苯并异噻唑类缺氧诱导因子2激动剂类化合物或其药学上可接受的盐、制备方法及应用
本发明涉及药物化学领域,尤其涉及一种苯并异噻唑类缺氧诱导因子2激动剂类化合物或其药学上可接受的盐,还涉及所述化合物的制备方法和应用。
背景技术:
贫血是一种常见疾病,其表现为血液中的红细胞水平不足以供给体内的氧气运输。成年人大约需要2~3×1013个红细胞以保证氧气输送,但红细胞寿命仅有100~120天。红细胞生成过程主要在骨髓中进行,血红蛋白是红细胞的主要组成成分,其由珠蛋白和亚铁血红素组成。在红细胞生成过程中,促红细胞生成素(erythropoietin,epo)是红细胞生成中最关键的激素,同时亚铁离子则是亚铁血红素的重要原料,两者的水平对于红细胞的生成都至关重要。
缺氧诱导因子(hypoxiainduciblefactor,hif)是一类响应缺氧的转录因子,其主要有3种亚型分别为hif-1、hif-2和hif-3。hif是一类异源二聚蛋白,其由氧敏感的缺氧诱导因子α(hypoxiainduciblefactorα,hif-α)与稳定表达的芳香烃受体核转位因子(arylhydrocarbonreceptornucleartranslocator,arnt)两个亚基组成。在常氧条件下,氧敏感的hif-α亚基中特定的脯氨酸残基(hif-1α:pro402和pro564;hif-2α:pro405和pro531;hif-3α:pro492)会被脯氨酰羟化酶(prolinehydroxylase,phd)羟基化。羟基化的hif-α被希佩尔-林道蛋白(vonhippel-lindau,vhl)识别,vhl招募e3泛素连接酶复合物使hif-α通过蛋白酶体途径降解。在缺氧环境中,由于phd的羟基化修饰过程需要氧气参与,氧气浓度的降低将导致phd对hif-α的羟基化修饰被抑制。vhl难以识别未羟基化的hif-α,这导致细胞中的hif-α含量上升,其与arnt二聚形成hif蛋白进入细胞核激活转录功能从而缓解缺氧。相比hif-1α、hif-3α,hif-2α由于其在肾脏细胞中的特异性表达,是一个理想的相关疾病治疗靶标。当机体缺氧时,hif-2转录激活其下游基因,诱发多种生理进程(如促进红细胞生成、加强氧代谢、促进新生血管生成等)促使机体适应缺氧。
成年人的epo生成主要在肾脏进行,其受到hif-2的直接调控(nat.rev.nephrol.11,394-410(2015))。提高hif-2转录活性可直接提升肾脏细胞中epo的合成和分泌。此外,hif-2同样能够调控十二指肠细胞色素b还原酶(dcytb)、二价金属转运蛋白1(dmt1)、膜铁转运蛋白(fpn1)等蛋白促进人体对食物中铁的摄取及铁在体内的转运,从而提高体内亚铁离子含量(j.biol.chem.286,4090-4097(2011))。
目前可提升hif-2活性的小分子最为人熟知的为phd抑制剂,其可抑制phd羟基化修饰活性以阻止hif-α降解,最终提升细胞中hif含量(j.med.chem.63,10045-10060(2020))。近年来已有5个phd抑制剂被批准上市治疗肾性贫血,分别为罗沙司他(roxadustat,fg-4592)、伐度司他(vadadustat,akb-6548)、达度司他(daprodustat,gsk1278863)、恩那司他(enarodustat,jtz-951)、莫立司他(molidustat,bay85-3934)。但伐度司他在后期临床试验中展现出心血管毒性。phd抑制剂在稳定hif-α时选择性差,其可同时提升三种hif亚型的含量,不利于对hif-2的特异性激活。
近年来发现了两个hif-2α直接激动剂(nat.chem.biol.15,367–376(2019)),分别是
技术实现要素:
发明目的:本发明针对现有hif-2α激动剂在激动hif-2时活性弱的问题,提供一种苯并异噻唑类缺氧诱导因子2激动剂类化合物或其药学上可接受的盐;还提供了上述化合物的制备方法及应用。
技术方案:本发明公开了一种苯并异噻唑类缺氧诱导因子2激动剂类化合物或其药学上可接受的盐,其通式如式i:
其中x代表-s-或
l代表-nh-或
r1和r2分别各自独立代表氢、卤素基、c1-c4卤素取代的烷基;
优选的,当l为
优选的,当l为-nh-时,ar代表
优选的,所述的盐包括可药用金属盐如钠、钾、锂、钙、镁、铝和锌盐;可药用金属阳离子如钠、钾、锂、钙、镁、铝和锌的碳酸盐和碳酸氢盐;可药用有机伯胺、仲胺和叔胺,包括脂肪胺、芳香胺、脂肪二胺和羟基烷基胺,如甲胺、乙胺、2-羟基乙基胺、二乙胺、三乙胺、乙二胺、乙醇胺、二乙醇。
本发明还公开了上述化合物的制备方法,
当通式(i)中l为-nh-,x为s时,化合物的制备方法包括以下步骤:
其中r1、r2、ar定义同前。
化合物2与芳胺发生取代反应得到化合物3。反应温度优选为20~60℃,反应溶剂可选丙酮、乙腈、n,n-二甲基甲酰胺、四氢呋喃、甲苯等。反应中还应加入碱,如氢氧化钠、氢氧化钾、氢化钠、氢化钾等。
