具有聚集诱导发光性质的化合物及其在手术导航领域的应用的制作方法
本发明属于聚集诱导发光性质化合物合成技术领域,涉及具有聚集诱导发光性质的化合物及其应用。
背景技术:
巴比妥酸类化合物除了可作为聚合催化剂、制取染料的原料和合成维生素b12等常用药品的中间体,其衍生物巴比妥类药物为临床上较早应用的长效神经类药物。因巴比妥类药物生物毒副作用小、作用缓慢、可维持时间长等优点,在临床上常用于镇静、催眠、抗惊厥及抗癫痫,还可用作麻醉前给药。目前,利用具有生物活性的基团充当电子给体(或受体)来合成长波长发射的aie分子的例子还很少,该工作成功利用了廉价的生物活性物质巴比妥酸,通过与aie单元的共轭连接,达到红移aie分子发光波长的目的,合成简单,成本较低,且目标分子性能优良,可有效应用于生物体内肿瘤切除手术的荧光导航。
本专利设计合成了基于1,3-二乙基硫代巴比妥酸为吸电子基团的2种荧光染料,丰富了aie家族,为分子设计提供了全新的思路,同时给出了其在治疗相关疾病的中的新应用,以解决现有技术中的问题。
技术实现要素:
本发明的目的之一是设计合成了基于1,3-二乙基硫代巴比妥酸为吸电子基团的2种荧光染料,丰富了aie家族;目的之二提供了所述化合物的新用途。
本发明公开了一种具有聚集诱导发光性质的化合物,为通式(i)所示分子结构:
或其衍生物、盐、水合物、螯合物、聚合物中的至少一种,其中,n为自然数,所述x采用o或s;
所述r1基团包括如下结构:
所述r2、r3基团包括如下结构:
中至少一种。
这样设计的目的在于,采取“d-π-a”的设计思路,以三维结构高度扭曲的四苯乙烯作为供体单元,通过进一步将噻吩插入其分子结构中,得到更强共轭和更红发射,合成了一系列合成简单、吸收和发射波长逐渐红移的aie分子。为了获得能隙更低的荧光染料,使其发射波长红移至远红/近红外区,以适用于手术导航。选择使用第ⅵa族的s原子替代1,3-二乙基巴比妥酸中的o原子,相较于二乙基巴比妥酸为供体单元的分子,合成的以1,3-二乙基硫代巴比妥酸为吸电子基团的荧光染料具有红移的最大发射波长,即具有更优异的光学穿透力,较小的光损伤,光散射更低,因此更适用于生物成像领域的研究。
优选的,所述r2基团与所述r3基团选用相同的基团。
优选的,所述n取值范围为6-12,进一步优选为8-10。
优选地,具有聚集诱导发光性质的化合物,采用如下所示分子结构:
或其衍生物、盐、水合物、螯合物、聚合物中的至少一种。
本发明还公开了所述具有聚集诱导发光性质的化合物作为手术导航标记试剂的应用。
优选的,所述具有聚集诱导发光性质的化合物用于肿瘤手术导航标记试剂。
本发明还公开了一种手术导航标记试剂组合物,包括所述具有聚集诱导发光性质的化合物。
本发明的有益效果在于:
1、提供了手术导航的新思路;
2、材料获取容易,成本低,易于制备;
3、生物毒性低;
4、发光波长长,受背景荧光干扰小,组织穿透性强。
附图说明
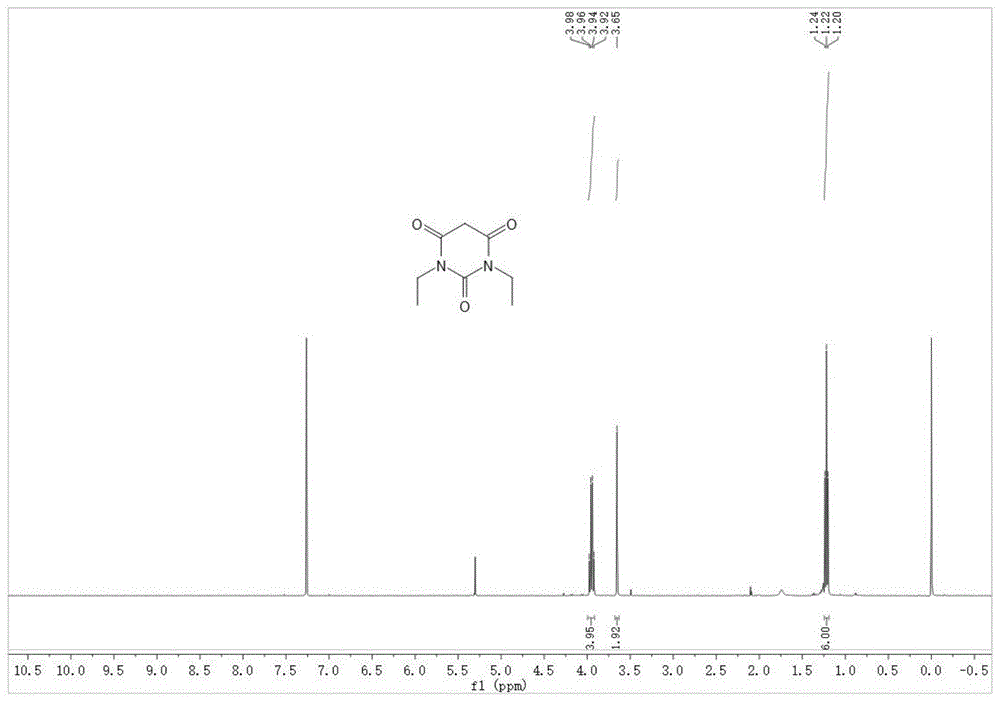
图1为化合物b8的h核磁谱图;
图2为化合物b11的h核磁谱图;
图3为化合物b5的h核磁谱图;
图4为化合物a1的h核磁谱图;
