一种联二萘骨架的环戊二烯铑络合物的制备方法
1.本发明属于有机化学中的有机合成技术领域,涉及一种联二萘骨架的环戊二烯铑络合物的制备方法。
背景技术:
2.近十年来,rh(iii)催化的c-h键官能团化反应得到了飞速的发展。2013年,cramer课题组从手性萘酚出发,合成了轴手性环戊二烯配体及其铑络合物。这类配体可以在联萘骨架上3,3
’‑
位进行修饰,拓宽了其应用范围,目前已经被多个课题应用发展新的不对称c-h键官能团化反应。但是铑络合物合成路线相对复杂繁琐,且基本每步都需要柱层析分离(见参考文献j.am.chem.soc.2003,125,5139.j.am.chem.soc.2013,135,636.),很难实现放大反应。
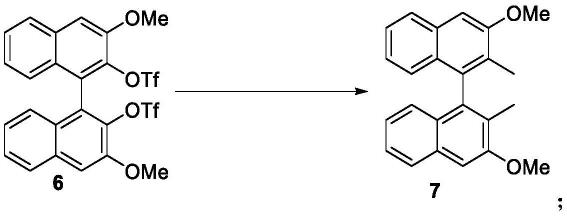
3.cramer课题组关于轴手性联萘骨架环戊二烯配体及其铑络合物的合成步骤如下:(1)以手性联二萘酚为原料,经过连续三步反应以88%的收率制备化合物3,产物需要柱层析分离纯化;(2)在碘甲烷作用下对羟基进行保护,紧接着在酸性条件下脱除mom保护,以87%的收率得到化合物5,产物需要柱层析分离纯化;(3)与三氟甲磺酸酐反应,以97%的收率制备化合物6,产物需要柱层析分离纯化;(4)在nicl2(dppp)催化下,和甲基溴化镁发生kumada反应,以88%的收率制备化合物7,产物需要柱层析分离纯化。(5)2,2
’‑
二甲基-3,3
’‑
二甲氧基化合物7发生自由基溴代,以76%的收率制备溴代化合物8,产物需要柱层析分离纯化;(6)溴代化合物8与环戊二烯基钠反应,以76%的收率制备配体9,配体9通过柱层析分离制得;(7)配体9与乙醇铊/乙烯基氯化铑二聚体反应,以87%收率得到rh-1。
4.
技术实现要素:
5.本发明所要解决的技术问题在于克服现有技术中合成轴手性环戊二烯配体时,全程须柱层析分离,且不能大量制备的缺陷;为此,本发明提供一种联二萘骨架的环戊二烯铑络合物的制备方法。
6.本发明提供一种化合物8的制备方法,其包括如下步骤:溶剂中,在光照下,在溴化试剂和引发剂的存在下化合物7经如下式所示的溴化反应得到所述化合物8;所述的溶剂为环烷烃类溶剂、氯代烷烃类溶剂或氯代芳烃类溶剂;
[0007][0008]
所述的化合物8的制备方法中,较佳地,所述的氯代烷烃类溶剂可为本领域此类反应常规的氯代烷烃类溶剂,例如四氯化碳。
[0009]
所述的化合物8的制备方法中,较佳地,所述的环烷烃类溶剂可为本领域此类反应常规的环烷烃类溶剂,例如环己烷。
[0010]
所述的化合物8的制备方法中,较佳地,所述的氯代芳烃类溶剂可为本领域此类反应常规的氯代芳烃类溶剂,例如氯苯。
[0011]
所述的化合物8的制备方法中,较佳地,所述的溴化试剂为本领域此类反应常规的溴化试剂,例如nbs(n-溴代琥珀酰亚胺)。
[0012]
所述的化合物8的制备方法中,较佳地,所述的引发剂为本领域此类反应常规的引发剂,例如偶氮二异丁腈。
[0013]
所述的化合物8的制备方法中,较佳地,所述的光照的光源为普通白炽灯。
[0014]
所述的化合物8的制备方法中,较佳地,所述的光照需要的时间与反应规模相关,例如大于等于20分钟。
[0015]
所述的化合物8的制备方法中,较佳地,所述的化合物7与所述的溴化试剂的摩尔比可为1:2.2。
[0016]
所述的化合物8的制备方法中,较佳地,所述的化合物7与所述的引发剂的摩尔比可为1:10。
[0017]
所述的化合物8的制备方法中,较佳地,所述的化合物7与所述的环烷烃类溶剂的摩尔体积比可为2mmol/ml-5mmol/ml;例如3.4mmol/ml、2mmol/ml或5mmol/ml。
[0018]
所述的化合物8的制备方法中,较佳地,所述的溴化反应温度为本领域此类反应常规反应温度,所述的溴化反应温度为75-135℃,例如85℃。
[0019]
所述的化合物8的制备方法中,较佳地,所述的溴化反应的时间与反应规模相关,例如8h。
[0020]
所述的化合物8的制备方法中,较佳地,所述的化合物8为
[0021]
所述的化合物8的制备方法中,较佳地,所述的化合物7为