化合物3经过硫单质参与的关环反应得到通式i化合物。反应温度优选为120~200℃,反应溶剂优选为二甲基亚砜、甲苯、n-甲基吡咯烷酮、n,n-二甲基甲酰胺等的混合溶液,反应还应加入碱,如碳酸钾、碳酸钠、碳酸铯、磷酸钾、磷酸二氢钾等。
当通式(i)中l为
其中r1、r2、ar定义同前。
化合物4与取代羧酸缩合得到通式(i)化合物。反应温度为20~60℃。反应所用溶剂可选二氯甲烷、三氯甲烷、丙酮、乙腈、二氧六环等。反应中还应加入有机碱或无机碱,如碳酸钾、碳酸钠、碳酸铯、三乙胺、吡啶等。反应中还应加入缩合剂,如二环己基碳二亚胺(dcc)、二异丙基碳二亚胺(dic)、1-(3-二甲胺基丙基)-3-乙基碳二亚胺(edci)、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸盐(hatu)、苯并三氮唑-n,n,n',n'-四甲基脲六氟磷酸盐(hbtu)等。
当通式(i)中x为
其中r1、r2、ar、l定义同前。
化合物5经过氧化反应得到通式(i)化合物。反应温度优选为-78~-10℃,反应溶剂优选为二氯甲烷、三氯甲烷、四氢呋喃、硫酸等,反应还应加入氧化剂,如间氯过氧苯甲酸、硫酸、硝酸等。
本发明还公开了上述苯并异噻唑类缺氧诱导因子2激动剂类化合物或其药学上可接受的盐在制备治疗缺血性疾病药物中的用途。
进一步的,所述缺血性疾病包括缺血引起的贫血症、脑卒、中风和心肌缺血,本发明的化合物临床所用剂量为0.0lmg-1000mg/天。
进一步的,还包括与脯氨酰羟化酶抑制剂联合使用制备治疗缺血性疾病药物,所述脯氨酰羟化酶抑制剂包括罗沙司他(roxadustat,fg-4592)
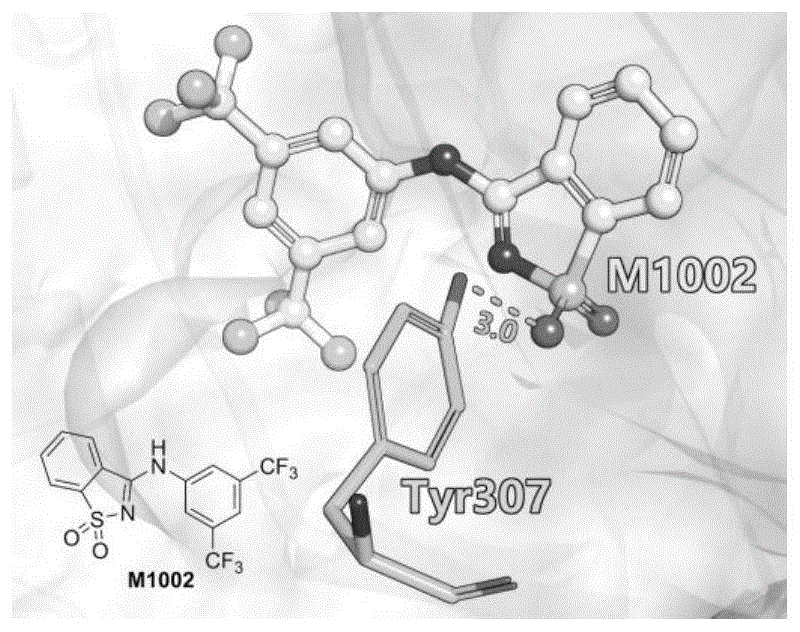
本发明设计合成了一类新型苯并异噻唑类hif-2α激动剂,如附图1所示,本发明通过分子对接方法分析了活性较优的hif-2α激动剂m1002与hif-2的结合模式,m1002的-so2n-结构中的氧原子可以作为氢键受体与hif-2α-pasb结构域中的tyr307形成氢键促进结合,这个氢键是m1002与蛋白的重要结合特征,但是仅有一侧的氧原子可与tyr307形成氢键提示,通过去除-so2n上的氧原子以降低小分子溶剂化能最终加强小分子与蛋白的结合,去除氧之后,异噻唑环上的硫与氮原子同样可与tyr307形成氢键;本发明去除苯并异噻唑环中-so2n-结构中的一个或者两个氧原子所带来的的特殊结构特征,能有效的激活hif-2信号通路,从而促进hif-2下游基因的转录激活。
本发明的苯并异噻唑类hif-2α激动剂或其药学上可接受的盐与phd抑制剂联合使用时,两者的使用剂量分别独立为0.01mg-1000mg/天,也可以根据病情轻重或剂型的不同偏离此范围;化合物与phd抑制剂可以分别给药、同时给药或制备成复合物同时给药。在体内外药效学实验中,该类化合物可与phd抑制剂联合使用在促进hif-2激活上起协同作用。phd抑制剂与hif-2α激动剂在提升hif-2转录活性中有着不同的机制:phd抑制剂通过抑制phd对hif-2α的羟基化修饰活性以阻止细胞中的hif-2α降解,最终提升hif-2复合物的含量以增强hif-2转录活性;hif-2α激动剂通过与hif-2α-pasb结构域结合以加强hif-2α与arnt两个亚基二聚化作用,最终提升hif-2水平以加强hif-2转录活性。将两者联用既可稳定hif-2α水平,又可提升hif-2α与arnt二聚能力,协同地提升hif-2复合物含量。
有益效果:与现有技术相比,本发明具有如下显著优点。
1、本发明所制备的苯并异噻唑类缺氧诱导因子2激动剂类化合物,在细胞水平具有良好的hif-2激动活性,代表化合物2和18分别可将hif-2转录活性提升至349%和359%,远优于现有报道最优的m1002的转录活性141%;
2、本发明将hif-2α激动剂与phd抑制剂联用以激活hif-2通路,相比单独使用phd抑制剂,将本发明的苯并异噻唑衍生物与phd抑制剂联用可更高效提升小鼠血液中epo水平,规避phd抑制剂亚型选择性的缺点;本发明化合物在肾性贫血、缺血性脑卒等缺血性疾病中有着一定的应用前景。
附图说明
图1:分子对接分析已有hif-2α激动剂m1002与hif-2的结合模式;
图2:荧光素酶报告基因实验测试化合物2与phd抑制剂对hif-2转录活性的协同作用;
图3:化合物2和phd抑制剂akb-6548联合给药对小鼠血浆epo的影响。
具体实施方式
下面结合附图对本发明的技术方案作进一步说明。
实施例1
n-(3,5-二溴苯基)苯并[d]异噻唑-3-胺(1)
(1)2-氯-n-(3,5-二溴苯基)苯甲酰胺的制备