图5为化合物a2的h核磁谱图;
图6为化合物a1的c核磁谱图;
图7为化合物a2的c核磁谱图;
图8为化合物a1(图a)和化合物a2(图b)在不同有机溶剂中的紫外吸收光谱;
图9为化合物a1(图a)和化合物a2(图b)在不同有机溶剂中的荧光发射光谱;
图10为化合物a1(图a)在不同体积比例下thf/水混合溶液中的归一化荧光发射光谱;化合物a2(图b)在不同体积比例下thf/水混合溶液中的归一化荧光发射光谱;化合物a1与化合物a2的聚集诱导发光曲线(图c)(i0为水体积为10%时的荧光强度)图;
图11为化合物a1和化合物a2的优化结构、最高占据分子轨道(homo)和最低未占据分子轨道(lumo)的分布图;
图12为化合物a1nps(图a)和化合物a2nps(图b)的动态光散射直径(dls);插图为相应纳米颗粒的透射电子显微镜图(tem);
图13为化合物a1和化合物a2的细胞活性测试(mtt法)图;
图14为共聚焦激光扫描显微镜(clsm)观察4t1乳腺癌细胞与化合物a2nps共孵育4h后的图像,细胞核用dapi(蓝色)染色图;
图15为肿瘤切除前小鼠腹腔的生物发光和荧光显像图(图a);荧光影像引导切除前后的腹腔荧光图像和肿瘤结节荧光显像(图b);未引导组和化合物a2nps引导组切除的肿瘤结节的生物发光图像(图c)。
具体实施方式
下面结合实施例对本发明的具体实施方式作进一步描述,以下实施例仅用于更加清楚地说明本发明的技术实施例,而不能以此来限制本发明的保护范围。
实施例1
化合物a1(tto)合成路径如下:
具有聚集诱导发光性质的化合物a1的制备方法,包括以下步骤:
步骤一,化合物b3的合成。在氮气氛围下,将化合物b1(溴代四苯乙烯,820mg,2mmol)、化合物b2(联硼酸频那醇酯,762mg,3mmol)、醋酸钾(588mg,6mmol)和pd(dppf)2cl2(87mg,0.12mmol)溶于甲苯溶液(10ml)中,置于25ml反应瓶,回流反应12h。反应结束后,冷却至室温,减压蒸馏除去反应溶剂,得粗产物。通过柱层析法(洗脱剂比例为石油醚:乙酸乙酯=10:1)分离提纯,得产物化合物b3(900mg,产率为98.3%)
步骤二,化合物b5的合成。在氮气氛围下,将化合物b3(900mg,2mmol)、化合物b4(5-溴噻吩-2-甲醛,573mg,3mmol)、四丁基溴化铵(tbab,65mg,0.2mmol)与四三苯基膦钯(pd(pph4)3,47mg,0.04mmol)溶于10ml甲苯溶剂中,置于50ml反应瓶中,加入6ml2m浓度的碳酸钾溶剂(1.65gk2co3,6mlddh2o),回流反应12h。反应结束后,用乙酸乙酯(15ml*3)萃取水相,减压蒸馏除去反应溶剂,得粗产物。通过柱层析法洗脱剂比例为石油醚:乙酸乙酯=8:1)分离提纯,得产物化合物b5(850mg,产率为96.15%)。