[0022]
本发明一些实施方案中,所述的化合物8的制备方法包括;溶剂中,在光照下,在nbs和偶氮二异丁腈的存在下,所述的化合物7经所述的溴化反应得到所述化合物8;所述的溶剂为四氯化碳、氯苯或环己烷。
[0023]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:化合物6与甲基化试剂进行如下式所示的甲基化反应得到化合物7;
[0024][0025]
本发明一些实施方案中,所述的甲基化反应的反应条件为本领域常规的甲基化的反应条件。
[0026]
本发明一些实施方案中,较佳地,所述的甲基化试剂为memgi。
[0027]
所述的化合物8的制备方法中,较佳地,所述的化合物6为
[0028]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:在乙醚中,在ni(dppp)2cl2存在下,所述的化合物6与memgi进行所述的甲基化反应得到所述的化合物7。
[0029]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:化合物5在碱存在下与三氟甲磺酸酐进行如下式所示的酚羟基酯化反应得到化合物6;
[0030][0031]
本发明一些实施方案中,所述的酚羟基酯化反应的反应条件为本领域常规的酚羟基酯化反应的反应条件。
[0032]
所述的化合物8的制备方法中,较佳地,所述的化合物5为
[0033]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:溶剂中,化合物4在酸存在下进行如下所示的脱保护反应a得到化合物5;
[0034][0035]
所述的脱保护反应a中,所述的脱保护反应a的反应条件为本领域常规的脱保护反应的反应条件。
[0036]
所述的脱保护反应a中,较佳地,所述的溶剂为醇类溶剂,例如甲醇。
[0037]
所述的化合物8的制备方法中,较佳地,所述的化合物4为
[0038]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:甲醇中,所述的化合物4在盐酸的存在下经所述的脱保护反应a;将所述的脱保护反应a的产物溶于二氯甲烷中,在吡啶存在下与三氟甲磺酸酐经所述的酚羟基酯化反应得到所述的化合物6。
[0039]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:溶剂中,化合物3在碱的存在下进行如下所示的脱保护反应b得到化合物4;
[0040][0041]
本发明一些实施方案中,所述的脱保护反应b的反应条件为本领域常规的此类脱保护反应的反应条件。
[0042]
所述的化合物8的制备方法中,较佳地,所述的化合物3为
[0043]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:溶剂中,化合物2在碱的存在下进行如下所示的羟基化反应得到化合物3;
[0044][0045]
所述的化合物8的制备方法中,较佳地,所述的化合物2为
[0046]
本发明一些实施方案中,所述的羟基化反应的反应条件为本领域常规的此类羟基化反应的反应条件。
[0047]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:将所述的化合物2溶于四氢呋喃中,依次滴加入正丁基锂和硼酸三甲酯,除去四氢呋喃,加入氯仿后滴加h2o2进行所述的羟基化反应;将所述的羟基化反应的产物溶于丙酮,依次加入k2co3和mei后进行所述的脱保护反应b得到化合物4。
[0048]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:溶剂中,化合物1和mom试剂在碱的存在下进行如下所示的保护反应得到化合物2;
[0049][0050]
本发明一些实施方案中,所述的保护反应的反应条件为本领域常规的此类保护反应的反应条件。
[0051]
本发明一些实施方案中,较佳地,所述的mom试剂为溴甲基甲基醚。
[0052]
所述的化合物8的制备方法中,较佳地,所述的化合物1为