在冰浴条件下,于邻氯苯甲腈(138.9mg,1mmol)的干燥的二甲亚砜(5ml)溶液中加入氢化钠(31.3mg,1.2mmol)和3,5-二溴苯胺(276.0mg,1.1mmol)。冰浴下搅拌反应0.5小时后反应逐渐恢复室温,保持室温反应24小时。反应结束后,反应液加水(5ml)淬灭。使用稀盐酸将反应液调至ph~3.0,使目标产物质子化溶于水相,二氯甲烷(3×10ml)洗涤水相洗去有机相杂质。使用饱和碳酸氢钠溶液将水相调至ph~7.0,使用二氯甲烷(3×10ml)萃取。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后得棕褐色油状粗品即为产物,其直接用于下一步反应无需进一步纯化。
(2)n-(3,5-二溴苯基)苯并[d]异噻唑-3-胺的合成
于2-氯-n-(3,5-二溴苯基)苯甲酰胺(388.5mg,1mmol)的二甲亚砜和甲苯的混合溶液(5ml,v:v=1:1)中加入单质硫(143.9mg,4.5mmol)和磷酸三钾(318.4mg,1.5mmol)。在氮气保护下160℃反应24小时。反应结束后,反应液中加入水(10ml)淬灭,抽滤除去单质硫,滤液依次使用乙酸乙酯萃取(15ml×3)、饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=10:1)得目标化合物为白色固体(107.7mg,28.2%)。mp165.3-166.2℃.1h-nmr(400mhz,dmso-d6)δ9.92(s,1h),8.43(d,j=8.1hz,1h),8.24(d,j=1.7hz,2h),8.10(d,j=8.1hz,1h),7.67–7.51(m,2h),7.36(d,j=1.8hz,1h).13c-nmr(101mhz,dmso-d6)δ154.68,150.31,144.28,129.12,127.60,125.78,125.10,123.02,122.85,121.15,119.12,40.60,40.39,40.19,39.98,39.77,39.56,39.35.hrms(esi):calcd.forc13h8br2n2s[m+h]+=382.8848,found382.8854.tr=10.564min,hplcpurity:96.78%。
实施例2
n-(3,5-二甲氧基苯基)苯并[d]异噻唑-3-胺(2)
(1)2-氯-n-(3,5-二甲氧基苯基)苯甲酰胺的制备
在冰浴条件下,于邻氯苯甲腈(1g,7.3mmol)的无水n,n-二甲基甲酰胺(5ml)溶液中加入3,5-二甲氧基苯胺(1.23g,8.0mmol)和氢化钠(0.524g,22.0mmol)。室温反应1小时。反应结束后,反应液加入冰水(25ml)淬灭。用稀盐酸将反应液调至ph~3.0。加入二氯甲烷(3×10ml)洗涤水相洗去有机相杂质。水相用饱和碳酸氢钠溶液调至ph~7.0,使用二氯甲烷(3×10ml)萃取。合并有机相,减压蒸除溶剂后得红色油状产物。无需进一步纯化直接进行下一步反应。
(2)n-(3,5-二甲氧基苯基)苯并[d]异噻唑-3-胺的制备
将2-氯-n-(3,5-二甲氧基苯基)苯甲酰胺(700mg,2.4mmol)溶于二甲亚砜和甲苯的混合溶液(12ml,v:v=1:1)中,依次加入沉降硫(2.15g,8.0mmol)、磷酸三钾(0.71g,3.6mmol)。在氮气保护下135℃加热反应36小时。反应结束后,将反应液抽滤并保留滤液。减压蒸除滤液中的甲苯,残留液体加入水(25ml)。混合液依次使用乙酸乙酯(3×25ml)萃取,饱和食盐水(15ml)洗涤。合并有机相,无水硫酸钠干燥。减压蒸除溶剂后经柱层析纯化(石油醚:乙酸乙酯=10:1)后得目标产物,其为白色固体(427.9mg,62.1%)。mp144.2-144.9℃.1h-nmr(400mhz,dmso-d6)δ9.62(s,1h),8.51(d,j=8.1hz,1h),8.08(d,j=7.9hz,1h),7.58(dt,j=26.8,7.2hz,2h),7.30(s,2h),6.19(s,1h),3.79(s,6h).13c-nmr(101mhz,dmso-d6)δ161.10,155.48,150.13,143.27,128.87,127.87,124.81,123.10,121.00,96.67,93.88,55.52.hrms(esi):calcd.forc15h14n2o2s[m+h]+=287.0849,found287.0854.tr=6.014min,hplcpurity:98.74%。
实施例3
n-(3,5-二甲基苯基)苯并[d]异噻唑-3-胺(3)
(1)2-氯-n-(3,5-二甲基苯基)苯甲酰胺的制备
在冰浴条件下,于邻氯苯甲腈(138.9mg,1mmol)的干燥的二甲亚砜(5ml)溶液中加入氢化钠(31.3mg,1.2mmol)和3,5-二甲基苯胺(133.3mg,1.1mmol)。反应液在冰浴条件下反应0.5小时后恢复室温,保持室温反应24小时。反应结束后,反应液加水(5ml)淬灭。用稀盐酸将反应液调至ph~3.0。加入二氯甲烷(3×10ml)洗涤水相洗去有机相杂质。水相用饱和碳酸氢钠溶液调至ph~7.0,使用二氯甲烷(3×10ml)萃取。合并有机相,减压蒸除溶剂得棕褐色油状粗品,其直接用于下一步反应无需进一步纯化。
