用于低阶煤成浆的二元面面吸附型分散剂及其制备方法
本发明属于分散剂领域,涉及一种用于低阶煤成浆的二元面面吸附型分散剂及其制备方法。
背景技术:
不粘煤、次烟煤和褐煤是在成煤初期已经受到氧化作用的低变质程度的煤种,由于其水分大,含氧量多,因此具有低发热量的特点,主要用作气化和发电用煤,也可作动力及民用燃料等。水煤浆因其具有高效、环保等优点,被认为是我国实现煤炭高效清洁利用的重要方式之一,水煤浆技术也是目前煤炭气化的主要手段。将不粘煤、次烟煤和褐煤制备成高质量水煤浆对于丰富水煤浆煤种、提高能源利用效率具有重要的现实意义。分散剂可以改善煤粒界面结构以及界面的相互作用,降低浆体中的结合水含量,从而提高水煤浆性能。目前,常规的水煤浆分散剂在高阶煤制浆中效果较好,但是不适用于低阶煤制浆。
因此,针对低阶煤的分子结构特性,开发低阶煤的高效分散剂,进而提高低阶煤的成浆性能,这仍是目前煤化工行业急需解决的重要问题。
技术实现要素:
本发明的目的在于克服上述现有技术中,水煤浆分散剂不适用于不粘煤、次烟煤和褐煤制浆的缺点,提供一种用于低阶煤成浆的二元面面吸附型分散剂及其制备方法。
为了达到上述目的,本发明采用以下技术方案予以实现:
一种用于低阶煤成浆的二元面面吸附型分散剂,所述面面吸附型分散剂的结构式为:
其中,r为亲水性接枝物。
优选地,亲水性接枝物为淀粉、丙烯酸或apeg。
一种用于低阶煤成浆的二元面面吸附型分散剂的制备方法,包括如下步骤:
s1:将nano3、浓h2so4及石墨按照(1~1.05):(40~41):1的质量比混合反应0.5~1h,得到第一混合溶液,在第一混合溶液中加入kmno4,之后依次进行低温反应、中温反应和高温反应,反应完成后加入蒸馏水和h2o2直至溶液颜色变为亮黄色,依次进行过滤、洗涤和干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:(30~50),冷冻干燥后得到中间体i;
s2:按照(0.5~1)g中间体i:(100~150)mlsocl2:(2~5)mldmf的配料比将中间体i、socl2和dmf混合,进行酰氯化反应,反应后除去溶剂,得到第二产物,将第二产物、三乙胺和丙烯酸羟乙酯按照摩尔比为1:0.08:0.08混合,搅拌酯化反应10~14h,反应结束后静置10~14h,依次经过抽滤、洗涤和干燥后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:(20~30),冷冻干燥后得到中间体ⅱ;
s3:将中间体ⅱ和亲水性接枝物加入蒸馏水中,得到第三混合溶液,蒸馏水的质量为第三混合溶液总质量的70~80wt%,并向第三混合溶液中加入乳化剂和引发剂,聚合反应6~8.5h后,依次经旋蒸、抽滤、洗涤和干燥后,得到用于低阶煤成浆的面面吸附型分散剂。
优选地,s3中,亲水性接枝物为淀粉、丙烯酸或apeg;
当亲水性接枝物为淀粉时,中间体ii和淀粉的质量比为1:(1~5);
当亲水性接枝物为丙烯酸时,中间体ii和丙烯酸的质量比为(1.2~2):(5~10.8);
当亲水性接枝物为apeg时,中间体ii和apeg的投料比为(2.1~2.5)g:(0.005~0.015)mol;
apeg为apeg500、apeg700、apeg1000、apeg2000、apeg2400和apeg3000中的任意一种。
优选地,s1所述的kmno4的添加量为石墨质量的2~3倍。
优选地,s1所述的石墨通过废石墨预处理制备而成,具体为:将废石墨与浓hcl按照1:(5~8)的质量比混合,再依次进行水洗和干燥,得到石墨。
优选地,s1中,
低温反应的温度为0~5℃,反应时间为2~2.5h;
中温反应的温度为35~40℃,反应时间为应2~2.5h;
高温反应的温度为85~90℃,反应时间为1~1.5h;
s1所述的高温反应中添加有蒸馏水;
高温反应开始时蒸馏水的添加量为中温反应后的体系总质量的60~70wt%;
高温反应结束后蒸馏水的添加量为高温反应后的体系总质量的50~60wt%。
优选地,s3中,
乳化剂的添加量为中间体ⅱ和亲水性接枝物总质量的0.2~0.4wt%;
引发剂的添加量为中间体ⅱ和亲水性接枝物总质量的0.2~0.4wt%;
乳化剂为十二烷基硫酸钠或op-10;
引发剂为过硫酸铵、过硫酸钾或过硫酸钠。
优选地,s1所述的洗涤是依次采用15%的稀hcl及蒸馏水将溶液洗至中性且溶液中无so42-。
优选地,s1和s2中所述的冷冻干燥的温度均为-15~-5℃;s3所述的干燥为70~80℃的真空干燥;s3所述的聚合反应的温度为50~85℃,聚合反应时间为6~8.5h;
s2中第二产物的制备过程具体为:首先将中间体i、socl2和dmf混合后超声处理1~1.5h,之后在温度为70~75℃的氮气气氛中酰氯化反应24~26h,冷却至室温后再加入与socl2相同体积的dmf,旋蒸法除去溶剂,得到第二产物。
与现有技术相比,本发明具有以下有益效果:
本发明提供了一种用于低阶煤成浆的二元面面吸附型分散剂,该分散剂具有与不粘煤、次烟煤和褐煤等低阶煤表面结构相似的中心疏水“锚固”基团,因此可以与煤表面通过π-π作用、氢键和疏水作用实现面面吸附,吸附作用力及吸附面积均较大。分散剂结构中引入了亲水性接枝物构成亲水链,该亲水链可以伸入水中,因此该分散剂可以有效改善不粘煤、次烟煤和褐煤与水的界面结构,降低界面极性相互作用能,增强不粘煤、次烟煤和褐煤颗粒之间的静电斥力和空间位阻,抑制不粘煤、次烟煤和褐煤的聚集及水分吸附,从而提高不粘煤、次烟煤和褐煤的成浆性能。