[0053]
本发明一些实施方案中,所述的化合物8的制备方法还包括如下步骤:将所述的化合物1溶于四氢呋喃中,依次添加nah和溴甲基甲基醚,经所述的保护反应得到所述的化合物2。
[0054]
本发明提供一种化合物9的制备方法,其包括如下步骤:
[0055]
(1)按照如前述所述的化合物8的制备方法制备得到化合物8;
[0056]
(2)步骤(1)制备得到的所述的化合物8在溶剂和碱存在下与cpna(环戊二烯基钠)进行偶联反应得到所述化合物9
[0057]
本发明一些实施方案中,所述的偶联反应的反应条件为本领域常规的此类偶联反应的反应条件。
[0058]
本发明一些实施方案中,较佳地,所述的化合物9为
[0059]
本发明一些实施方案中,所述的化合物9的制备方法还包括如下步骤:向四氢呋喃和nah的溶液中滴加cpna后,再加入步骤(1)制备得到的所述的化合物8,经所述的偶联反应得到所述的化合物9。
[0060]
本发明提供一种rh-1铑络合物的制备方法,其包括如下步骤:
[0061]
(i)按照如前述所述的化合物9的制备方法制备得到化合物9;
[0062]
(ii)步骤(i)制备得到的化合物9在溶剂和tloet存在下与[rh(c2h4)2cl]2进行如下所示的配位反应得到如式rh-1所示的铑络合物;所述rh-1铑络合物为
[0063]
本发明一些实施方案中,较佳地,所述的rh-1铑络合物为
[0064]
本发明一些实施方案中,所述的rh-1铑络合物的制备方法还包括如下步骤:将步骤(i)制备得到所述的化合物9在脱气苯中溶解,避光加入tloet,在80℃反应后,室温下加入[rh(c2h4)2cl]2,经所述的配位反应得到所述的rh-1铑络合物。
[0065]
本发明一些实施方案中,所述的配位反应的反应条件为本领域常规的此类配位的反应条件。
[0066]
本发明中,“室温”是指10℃-40℃。
[0067]
本发明中,“h”是指小时。
[0068]
本发明中,各个反应的反应温度为外温加热温度,如外温温度超过溶剂沸点,实际反应温度为溶剂沸点的温度。
[0069]
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0070]
本发明所用试剂和原料均市售可得。
[0071]
本发明的积极进步效果在于:
[0072]
本发明提供了一种化合物8的制备方法,本发明的化合物8的制备方法可以以较高产率制备得到化合物8,且本发明制备方法可适用于简单的后处理步骤,制备得到的化合物8无需通过柱层析分离就能用于制备铑络合物。
具体实施方式
[0073]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0074]
本发明实施例仅以一个构型为例,说明各个化合物的制备过程。
[0075]
实施例1
[0076][0077]
在一个干燥的1l反应瓶中加入(s)-联二萘酚1(57.3g,0.2mol),加入350ml的thf溶解,冰浴下向反应液中缓慢加入nah(32.0g,0.8mol,4.0equiv)。待加入完毕,将反应液回温至室温。反应5小时后,在冰浴下,向该反应液中缓慢加入另一原料溴甲基甲基醚(65.3ml,800mmol,4.0equiv),室温反应12小时后,冰浴下加入饱和nahco3水溶液淬灭反应,然后用水和乙酸乙酯萃取反应液,有机相用无水na2so4干燥,过滤,然后减压浓缩干燥,
得产物化合物2(74.9g,定量收率。)
[0078]1h nmr(400mhz,cdcl3)δ7.98(d,j=9.2hz,2h),7.90(d,j=8.0hz,2h),7.60(d,j=9.2hz,2h),7.34(t,j=8.0hz,2h),7.25
–
7.20(m,2h),7.15(d,j=8.4hz,2h),5.09(d,j=6.8hz,2h),4.97(d,j=6.8hz,2h),3.14(s,6h).
13
cnmr(101mhz,cdcl3):δ152.8,134.2,130.1,129.6,128.1,126.4,125.7,124.3,121.5,117.5,95.4,56.0.