(2)n-(3,5-二甲基苯基)苯并[d]异噻唑-3-胺的合成
于2-氯-n-(3,5-二甲基苯基)苯甲酰胺(258.7mg,1mmol)的二甲亚砜与甲苯(5ml,v:v=1:1)的混合溶液中依次加入单质硫(143.9mg,4.5mmol)和磷酸三钾(318.4mg,1.5mmol)。反应液在氮气保护下160℃反应24小时。反应完毕后,反应液中加入水(10ml)淬灭。将反应液抽滤除去硫,滤液依次使用乙酸乙酯萃取(15ml×3)萃取、饱和食盐水(10ml)洗涤。合并有机相、无水硫酸钠干燥。减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=10:1~5:1)得目标化合物,其为白色固体(143.6mg,56.5%)。mp123.0-123.8℃.1h-nmr(300mhz,dmso-d6)δ9.44(s,1h),8.48(d,j=8.2hz,1h),8.06(d,j=8.1hz,1h),7.64–7.57(m,3h),7.51(t,j=7.2hz,1h),6.63(s,1h),2.28(s,6h).13c-nmr(75mhz,dmso-d6)δ155.63,150.14,141.59,138.03,128.79,127.96,124.76,123.36,123.20,120.96,115.94,40.79,40.51,40.23,39.96,39.68,39.40,39.12,21.77.hrms(esi):calcd.forc15h14n2s[m+h]+=255.0950,found255.0953.tr=5.119min,hplcpurity:99.17%。
实施例4
n-(3,5-二氟苯基)苯并[d]异噻唑-3-胺(4)
(1)2-氯-n-(3,5-二氟苯基)苯甲酰胺的制备
在冰浴条件下,于邻氯苯甲腈(138.9mg,1mmol)干燥的二甲亚砜(5ml)溶液中加入氢化钠(31.3mg,1.2mmol)和3,5-二氟苯胺(142.0mg,1.1mmol)。在冰浴条件下,反应0.5小时后自然恢复室温,保持室温反应24小时。反应液加水(5ml)淬灭。然后用稀盐酸将反应液调至ph~3.0,使目标产物质子化溶于水相。使用二氯甲烷(3×10ml)洗涤水相洗去有机相杂质。用饱和碳酸氢钠溶液将水相调至ph~7.0,使用二氯甲烷(3×10ml)萃取。合并有机相,减压蒸除溶剂后得棕褐色油状粗品,其直接用于下一步反应无需进一步纯化。
(2)n-(3,5-二氟苯基)苯并[d]异噻唑-3-胺的合成
于2-氯-n-(3,5-二氟苯基)苯甲酰胺(266.0mg,1mmol)的二甲亚砜和甲苯的混合溶液(5ml,v:v=1:1)中依次加入单质硫(143.9mg,4.5mmol)和磷酸三钾(318.4mg,1.5mmol)。在氮气保护下160℃反应24小时。反应结束后,反应液中加入水(10ml)淬灭。抽滤除去单质硫,滤液依次使用乙酸乙酯萃取(15ml×3)、饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=15:1)得目标化合物为白色固体(123.4mg,47.1%)。mp159.2-160.4℃.1h-nmr(300mhz,dmso-d6)δ10.03(s,1h),8.45(d,j=8.0hz,1h),8.10(d,j=8.1hz,1h),7.71(dd,j=10.5,2.2hz,2h),7.67–7.51(m,2h),6.77(tt,j=9.3,2.3hz,1h).13c-nmr(75mhz,dmso-d6)δ164.84,164.63,161.63,161.42,154.88,150.31,144.34,144.15,143.96,129.08,127.57,125.03,123.04,121.13,100.93,100.54,96.82,96.47,96.12,40.79,40.51,40.24,39.96,39.68,39.40,39.13.hrms(esi):calcd.forc13h8f2n2s[m+h]+=263.0449,found263.0451.tr=5.613min,hplcpurity:99.05%。
实施例5
n-(3,5-二氯苯基)苯并[d]异噻唑-3-胺(5)
(1)2-氯-n-(3,5-二氯苯基)苯甲酰胺的制备