本发明公开了一种用于低阶煤成浆的二元面面吸附型分散剂的制备方法,首先对石墨进行氧化超声处理得到氧化石墨,接着对氧化石墨边缘的羧基依次进行酰氯化和酯化,再用乳液聚合法接枝亲水链,得到用于不粘煤、次烟煤和褐煤成浆的面面吸附型分散剂。该分散剂可以有效改善不粘煤、次烟煤和褐煤与水的界面结构,降低界面极性相互作用能,增强不粘煤颗粒之间、次烟煤颗粒之间和褐煤颗粒之间的静电斥力和空间位阻,抑制不粘煤、次烟煤和褐煤聚集及水分吸附,从而提高不粘煤、次烟煤和褐煤的成浆性能。
进一步地,该分散剂以废石墨作为原料,经过纯化及改进的hummers法对其进行氧化处理获得纳米级片状的氧化石墨,从而能够得到具有中心疏水边缘亲水结构的氧化石墨纳米片。
进一步地,s1中依次采用低温反应、中温反应和高温反应,从而有利于实现在石墨碳层结构中引入羧基、羟基及环氧基。
进一步地,s1所述的洗涤是依次采用15%的稀hcl及蒸馏水将溶液洗至中性且无so42-,是为了除去过量的h2so4和kmno4。
进一步地,s2中第二产物的制备过程酰氯化反应24h,是为了活化第一产物分子结构边缘的羧基,为酯化反应提供条件。
进一步地,s1和s2中均采用冷冻干燥,是为了防止由于聚集而影响s3中的乳液聚合过程。
附图说明
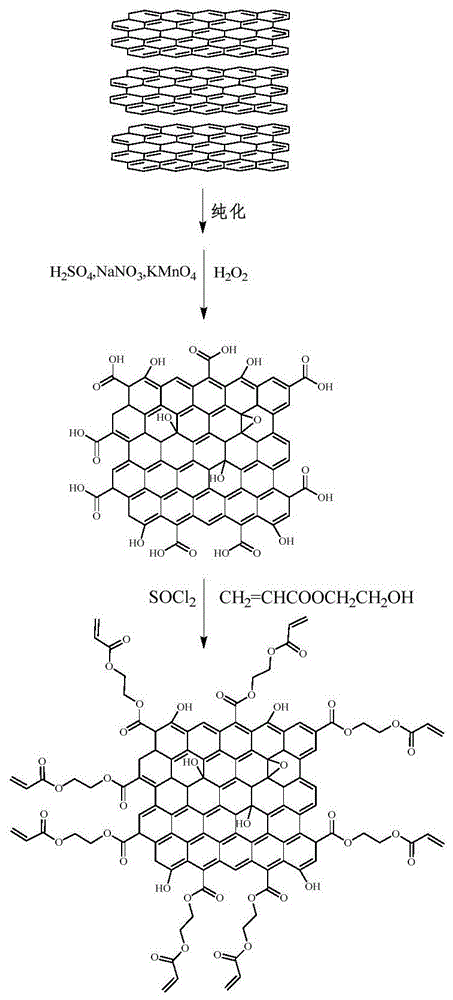
图1为本发明中中间体ii的合成路线图;
图2为本发明制备的用于低阶煤成浆的二元面面吸附型分散剂的分子结构图;其中,(a)为亲水性接枝物为淀粉时的分散剂的分子结构图;(b)为亲水性接枝物为丙烯酸时的分散剂的分子结构图;(c)为亲水性接枝物为apeg时的分散剂的分子结构图;
图3为本发明制备的用于低阶煤成浆的二元面面吸附型分散剂的红外光谱图;
图4为实施例3中亲水性接枝物为淀粉时制备的用于低阶煤成浆的二元面面吸附型分散剂与现有的两种分散剂对于不粘煤水煤浆的降粘效果图;
图5为实施例6中亲水性接枝物为丙烯酸时制备的用于低阶煤成浆的二元面面吸附型分散剂与现有的两种分散剂对于次烟煤水煤浆的降粘效果图;
图6为实施例10中亲水性接枝物为apeg时制备的用于低阶煤成浆的二元面面吸附型分散剂与现有的两种分散剂对于褐煤水煤浆的降粘效果图。
具体实施方式
下面结合附图对本发明做进一步详细描述:
实施例1
一种用于不粘煤成浆的二元面面吸附型分散剂的制备方法,包括如下步骤:
步骤1),将5g废石墨经25g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1:40加入到干燥的三口烧瓶中,在0℃条件下搅拌0.5h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨2倍质量的kmno4,待kmno4全部加入后在0℃搅拌下低温反应2h,随后升温至35℃进行中温反应,反应2h,最后往体系中缓慢加入质量为当前体系总质量60wt%的蒸馏水,在85℃下进行高温反应,反应1h。反应完成后向当前体系中加入质量为当前体系总质量50wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用质量分数为15%的稀hcl及蒸馏水充分洗涤,直至过滤后的滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:30,超声分散30min,之后-5℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和5mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌12h,静置12h。所得产物稀释、抽滤、洗涤、干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:20,超声处理30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与淀粉按质量比1:5的比例称取,将中间体ⅱ和淀粉溶解在水中,水的质量为体系总质量的70wt%,加入中间体ⅱ和淀粉总质量的0.32wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和淀粉总质量的0.4wt%的过硫酸铵引发剂,加入完毕后升温至50℃,反应7h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
该分散剂的结构式如图2(a)所示,其中,n为淀粉分子的聚合度,n的取值范围为50~2000。
