的式i化合物无定型加入到样品瓶中,加入2ml甲醇和12ml无水乙醇中,升温至55-60℃打浆1h,缓慢降温至0-10℃搅拌析晶2h,抽滤、淋洗,70℃-80℃真空干燥4h。该晶体经xrpd、dsc和tga进行固体形态的表征,分别得到如图4、5、6所示的图谱。
[0114]
实施例2b式i化合物的晶型iii的制备
[0115]
称取1g参考实施例1中制备得到的式i化合物无定型加入到样品瓶中,加入2ml甲醇和12ml无水乙醇中,升温至55-60℃打浆1h,缓慢降温至0-10℃搅拌析晶2h,抽滤、淋洗,70℃-80℃真空干燥6h。经xrpd表征,该晶体为晶型iii。
[0116]
实施例2c式i化合物的晶型iii的制备
[0117]
称取100g参考实施例1中制备得到的式i化合物无定型加入到样品瓶中,加入1l乙醇和0.1l甲醇,升温至55℃-60℃,保温搅拌1h,降至20℃-30℃,保温搅拌2h,再降至0℃-10℃,保温搅拌2h,过滤,0.2l乙醇淋洗滤饼,70℃-80℃真空干燥6h。经xrpd表征,该晶体为晶型iii。
[0118]
实施例3式i化合物的晶型iii的制备
[0119]
将10g式i化合物无定型固体加入60ml甲醇中,升温溶解、过滤得到澄清溶液,在25℃条件下边搅拌边加入180ml异丙醇,滴加完毕后保温搅拌24h,抽滤、淋洗、70-80℃干燥得到晶型iii。经xrpd表征,该晶体为晶型iii。
[0120]
实施例4溶剂残留量测试
[0121]
针对参考实施例2-4制备得到的式i化合物的晶型a和实施例2a-2c制备得到的式i化合物的晶体iii利用气相色谱法进行了溶剂残留量检测。
[0122]
仪器条件:
[0123][0124]
供试品溶液的配制:取各个批次的晶型a或晶型iii约100mg,精密称定,置顶空瓶中,精密加入溶剂1ml使溶解,密封,作为供试品溶液。
[0125]
对照品溶液的配制:取甲醇、乙醇、二氯甲烷、乙酸乙酯、正己烷、四氢呋喃、吡啶和甲苯各适量,精密称定,加溶剂溶解并稀释制成每1ml中约含甲醇0.3mg、乙醇0.5mg、二氯甲烷0.06mg、乙酸乙酯0.5mg、正己烷0.029mg、四氢呋喃0.072mg、吡啶0.020mg和甲苯0.089mg的混合溶液,精密量取1ml,置顶空瓶中,密封,作为对照品溶液。
[0126]
采用上述的气象色谱法测试晶型a与晶型iii中溶剂残留量,按外标法以峰面积计算。
[0127][0128]
从上表看出,不同方法制备的晶型iii的溶剂残留结果均未超过限度,而不同方法制备的晶型a的溶剂残留均超过限度。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1