在冰浴条件下,于邻氯苯甲腈(138.9mg,1mmol)的干燥的二甲亚砜(5ml)溶液中加入氢化钠(31.3mg,1.2mmol)和3,5-二氯苯胺(176.0mg,1.1mmol)。冰浴条件下反应0.5小时后逐渐恢复室温,保持室温反应24小时。反应完毕后,反应液加水(5ml)淬灭反应。使用稀盐酸将反应液调至ph~3.0,使目标产物质子化溶于水相,二氯甲烷(3×10ml)洗涤水相洗去有机相杂质。使用饱和碳酸氢钠溶液将水相调至ph~7.0,水相使用二氯甲烷(3×10ml)萃取。合并有机相,无水硫酸钠干燥,减压蒸除溶剂得目标产物为棕褐色油状粗品即为产物,其直接用于下一步反应无需进一步纯化。
(2)n-(3,5-二氯苯基)苯并[d]异噻唑-3-胺的合成
于2-氯-n-(3,5-二氯苯基)苯甲酰胺(299.6mg,1mmol)溶于二甲亚砜和甲苯的混合溶液(5ml,v:v=1:1)中加入单质硫(143.9mg,4.5mmol)和磷酸三钾(318.4mg,1.5mmol)。在氮气保护下160℃反应24小时。反应完毕后,反应液中加入水(10ml)淬灭,抽滤除去单质硫,滤液依次使用乙酸乙酯萃取(15ml×3)、饱和食盐水(10ml)洗涤。合并有机相、无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=10:1~5:1)得目标化合物为白色固体(150.5mg,51.2%)。mp175.4-176.3℃.1h-nmr(300mhz,dmso-d6)δ9.93(s,1h),8.41(d,j=8.1hz,1h),8.12–8.00(m,3h),7.56(dt,j=25.7,7.2hz,2h),7.08(s,1h).13c-nmr(75mhz,dmso-d6)δ155.24,150.83,144.44,135.03,129.57,128.12,125.55,123.54,121.60,120.98,116.45,41.33,41.05,40.77,40.49,40.21,39.94,39.66.hrms(esi):calcd.forc13h8cl2n2s[m+h]+=294.9858,found294.9853.tr=2.218min,hplcpurity:100%。
实施例6
3-((3,5-二甲基苯基)氨基)苯并[d]异噻唑1-氧化物(6)
在-70℃条件下,于n-(3,5-二甲基苯基)苯并[d]异噻唑-3-胺(100mg,0.39mmol,1.0eq)的二氯甲烷(5ml)溶液中,缓慢滴加间氯过氧苯甲酸(87.89mg,0.43mmol,1.1eq)的二氯甲烷(10ml)溶液。滴加完毕后反应恢复室温,搅拌反应0.5小时。反应结束后,加入饱和碳酸氢钠水溶液将反应液ph值调至中性,使用二氯甲烷萃取(3×15ml)。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=3:1)得目标化合物为白色固体(42.5mg,40.3%)。mp236.7-237.6℃.1h-nmr(400mhz,dmso-d6)δ10.25(s,1h),8.48(s,1h),8.10(d,j=7.7hz,1h),7.81(t,j=11.5hz,2h),7.64(s,2h),6.85(s,1h),2.32(s,6h).hrms(esi):calcd.forc15h14n2os[m+h]+=271.0900,found271.0899.tr=6.377min,hplcpurity:100%。
实施例7
n-(3,5-双(三氟甲基)苯基)苯并[d]异噻唑-3-胺(7)
(1)2-氯-n-(3,5-二三氟甲基苯基)苯甲酰胺的制备
在冰浴条件下,于邻氯苯甲腈(138.9mg,1mmol)的干燥的二甲亚砜(5ml)溶液中加入氢化钠(31.3mg,1.2mmol)和3,5-双三氟甲基苯胺(252.0mg,1.1mmol)。冰浴条件下反应0.5小时后逐渐恢复室温,保持室温反应24小时后。反应结束后,反应液加水(5ml)淬灭。使用稀盐酸将反应液调至ph~3.0,使目标产物质子化溶于水相,二氯甲烷(3×10ml)洗涤水相洗去有机相杂质。使用饱和碳酸氢钠溶液将水相调至ph~7.0,水相用二氯甲烷(3×10ml)萃取。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后得棕褐色油状粗品即为产物,其直接用于下一步反应无需进一步纯化。
(2)n-(3,5-双三氟甲基苯基)苯并[d]异噻唑-3-胺的合成