步骤三,化合物b8的合成。将化合物b6(丙二酸,208mg,2mmol)与化合物b7(1,3-二乙基脲,232mg,2mmol)溶于2ml冰乙酸置于25ml反应瓶中,加热至60℃后,再加入2ml乙酸酐,反应6h。再加入超纯水1ml,将反应升温至70℃,反应0.5h。反应结束后,将反应冷却至室温,用乙酸乙酯(10ml*3)萃取后得有机相,减压蒸馏除去溶剂,得粗产物。通过柱层析法(洗脱剂比例为石油醚:乙酸乙酯=2:1)分离提纯,得产物化合物b8(360mg,产率为97%)。
步骤四,化合物a1的合成。在氮气氛围下,将化合物b5(226mg,0.6mmol)与化合物b8(120mg,0.66mmol)溶于6ml的乙腈溶液中,滴加2滴哌啶,回流温度下反应8h。反应结束后,将反应冷却至室温,过滤,洗涤,得橙红色固体(350mg,产率为96%)。
实施例2
化合物a2(tts)合成路径如下:
本实施例与实施例1相比较,仅仅是合成步骤三、步骤四不同。
具体如下:
步骤一,化合物b3的合成。在氮气氛围下,将化合物b1(溴代四苯乙烯,820mg,2mmol)、化合物b2(联硼酸频那醇酯,762mg,3mmol)、醋酸钾(588mg,6mmol)和pd(dppf)2cl2(87mg,0.12mmol)溶于甲苯溶液(10ml)中,置于25ml反应瓶,回流反应12h。反应结束后,冷却至室温,减压蒸馏除去反应溶剂,得粗产物。通过柱层析法(洗脱剂比例为石油醚:乙酸乙酯=10:1)分离提纯,得产物化合物b3(900mg,产率为98.3%)
步骤二,化合物b5的合成。在氮气氛围下,将化合物b3(900mg,2mmol)、化合物b4(5-溴噻吩-2-甲醛,573mg,3mmol)、四丁基溴化铵(tbab,65mg,0.2mmol)与四三苯基膦钯(pd(pph4)3,47mg,0.04mmol)溶于10ml甲苯溶剂中,置于50ml反应瓶中,加入6ml2m浓度的碳酸钾溶剂(1.65gk2co3,6mlddh2o),回流反应12h。反应结束后,用乙酸乙酯(15ml*3)萃取水相,减压蒸馏除去反应溶剂,得粗产物。通过柱层析法洗脱剂比例为石油醚:乙酸乙酯=8:1)分离提纯,得产物化合物b5(850mg,产率为96.15%)。
步骤三,化合物b11的合成。将化合物b10(丙二酸二乙酯,264mg,2mmol)与化合物b9(1,3二乙基硫脲,264mg,2mmol)溶于2ml冰乙酸置于25ml反应瓶中,升温至70℃,再加入1ml乙酸酐,反应13h。13h后,升温至回流温度,再反应1h。反应结束后,减压蒸馏除去反应溶剂,得粗产物。通过柱层析法(洗脱剂比例为石油醚:乙酸乙酯=2:1)分离提纯,得产物化合物b11(360mg,产率为96%)。
步骤四,化合物a2的合成。在氮气氛围下,将化合物b5(200mg,0.46mmol)与化合物b11(100mg,0.5mmol)溶于6ml的乙腈溶液中,滴加2滴哌啶,回流温度下反应8h。反应结束后,将反应冷却至室温,过滤,洗涤,得红色固体(285mg,产率为95%)。
为了证明实施例1-2所述的合成路径中化合物(a1)与化合物(a2)被成功,特进行如下测试:
如附图1所示,化合物b8的核磁吸收峰如下:1hnmr(400mhz,chloroform-d)δ3.95(q,j=7.1hz,4h),3.65(s,2h),1.22(t,j=7.1hz,1h).