[0079]
实施例2
[0080]
在一个干燥的1l反应瓶中加入nah(32g,0.8mol,4.0equiv),氩气置换三次,氩气保护,冰浴下加入thf(350ml)溶解,氩气保护,冰浴下,缓慢加入(s)-联二萘酚(57.3g,0.2mol,1.0equiv),加入完毕后,恢复至室温搅拌0.5小时。再加入少量nah(约0.2g即可)如有气泡产生,则室温下继续搅拌,若无气泡产生,则缓慢加入溴甲基甲基醚(49.0ml,0.6mol,3.0equiv),反应12小时。其他操作同实施例1,得产物化合物2,乳白色固体,定量收率。其结构鉴定数据同实施例1。
[0081]
实施例3
[0082][0083]
干燥的1l反应瓶中加入将实施例1制备得到的化合物2(87.9g,0.2mol),氩气置换三次,氩气下加入300ml thf,使用机械搅拌,搅拌溶解,降温至-78℃,向反应液中滴加nbuli(192ml,0.48mmol,2.4equiv)。滴加完成后稳定十分钟,将反应液升至0℃反应6小时。将反应再次降至-78℃,滴加入硼酸三甲酯(67ml,0.6mol,3.0equiv),滴加完成后稳定10分钟,将反应液恢复至室温反应12小时。减压除去反应液中溶剂thf,加入氯仿(300ml),冰浴冷却下滴加h2o2(60ml,0.6mol,3.0equiv),用时约10分钟。滴加加毕后,将该反应升温至70℃反应10小时。待反应完成后,在冰浴下滴加饱和na2so3水溶液淬灭反应。然后用二氯甲烷萃取反应液,有机相用饱和食盐水洗涤,无水na2so4干燥,过滤,减压浓缩,得黄褐色油状液体3。在1l反应瓶中,用300ml的丙酮溶解上述黄褐色油状物,依次加入k2co3(82.9g,0.6mol,3.0equiv),加入mei(37.4ml,0.6mol,3.0equiv),将反应升至65℃反应约10h。待反应完成后,冷至室温,向反应液中加入水淬灭反应,然后用乙酸乙酯萃取,饱和食盐水洗涤,有机相用无水na2so4干燥,过滤,减压浓缩。得到的固体用200ml(石油醚/乙酸乙酯=10:1)溶液打浆,过滤,得白色固体粉末,其余大颗粒固体继续用100ml(石油醚/乙酸乙酯=10:1)溶液打浆,过滤,重复多次,分离得产物化合物4,白色固体62.6g,从化合物2到化合物4收率为72%。
[0084]1h nmr(400mhz,cdcl3)δ7.76(d,j=8.0hz,2h),7.35(t,j=12.0hz,2h),7.29(s,2h),7.14(dd,j=4.0hz,8.0hz,2h),4.95(d,j=8.0hz,2h),4.83(d,j=8.0hz,2h),4.02(s,6h),2.57(s,6h).
13
c nmr(100mhz,cdcl3)δ147.1,143.6,129.0,128.8,126.8,124.6,124.5,124.0,114.4,106.2,56.0.
[0085]
实施例4
[0086][0087]
将实施例3制备得到的化合物4(62.6g,144mmol),加入720ml甲醇,放入70℃加热搅拌溶解,向反应液中滴加浓hcl(12ml,144mmol,1.0equiv),滴加完毕后反应约0.5小时。待反应完成后,冷却至室温,旋干溶剂,并用二氯甲烷萃取,有机相用无水na2so4干燥,过滤,减压浓缩,得到黄褐色油状物。将上述油状物溶于300ml二氯甲烷中,向反应液中加入吡啶(46.4ml,0.57mol,4.0equiv)。在-78℃下稳定5分钟,向反应液中加入三氟甲磺酸酐(53.3ml,316.8mmol,2.2equiv),滴加完毕后,反应30分钟,然后将该反应升至室温,反应12小时。待反应完成后,向反应液中加入1.0n hcl溶液调节ph至酸性,并用二氯甲烷萃取,有机相用饱和nacl水溶液洗涤,无水na2so4干燥,过滤,减压浓缩,在5cm硅胶短柱上用(石油醚:乙酸乙酯=10:1)淋洗,所得溶液浓缩得化合物6,淡白色固体51.6g,从化合物4到化合物6收率为59%。
[0088]1h nmr(300mhz,cdcl3)δ7.87(2h,d,j=8.4hz),7.52(2h,ddd,j=8.4,6.9,1.2hz),7.49(2h,s),7.24(2h,ddd,j=7.8,6.9,1.2hz),7.14(2h,d,j=7.8hz),4.12(6h,s).