于2-氯-n-(3,5-双三氟甲基苯基)苯甲酰胺(366.7mg,1mmol)的二甲亚砜和甲苯的混合溶液(5ml,v:v=1:1)中加入单质硫(143.9mg,4.5mmol)和磷酸三钾(318.4mg,1.5mmol)。在氮气保护下160℃油浴加热搅拌回流反应24小时。反应结束后,反应液中加入水(10ml)淬灭。抽滤除去单质硫,滤液依次使用乙酸乙酯萃取(15ml×3)、饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=10:1~5:1)得目标化合物为白色固体(193.3mg,53.4%)。mp163.4-164.8.3℃.1h-nmr(300mhz,dmso-d6)δ10.36(s,1h),8.74–8.68(m,2h),8.50(dt,j=8.1,1.0hz,1h),8.17(dt,j=8.1,1.0hz,1h),7.72–7.58(m,3h).13c-nmr(75mhz,dmso-d6)δ154.74,150.33,143.39,131.82,131.39,130.96,130.53,129.18,127.50,125.76,125.16,122.99,122.14,121.17,117.37,117.33,113.81,40.78,40.51,40.23,39.95,39.67,39.39,39.11.hrms(esi):calcd.forc15h8f6n2s[m+h]+=363.0385,found363.0390.tr=3.225min,hplcpurity:97.53%。
实施例8
3-((3,5-二溴苯基)氨基)苯并[d]异噻唑1-氧化物(8)
在-30℃条件下,于n-(3,5-二溴苯基)苯并[d]异噻唑-3-胺(100mg)的浓硫酸(2ml)溶液中缓慢加入冷的浓硫酸(225μl)和浓硝酸(225μl)的混合溶液。滴加完毕后,反应液立即倒入冰水中淬灭。缓慢加入饱和碳酸钠水溶液将反应液ph调至中性,依次使用乙酸乙酯(3×20ml)萃取、饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=3:1)得目标化合物为白色固体(58.1mg,51.2%)。mp216.2-217.4℃.1h-nmr(300mhz,dmso-d6)δ11.09(s,1h),8.13(d,j=7.4hz,1h),8.05–7.81(m,4h),7.69(s,1h).hrms(esi):calcd.forc13h8br2n2os[m+na]+=467.8447,found467.8452.tr=4.211min,hplcpurity:100%。
实施例9
3-((3,5-双(三氟甲基)苯基)氨基)苯并[d]异噻唑1-氧化物(9)
在-30℃条件下,于化合物n-(3,5-双三氟甲基苯基)苯并[d]异噻唑-3-胺(100mg)的浓硫酸(2ml)溶液中缓慢加入冷的浓硫酸(225μl)和浓硝酸(225μl)的混合溶液。滴加完毕后,反应液立即倒入冰水中淬灭。加入饱和碳酸钠水溶液将其ph调至中性,依次使用乙酸乙酯萃取(3×20ml)、饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥后,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=5:1)得目标化合物为白色固体(56.0mg,53.6%)。mp269.4-270.5℃.1h-nmr(400mhz,dmso-d6)δ10.89(s,1h),8.74(s,2h),8.43(d,j=7.6hz,1h),8.16(d,j=7.4hz,1h),7.94–7.80(m,3h).hrms(esi):calcd.forc15h8f6n2os[m+h]+=379.0344,found379.0334.tr=12.833min,hplcpurity:100%。
实施例10
3-((3,5-二甲氧基苯基)氨基)苯并[d]异噻唑1-氧化物(10)
在-70℃条件下,于n-(3,5-二甲氧基苯基)苯并[d]异噻唑-3-胺(57.2mg,0.2mmol,1.0eq)的二氯甲烷(5ml)溶液中缓慢滴加间氯过氧苯甲酸(39.0mg,0.21mmol,1.1eq)的二氯甲烷(5ml)溶液。滴加完毕后,反应液自然恢复室温,搅拌反应0.5小时。反应结束后,加入饱和碳酸氢钠水溶液将反应液ph调至中性,使用二氯甲烷(3×15ml)萃取。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后柱层析纯化(石油醚:乙酸乙酯=3:1)得目标化合物为白色固体(45.1mg,74.6%)。mp235.5-235.6℃.1h-nmr(300mhz,dmso-d6)δ10.28(s,1h),8.48(d,j=7.3hz,1h),8.11(d,j=7.0hz,1h),7.93–7.75(m,2h),7.29(s,2h),6.37(s,1h),3.79(s,6h)。
实施例11
n-(苯并[d]异噻唑-3-基)呋喃-2-甲酰胺(11)
1)1-(2-氯苯基)乙烷-1-亚胺的制备
在冰浴条件下,于2-氯苯甲腈(5g,0.036mol)的乙醚(60ml)溶液中缓慢滴加二(三甲基硅基)氨基锂(1minthf,60ml,0.06mol)。滴加完毕后,反应液逐渐恢复室温,保持室温反应12小时。反应结束后,将反应液缓慢倒入冰水(200ml)中淬灭,悬浊液依次使用乙酸乙酯(3×100ml)萃取,饱和食盐水(50ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后得红色油状产物,其无需进一步纯化直接进行下一步反应。