实施例2用于不粘煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经40g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1:41加入到干燥的三口烧瓶中,在3℃下搅拌1h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨2.5倍质量的kmno4,待kmno4全部加入后在5℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量70wt%的蒸馏水,在90℃下进行高温反应,反应1h。反应完成后加入体系总质量60wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:32,超声30min,-15℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和2.52mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至75℃,保温反应26h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌10h,静置14h。所得产物稀释、抽滤、洗涤干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:21,超声处理30min,-15℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与淀粉按质量比1:3比例称取,将中间体ⅱ和淀粉溶解在水中,水的质量为体系总质量的68wt%,加入中间体ⅱ和淀粉总质量的0.35wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和淀粉总质量的0.37wt%的过硫酸铵引发剂,加入完毕后升温至60℃,反应6h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例3用于不粘煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经30g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.05:41加入到干燥的三口烧瓶中,在0℃下搅拌0.5h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨3倍质量的kmno4,待kmno4全部加入后在0℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2h,最后往体系中缓慢加入体系总质量65wt%的蒸馏水,在85℃下进行高温反应,反应1.5h。反应完成后加入体系总质量60wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:45,超声处理30min,-8℃下冷冻干燥得到中间体ⅰ。
步骤2),将1.0g中间体ⅰ、150mlsocl2和2mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入150mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌14h,放置10h。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:25,超声处理30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与淀粉按质量比1:4比例称取,将中间体ⅱ和淀粉溶解在水中,水的质量为体系总质量的72wt%,加入中间体ⅱ和淀粉总质量的0.2wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和淀粉总质量的0.3wt%的过硫酸铵引发剂,加入完毕后升温至70℃,反应8.5h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例4用于不粘煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经35g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.05:42加入到干燥的三口烧瓶中,在5℃下搅拌1h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨2.5倍质量的kmno4,待kmno4全部加入后在3℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量65wt%的蒸馏水,在88℃下进行高温反应,反应1.5h。反应完成后加入体系总质量55wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:50,超声30min,-12℃下冷冻干燥得到中间体ⅰ。