[0089]
实施例5
[0090][0091]
在干燥1l反应瓶中,加入实施例4制备得到的化合物6(48.3g,79mmol),ni(dppp)2cl2(2.15g,3.96mmol,5mol%)置换氩气3次,加入500ml乙醚,搅拌溶解。冰浴下,向反应液中加入memgi(3m,132ml,5.0equiv),每50ml约5min)。滴加完毕后,将反应液升温至40℃反应12h。待反应完成后,冰浴下缓慢加入1.0n hcl溶液淬灭,并用乙酸乙酯萃取,有机相用饱和nacl水溶液洗涤,无水na2so4干燥,过滤,活性炭脱色,然后减压浓缩至干得化合物7,白色固体23.1g,收率85%。
[0092]1h nmr(300mhz,cdcl3)δ7.87(d,j=8.4hz,2h),7.52(ddd,j=8.4,6.9,1.2hz,2h),7.49(s,2h),7.24(ddd,j=7.8,6.9,1.2hz,2h),7.14(d,j=7.8hz,2h),4.12(s,6h).
13
c nmr(100mhz,cdcl3)δ149.1,137.6,132.9,127.7,127.6,126.8,126.6,125.4,124.6,118.0(q,j
c-f
=321hz),109.4,56.4.
[0093]
实施例6
[0094][0095]
在干燥250ml反应瓶中依次加入实施例5制备得到的7(15.2g,44.4mmol),重结晶的偶氮二异丁腈(729.1mg,4.4mmol,0.1equiv),n-溴代琥珀酰亚胺(17.4g,97.7mmol,2.2equiv)置换氩气3次,加入150ml环己烷,升温至85℃反应,同时使用白炽灯光照(边升温,边光照)至少20分钟,反应8h。待反应完成后,冷却至室温加水淬灭,并用乙酸乙酯萃取反应液,有机相经饱和nacl水溶液洗涤,无水na2so4干燥,过滤,减压浓缩至干,再加入10ml乙酸乙酯,体系中有大量固体析出,过滤得化合物8,白色固体22.0g,产率99.0%。(纯度大于98%)。
[0096]1h-nmr(400mhz,cdcl3):δ7.82(d,j=8.3hz,2h),7.44(ddd,j=8.1,6.8,1.3hz,2h),7.34(s,2h),7.11(ddd,j=8.2,6.8,1.3hz,2h),7.03-6.96(m,2h),4.38-4.26(m,4h),4.12(s,6h).
13
cnmr(100mhz,cdcl3)δ155.5,136.4,134.6,127.7,127.3,127.2,126.7,126.6,124.2,106.4,55.8,27.8.
[0097]
实施例7
[0098]
加入88.8ml环己烷,其他条件同实施例6。获得化合物8 21.0g,产率95.0%,纯度大于98%。
[0099]
实施例8
[0100]
加入222ml环己烷,其他条件同实施例6。获得化合物8 21.1g,产率95.0%,纯度大于98%。
[0101]
实施例9,使用150ml四氯化碳作为反应溶剂,其他条件同实施例6。获得化合物8 21.3g,产率96.0%,纯度大于98%。
[0102]
实施例10,使用150ml氯苯作为反应溶剂,其他条件同实施例6。获得化合物8 21.1g,产率95.0%,纯度大于98%。
[0103]
实施例11
[0104][0105]
在干燥500ml反应瓶中加入nah(1.4g,35.4mmol,1.5equiv),置换氩气3次,-20℃下,加入180ml thf溶解,向反应液中滴加cpna(2m,17.7ml,1.5equiv),-20℃下稳定20分钟,再向反应液中加入实施例6制得到的备8(11.8g,23.6mmol,1.0equiv)的thf(100ml)溶液,随后加入15-crown-5(9.4ml,47.3mmol,2.0equiv),-20℃下稳定20分钟,将反应液恢复至室温搅拌2小时。待反应完成后,冰浴下滴加水淬灭,并用乙醚萃取反应液,有机相经饱和
nacl水溶液洗涤,无水na2so4干燥,过滤,活性炭脱色,减压浓缩,加入18ml乙酸乙酯,析出固体,过滤;所得滤液浓缩,加入9ml乙酸乙酯,析出固体,过滤,反复多次,共得淡黄色化合物9(5.4g,产率56.6%)。
[0106]1h-nmr(400mhz,c6d6):δ(ppm)=7.78-7.71(m,1.92h),7.52(d,j=8.4hz,0.77h),7.46(dd,j=8.4,1.2hz,1.08h),7.33 7.24(m,1.95h),7.08(s,0.4h),7.06(s,1.46h),7.02-6.94(m,2h),6.52(dt,j=4.8,1.2hz,0.4h),6.18-6.12(m,1.5h),4.38(d,j=13.9hz,1.09h),4.20(d,j=14.3hz,0.41h),4.06(d,j=14.4hz,0.41h),3.44(s,3.16h),3.41(s,1.2h),3.37(s,1.14h),3.29 3.22(m,1.14h),3.15-2.98(m,1.28h),2.82(d,j=22.0hz,0.4h),2.68-2.62(m,1.11h).