2)苯并[d]异噻唑-3-胺的制备
于1-(2-氯苯基)乙烷-1-亚胺(278.3mg,1.8mmol)的二甲亚砜和甲苯的混合溶液(10ml.v:v=1:1)中加入单质硫(256mg,8mmol)、k3po4(764.1mg,3.6mmol)。在氮气保护下135℃反应36小时。反应结束后,抽滤除去硫,滤液减压蒸除甲苯。残余液加入水(20ml)后依次使用乙酸乙酯萃取(3×20ml),饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后经柱层析纯化(石油醚:乙酸乙酯=5:1)后得目标产物为棕色固体(247.1mg,91.5%)。mp℃.1h-nmr(300mhz,dmso-d6)δ8.10(dt,j=8.0,1.0hz,1h),7.92(dt,j=8.1,0.9hz,1h),7.51(ddd,j=8.1,7.0,1.2hz,1h),7.38(ddd,j=8.0,7.0,1.0hz,1h),6.81(s,2h)。
3)n-(苯并[d]异噻唑-3-基)呋喃-2-甲酰胺的制备
于苯并[d]异噻唑-3-胺(100mg,0.67mmol)的二氯甲烷(5ml)溶液中依次加入呋喃-2-羧酸(75.1mg,0.67mmol)、1-羟基苯并三唑(135.1mg,1mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(191.7mg,1mol)及三乙胺(202.3mg,2mmol)。室温反应12小时。反应结束后,反应液抽滤,滤液依次使用二氯甲烷萃取(3×20ml),饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后经柱层析纯化(石油醚:乙酸乙酯=10:1)得目标产物为白色固体(146.3mg,75.6%)。mp256.1-257.0℃.1h-nmr(300mhz,dmso-d6)δ11.16(s,1h),8.19(d,j=8.2hz,1h),8.00(d,j=7.6hz,2h),7.63(s,1h),7.56–7.42(m,2h),6.75(d,j=4.1hz,1h).13c-nmr(75mhz,dmso-d6)δ157.20,153.26,152.25,147.03,147.01,130.01,128.73,125.31,121.26,116.47,112.75.hrms(esi):calcd.forc12h8n2o2s[m+na]+=267.0199,found.267.0202tr=2.660min,hplcpurity:99.21%。
实施例12
n-(苯并[d]异噻唑-3-基)-1h-吡咯-2-羧酰胺(12)
于苯并[d]异噻唑-3-胺(100mg,0.67mmol)的二氯甲烷溶液(5ml)中依次加入吡咯-2-羧酸(75.0mg,0.67mmol)、1-羟基苯并三唑(135.1mg,1mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(191.7mg,1mol)及三乙胺(202.3mg,2mmol)。室温反应12小时。反应结束后,反应液抽滤,滤液依次使用二氯甲烷萃取(3×20ml),饱和食盐水(10ml)洗涤。合并有机相,无水硫酸钠干燥,减压蒸除溶剂后经柱层析纯化(石油醚:乙酸乙酯=5:1)得目标产物为白色固体(131.1mg,81.1%)。mp>300℃.1h-nmr(400mhz,dmso-d6)δ11.44(s,1h),9.32(s,1h),8.99(d,j=2.5hz,1h),8.87(t,j=2.0hz,1h),8.22(d,j=8.2hz,1h),8.06(d,j=8.2hz,1h),7.66(s,1h),7.52(s,1h).hrms(esi):calcd.forc12h10n3os[m+na]+=267.0359,found.279.0323tr=8.900min,hplcpurity:96.85%。
实施例13
n-(苯并[d]异噻唑-3-基)噻吩-2-羧酰胺(13)
按照实施例12的方法,用噻吩-2-羧酸替换吡咯-2-羧酸,得目标产物为白色固体(136.0mg,78.1%)。mp276.4-177.2℃.1h-nmr(300mhz,dmso-d6)δ11.27(s,1h),8.23–8.11(m,2h),8.02–7.91(m,2h),7.65(d,j=1.1hz,1h),7.51(dd,j=8.1,1.1hz,1h),7.32–7.25(m,1h).hrms(esi):calcd.forc12h9n2os2[m+h]+=261.0151,found.261.0148.tr=15.167min,hplcpurity:97.93%。
实施例14
n-(苯并[d]异噻唑-3-基)-2-氟苯甲酰胺(14)