步骤2),将1g中间体ⅰ、100mlsocl2和4mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌12h,放置过夜。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:30,超声处理30min,-12℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与淀粉按质量比1:1比例称取,将中间体ⅱ和淀粉溶解在水中,水的质量为体系总质量的73wt%,加入中间体ⅱ和淀粉总质量的0.2wt%的op-10乳化剂,升温至35℃缓慢加入中间体ⅱ和淀粉总质量的0.4wt%的过硫酸铵引发剂,加入完毕后升温至65℃,反应8h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例5用于次烟煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经25g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.03:41.5加入到干燥的三口烧瓶中,在0℃条件下搅拌0.5h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨1.5倍质量的kmno4,待kmno4全部加入后在0℃搅拌下低温反应2h,随后升温至35℃进行中温反应,反应2h,最后往体系中缓慢加入质量为当前体系总质量70wt%的蒸馏水,在85℃下进行高温反应,反应1h。反应完成后向当前体系中加入质量为当前体系总质量52wt%的蒸馏水和h2o2(质量分数为30%),直到溶液为亮黄色,趁热过滤,采用质量分数为15%的稀hcl及蒸馏水充分洗涤,直至过滤后的滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:40,超声30min,-5℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和2.52mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.008mol三乙胺和0.008mol丙烯酸羟乙酯,室温下磁力搅拌12h,静置12h。所得产物稀释、抽滤、洗涤、干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:26,超声处理30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与丙烯酸按照质量比1.2:5的比例称取,将中间体ⅱ和丙烯酸溶解在蒸馏水中,蒸馏水的质量为体系总质量的76wt%,加入中间体ⅱ和丙烯酸总质量的0.25wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和丙烯酸总质量的0.38wt%的过硫酸铵引发剂,加入完毕后升温至78℃,反应6h后,采用0.1m的naoh溶液调节ph至8,旋蒸、抽滤、洗涤和83℃真空干燥处理后,得到用于次烟煤成浆的面面吸附型分散剂。
该分散剂具有以下结构式,如图2(b)所示:
其中,a为重复单元,a的取值范围为200~500。
实施例6用于次烟煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经40g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.02:40.8加入到干燥的三口烧瓶中,在冰水浴条件下搅拌1h。控制反应体系温度低于5℃下缓慢多次加入质量为预处理后的石墨1.8倍质量的kmno4,待kmno4全部加入后在0℃下搅拌低温反应2.5h,随后升温至35℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量70wt%的蒸馏水,在85℃下进行高温反应,反应1h。反应完成后加入体系总质量55wt%的蒸馏水和h2o2(质量分数为30%)直到溶液为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:35,超声30min,-5℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和2.52mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌10h,静置14h。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:28,超声处理30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与丙烯酸按质量比1.2:10.8比例称取,将中间体ⅱ和丙烯酸溶解在水中,水的质量为体系总质量的78wt%,加入中间体ⅱ和丙烯酸总质量的0.22wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和丙烯酸总质量的0.38wt%的过硫酸铵引发剂,加入完毕后升温至75℃,反应6.5h后,采用0.5m的naoh溶液调节ph至8,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例7用于次烟煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经30g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.07:40.7加入到干燥的三口烧瓶中,在冰水浴条件下搅拌0.5h。