如附图2所示,化合物b11的核磁吸收峰如下:1hnmr(400mhz,chloroform-d)δ4.55(dq,j=12.0,7.0hz,1h),2.74(s,0h),1.30(dt,j=10.4,7.0hz,1h).
如附图3所示,化合物b5的核磁吸收峰如下:1hnmr(400mhz,chloroform-d)δ9.87(s,1h),7.71(d,j=4.0hz,1h),7.43(d,j=8.4hz,2h),7.35(d,j=4.0hz,1h),7.16–7.10(m,10h),7.09–7.00(m,7h).
如附图4所示,化合物a1的h核磁吸收峰如下:1hnmr(400mhz,chloroform-d)δ8.64(s,1h),7.82(d,j=4.2hz,1h),7.54(d,j=8.4hz,2h),7.44(d,j=4.2hz,1h),7.16–7.10(m,10h),7.08–7.03(m,7h),4.11–4.03(m,4h),1.32–1.24(m,6h).
如附图5所示,化合物a2的h核磁吸收峰如下:1hnmr(400mhz,chloroform-d)δ8.30(d,j=15.2hz,1h),8.11(d,j=15.2hz,1h),7.41–7.36(m,3h),7.27(s,1h),7.14–7.09(m,9h),7.08–7.00(m,7h),4.60–4.51(m,4h),1.31(tt,j=8.0,4.0hz,6h).
如附图6所示,化合物a1的c核磁吸收峰如下:13cnmr(101mhz,cdcl3)δ162.45,161.63,160.22,150.61,148.46,146.76,145.81,143.43,143.25,142.21,140.02,136.13,132.20,131.43,131.34,131.10,127.96,127.89,127.74,126.93,126.78,126.73,126.06,124.36,109.76,37.49,36.73,13.50,13.41.
如附图7所示,化合物a2的c核磁吸收峰如下:13cnmr(101mhz,cdcl3)δ182.95,176.13,166.81,157.99,149.82,143.79,142.45,142.29,140.91,139.04,138.74,138.61,134.09,131.08,130.37,130.29,126.87,126.79,126.66,125.77,125.66,125.60,124.29,123.45,117.76,42.45,42.09,13.09.
应用例1
选用实施例2作为合成的化合物a2作为手术导航试剂。
为了进一步说明本发明的有益效果,特设置如下测试:
光学性质测试
如附图8所示,可以看出化合物a1与化合物a2在不同有机溶剂中均具有显著的紫外吸收峰。
如附图9所示,可以看出化合物a1与化合物a2在不同有机溶剂中均具能够发射荧光。
如附图10所示,附图10(a)、附图10(b)表明化合物a1与化合物a2在不同体积比例下的thf/水混合溶液下均能够发射荧光,证明包括化合物a1和/或化合物a2的组合物也能够做手术导航指示试剂;
附图10(c)中聚集诱导发光曲线表明:首先,化合物a1与化合物a2均具有聚集诱导发光性能;其次,化合物a2性能优于化合物a1的性能,这一变化是s取代o所带来的。
如附图13所示,证明化合物a1与化合物a2具备细胞活性。
如附图14所示,化合物a2能够在细胞尺度上染色。
如附图15所示,化合物a2能够在宏观组织尺度上染色,实现手术导航。
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
- 还没有人留言评论。精彩留言会获得点赞!