13
c-nmr(100mhz,cdcl3)δ156.7,156.0,155.9,144.5,139.9,138.5,138.3,138.1,137.7,137.4,134.2,133.8,133.8,130.0,129.2,129.0,128.6,128.5,128.4,128.3,128.1,127.6,127.4,127.2,127.2,126.2,126.1,126.1,124.3,124.3,106.1,106.0,105.8,55.1,55.1,55.0,46.8,39.0,29.2,26.9,26.7.
[0107]
实施例12
[0108][0109]
干燥250ml封管中加入实施例11制备得到的9(2.02g,5.0mmol),置换氩气3次,加入60ml脱气苯溶解,冻抽三次,避光加入tloet(乙醇铊)(1.5g,6.0mmol,1.2equiv)。将反应液封上锡纸放入80℃反应24小时。随后恢复至室温,氩气保护下,加入[rh(c2h4)2cl]2(1.17g,3.0mmol,0.6equiv),室温下搅拌24小时。待反应完毕后,用苯淋洗真空过滤,滤液抽干得到产品淡黄色化合物rh-1(2.74g,收率97%)。
[0110]1h-nmr(400mhz,c6d6):δ7.74(dd,j=19.6,8.1hz,2h),7.37(dd,j=12.5,8.5hz,2h),7.30-7.24(m,2h),7.19(s,1h),7.02(s,1h),6.98-6.90(m,2h),5.42(t,j=2.4hz,1h),4.68(t,j=2.7hz,1h),4.37-4.33(m,1h),4.25(d,j=14.3hz,1h),3.67(d,j=14.8hz,1h),3.60(s,3h),3.41(s,3h),3.04(d,j=14.4hz,1h),2.83-2.70(m,2h),2.50(d,j=13.5hz,1h),2.18-2.06(m,2h),1.31-1.20(m,2h),0.98-0.88(m,2h).
13
c-nmr(150mhz,c6d6)δ(ppm)=156.70,155.84,137.82,137.12,133.95,133.91,129.33,129.16,128.59,128.35,127.47,127.33,127.21,127.14,126.27,126.23,124.39,124.29,106.03(d,j
rh-c
=3.8hz),106.01,105.52,98.30(d,j
rh-c
=3.5hz),89.95(d,j
rh-c
=3.9hz),88.93(d,j
rh-c
=3.9hz),84.31(d,j
rh-c
=4.1hz),55.11,55.08,40.43(d,j
rh-c
=13.7hz),36.65(d,j
rh-c
=13.3hz),26.09,24.04.
[0111]
真空过滤:真空过滤装置,下层中性氧化铝(约20cm),上层硅藻土(约2cm),放入烘箱干燥2h,搭好装置,下口接抽气上口接充气(直接与双排相连);关闭上口,打开下口抽冷;真空过滤时先关抽气,再打开充气,充气一段时间后再加需要过滤的液体,再关闭充气,开启抽气,重复上述操作。
[0112]
硅藻土过滤:从下到上依次为砂芯漏斗,两层滤纸,硅藻土约2cm,石英砂约0.5cm。
[0113]
对比例1
[0114]
在干燥250ml反应瓶中依次加入化合物7(15.2g,44.4mmol),重结晶的偶氮二异丁腈(729.1mg,4.4mmol,0.1equiv),n-溴代琥珀酰亚胺(17.4g,97.7mmol,2.2equiv)置换氩气3次,加入150ml苯,升温至80℃反应,反应8h。待反应完成后,冷却至室温加水淬灭,并用乙酸乙酯萃取反应液,有机相经饱和nacl水溶液洗涤,无水na2so4干燥,过滤,减压浓缩至干,再加入10ml乙酸乙酯,体系中有大量固体析出,过滤得化合物8,白色固体,20.0g;产率90%;产品纯度>98%。
[0115]
对比例2
[0116]
使用实施例7的条件在不加光源条件反应,反应产生单/双取代均存在的混合体系,反应产率为60%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1