控制反应体系温度低于5℃下缓慢多次加入质量为预处理后的石墨1.6倍质量的kmno4,待kmno4全部加入后在0℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量69wt%的蒸馏水,在85℃下进行高温反应,反应1.5h。反应完成后加入体系总质量53wt%的蒸馏水和h2o2(质量分数为30%)直到溶液为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:38,超声30min,-5℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和2.52mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌14h,放置10h。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:21,超声30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与丙烯酸按质量比2:5.1比例称取,将中间体ⅱ和丙烯酸溶解在水中,水的质量为体系总质量的80wt%,加入中间体ⅱ和丙烯酸总质量的0.4wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和丙烯酸总质量的0.28wt%的过硫酸铵引发剂,加入完毕后升温至73℃,反应6.3h后,采用1m的naoh溶液调节ph至8.5,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例8用于次烟煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经35g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.1:42加入到干燥的三口烧瓶中,在冰水浴条件下搅拌1h。控制反应体系温度低于5℃下缓慢多次加入质量为预处理后的石墨1.7倍质量的kmno4,待kmno4全部加入后在3℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量68wt%的蒸馏水,在88℃下进行高温反应,反应1.5h。反应完成后加入体系总质量51wt%的蒸馏水和h2o2(质量分数为30%)直到溶液为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:50,超声30min,-5℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和2.52mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌12h,放置过夜。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:29,超声30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与丙烯酸按质量比2:10.8的比例称取,将中间体ⅱ和丙烯酸溶解在水中,水的质量为体系总质量的75wt%,加入中间体ⅱ和丙烯酸总质量的0.32wt%的op-10乳化剂,升温至35℃缓慢加入中间体ⅱ和丙烯酸总质量的0.38wt%的过硫酸铵引发剂,加入完毕后升温至70℃,反应8h后,采用0.3m的naoh溶液调节ph至8.5,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例9用于褐煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经25g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.1:40加入到干燥的三口烧瓶中,在0℃条件下搅拌0.5h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨2倍质量的kmno4,待kmno4全部加入后在0℃搅拌下低温反应2h,随后升温至35℃进行中温反应,反应2h,最后往体系中缓慢加入质量为当前体系总质量66wt%的蒸馏水,在85℃下进行高温反应,反应1h。反应完成后向当前体系中加入质量为当前体系总质量57wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用质量分数为15%的稀hcl及蒸馏水充分洗涤,直至过滤后的滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:30,超声30min,-3℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和5mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌12h,静置12h。所得产物稀释、抽滤、洗涤、干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:23,超声30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与apeg500按2.1g:0.005mol的比例称取,将中间体ⅱ和apeg500溶解在水中,水的质量为体系总质量的65wt%,加入中间体ⅱ和apeg500总质量的0.2wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和apeg500总质量的0.2wt%的过硫酸铵引发剂,加入完毕后升温至80℃,反应8.1h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
该分散剂具有以下结构式,如图2(c)所示:
其中a为聚合度,取值范围为1000~10000;m为(apeg)的重复单元,数值由其原料分子量决定(apeg500、apeg700、apeg1000、apeg2000、apeg2400、apeg3000)。
实施例10用于褐煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经40g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.05:42加入到干燥的三口烧瓶中,在3℃下搅拌1h。控制反应体系温度低于5℃下缓慢多次加入质量为预处理后的石墨2.5倍质量的kmno4,待kmno4全部加入后在5℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量68wt%的蒸馏水,在90℃下进行高温反应,反应1h。反应完成后加入体系总质量54wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:37,超声30min,-15℃下冷冻干燥得到中间体ⅰ。
步骤2),将0.5g中间体ⅰ、100mlsocl2和2.52mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至75℃,保温反应26h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌10h,静置14h。所得产物稀释、抽滤、洗涤干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:20,超声30min,-15℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与apeg700按2.5g:0.005mol的比例称取,将中间体ⅱ和apeg700溶解在水中,水的质量为体系总质量的69.5wt%,加入中间体ⅱ和apeg700总质量的0.3wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和apeg700总质量的0.2wt%的过硫酸铵引发剂,加入完毕后升温至85℃,反应8.1h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例11用于褐煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经30g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.05:41加入到干燥的三口烧瓶中,在0℃下搅拌0.5h。控制反应体系温度低于5℃下缓慢多次加入质量为预处理后的石墨3倍质量的kmno4,待kmno4全部加入后在0℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2h,最后往体系中缓慢加入体系总质量66wt%的蒸馏水,在85℃下进行高温反应,反应1.5h。反应完成后加入体系总质量60wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:48,超声30min,-8℃下冷冻干燥得到中间体ⅰ。
步骤2),将1.0g中间体ⅰ、150mlsocl2和2mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入150mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌14h,放置10h。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:22,超声30min,-10℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与apeg1000按2.5g:0.015mol的比例称取,将中间体ⅱ和apeg1000溶解在水中,水的质量为体系总质量的67wt%,加入中间体ⅱ和apeg1000总质量的0.3wt%的十二烷基硫酸钠乳化剂,升温至30℃缓慢加入中间体ⅱ和apeg1000总质量的0.3wt%的过硫酸铵引发剂,加入完毕后升温至83℃,反应8.5h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
实施例12用于褐煤成浆的二元面面吸附型分散剂的制备方法
步骤1),将5g废石墨经35g浓hcl酸化、水洗、干燥后,取预处理后的石墨、nano3及浓h2so4质量比1:1.05:40加入到干燥的三口烧瓶中,在5℃下搅拌1h。控制反应体系温度低于5℃下缓慢多次加入质量为石墨2.5倍质量的kmno4,待kmno4全部加入后在3℃下搅拌低温反应2.5h,随后升温至40℃进行中温反应,反应2.5h,最后往体系中缓慢加入体系总质量67wt%的蒸馏水,在88℃下进行高温反应,反应1.5h。反应完成后加入体系总质量56wt%的蒸馏水和h2o2(质量分数为30%),直至溶液颜色变为亮黄色,趁热过滤,采用15%稀hcl及蒸馏水充分洗涤产物直至滤液呈中性且无so42-(利用bacl2检测至无沉淀),干燥,得到第一产物,然后将第一产物溶解于蒸馏水中,第一产物与蒸馏水的质量比为1:32,超声30min,-12℃下冷冻干燥得到中间体ⅰ。
步骤2),将1g中间体ⅰ、100mlsocl2和4mldmf加入到三口烧瓶中,超声处理1h,氮气保护下,升温至70℃,保温反应24h,冷却至室温加入100mldmf,采用旋蒸除去溶剂将产物转移至三口烧瓶中,加入0.808g(0.008mol)三乙胺和0.928g(0.008mol)丙烯酸羟乙酯,室温下磁力搅拌12h,放置过夜。所得产物稀释、抽滤、洗涤和干燥处理后,得到第三产物,然后将第三产物溶解于蒸馏水中,第三产物和蒸馏水的质量比为1:30,超声30min,-12℃下冷冻干燥后得到中间体ⅱ。
步骤3),中间体ⅱ与apeg2000按2.1g:0.015mol的比例称取,将中间体ⅱ和apeg2000溶解在水中,水的质量为体系总质量的70.5wt%,加入中间体ⅱ和apeg2000总质量的0.35wt%的op-10乳化剂,升温至35℃缓慢加入中间体ⅱ和apeg2000总质量的0.3wt%的过硫酸铵引发剂,加入完毕后升温至80℃,反应8.1h后,旋蒸、抽滤、洗涤处理后,得到该分散剂。
性能表征
以实施例10为例,对上述实施例10制备的分散剂进行红外光谱表征,结果如图3所示,由图3的结果可以看出,在2926cm-1和2871cm-1处出现了c–h键的伸缩振动吸收峰,1355cm-1和1470cm-1处吸收峰由c–h键的弯曲振动产生;1732cm-1处吸收峰由-c=o键的伸缩振动吸收产生;1101cm-1处吸收峰由c-o键伸缩振动产生。根据以上结果可以推断,apeg成功引入到中间体ⅱ上,二元面面吸附型分散剂成功合成。
为了表征一种用于低阶煤成浆的二元面面吸附型分散剂对于各种水煤浆的分散效果,利用实施例3、实施例6、实施例10所制备的分散剂与传统的木质素磺酸钠和萘磺酸甲醛缩合物进行比较,固定浆体浓度为60wt%,分散剂用量为0.3wt%,浆体表观粘度随剪切速率变化的结果如图4、图5和图6所示。
从图4、图5和图6中可以看出,采用木质素磺酸钠和萘磺酸甲醛缩合物作为对比,当采用实施例3、实施例6和实施例10所制备的分散剂时,不粘煤水煤浆浆体、次烟煤水煤浆浆体和褐煤水煤浆浆体的表观粘度随着剪切速率的增加先下降后趋于平缓,表现出假塑性流体特征。当剪切速率为100s-1时,实施例3制备的不粘煤水煤浆表观粘度下降至334mpa·s,明显低于木质素磺酸钠制备的不粘煤水煤浆。实施例6制备的次烟煤水煤浆的表观粘度下降至371mpa·s,明显低于木质素磺酸钠制备的次烟煤水煤浆。实施例10制备的褐煤水煤浆表观粘度下降至425mpa·s,明显低于木质素磺酸钠制备的褐煤水煤浆。萘磺酸甲醛缩合物制备的不粘煤水煤浆、次烟煤水煤浆和褐煤水煤浆由于稳定性太差,煤粒沉降速率过快,导致测定过程中容器底部出现硬沉淀,以致表观粘度变化无规律。
为了表征本发明的二元面面吸附型分散剂对于不粘煤水煤浆、次烟煤水煤浆和褐煤水煤浆的稳定效果,将上述对比例和上述实施例制备的水煤浆分别搅拌均匀后倒入在100ml量筒(直径3cm)中,然后将量筒口密封并在室温下放置3天,通过注射器吸出量筒内浆体的上层清液并称量,记为m1,上层清液的质量与浆体总质量(m0)比即为析水率。析水率的计算采用下述公式计算,析水率(wt%)=m1/m0×100%。
表1.对比例及实施例制备的分散剂的析水率对比
从表1的结果可以看出,本发明实施例所制备的分散剂对于不粘煤水煤浆、次烟煤水煤浆和褐煤水煤浆的析水率均远小于3.96wt%,明显低于其他两种分散剂(木质素磺酸钠或萘磺酸甲醛缩合物)制备的水煤浆,具有优异的稳定性。同时结合图4~图6可以看出,本发明制备的分散剂可以大幅提高不粘煤水煤浆的分散稳定性,具有较好的制浆能力。
本发明的用于低阶煤成浆的二元面面吸附分散剂的合成原理参见图1。具体为:利用废石墨的疏水碳层结构,首先利用改进的hummers法对预处理后的废石墨进行氧化,得到含有羧基、羟基和环氧基等含氧官能团的氧化石墨纳米片,随后对预处理后的石墨进行酰氯化反应从而活化边缘羧基,利用和丙烯酸羟乙酯的酯化反应获得双键化的石墨,最后将淀粉、丙烯酸或apeg亲水链利用乳液聚合接枝在石墨的边缘,从而构建具有与不粘煤、次烟煤和褐煤表面结构相似的“锚固”基团,从而得到用于低阶煤成浆的二元面面吸附的分散剂。
以上内容仅为说明本发明的技术思想,不能以此限定本发明的保护范围,凡是按照本发明提出的技术思想,在技术方案基础上所做的任何改动,均落入本发明权利要求书的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!