按照实施例12的方法,用2-氟苯甲酸替换吡咯-2-羧酸,得目标化合物为白色固体(118.3mg,65.8%)。mp231.1-231.9℃.1h-nmr(400mhz,dmso-d6)δ11.26(s,1h),8.20(d,j=8.2hz,1h),8.11(d,j=8.2hz,1h),7.78(td,j=7.4,1.8hz,1h),7.67–7.61(m,2h),7.55–7.51(m,1h),7.41–7.35(m,2h).13c-nmr(100mhz,dmso-d6)δ164.03,160.94,158.46,153.02,152.06,133.61,130.71,130.68,129.53,128.82,125.40,125.10,125.07,124.72,121.30,116.81,116.59.hrms(esi):calcd.forc14h10fn2os[m+h]+=273.0492,found.273.0492tr=7.180min,hplcpurity:96.29%。
实施例15
n-(苯并[d]异噻唑-3-基)-3-氟苯甲酰胺(15)
按照实施例12的方法,用3-氟苯甲酸替换吡咯-2-羧酸,得目标产物为白色固体(129.3mg,70.8%)。mp>300℃.1h-nmr(400mhz,dmso-d6)δ11.29(s,1h),8.21(d,j=8.2hz,1h),8.01(d,j=8.2hz,1h),7.93(d,j=7.7hz,1h),7.87(d,j=9.8hz,1h),7.65(t,j=7.7hz,2h),7.56–7.47(m,2h).hrms(esi):calcd.forc14h10fn2os[m+h]+=273.0492,found.273.0496tr=7.156min,hplcpurity:100%。
实施例16
n-(苯并[d]异噻唑-3-基)-4-氟苯甲酰胺(16)
按照实施例12的方法,用4-氟苯甲酸替换吡咯-2-羧酸,得目标产物为白色固体(141.1mg,77.4%)。mp278.1-279.1℃.1h-nmr(300mhz,dmso-d6)δ11.23(s,1h),8.21(dt,j=8.2,0.9hz,1h),8.15(dd,j=8.9,5.5hz,2h),7.99(dt,j=8.2,1.0hz,1h),7.64(ddd,j=8.2,7.0,1.1hz,1h),7.49(ddd,j=8.1,6.9,1.0hz,1h),7.42(t,j=8.9hz,2h).hrms(esi):calcd.forc14h10fn2os[m+h]+=273.0492,found.273.0497tr=3.301min,hplcpurity:97.31%。
实施例17
n-(苯并[d]异噻唑-3-基)-2-甲氧基苯甲酰胺(17)
按照实施例12的方法,用2-甲氧基苯甲酸替换吡咯-2-羧酸,得目标产物为白色固体(127.6mg,67.8%)。mp161.5-162.1℃.1h-nmr(300mhz,dmso-d6)δ10.84(s,1h),8.18(s,0h),8.07(s,0h),7.77(d,j=7.6hz,1h),7.65(t,j=7.5hz,1h),7.55(dt,j=14.8,7.9hz,2h),7.23(d,j=8.4hz,1h),7.11(t,j=7.5hz,1h),3.92(s,3h).hrms(esi):calcd.forc15h13n2o2s[m+h]+=285.0692found.285.0693.tr=3.581min,hplcpurity:98.36%。
实施例18
n-(苯并[d]异噻唑-3-基)-4-溴苯甲酰胺(18)
按照实施例12的方法,用4-溴苯甲酸替换吡咯-2-羧酸,得目标产物为白色固体(128.6mg,58.1%)。mp277.1-279.3℃.1h-nmr(300mhz,dmso-d6)δ11.30(s,1h),8.21(d,j=8.3hz,1h),8.06–7.95(m,3h),7.80(d,j=8.6hz,2h),7.64(s,1h),7.50(s,1h).13c-nmr(75mhz,dmso-d6)δ165.78,153.87,152.20,132.92,132.04,130.74,129.96,128.73,126.63,125.30,125.28,121.30.hrms(esi):calcd.forc14h10brn2os[m+h]+=332.9692,found.332.9688tr=4.632min,hplcpurity:97.77%。
实施例19
n-(苯并[d]异噻唑-3-基)-4-溴-2-氟苯甲酰胺(19)
按照实施例12的方法,用2-氟-4-溴苯甲酸替换呋喃-2-羧酸,得目标产物为淡黄色固体(184.9mg,78.6%)。mp192.3-193.1℃.1h-nmr(300mhz,dmso-d6)δ11.35(s,1h),8.20(d,j=8.1hz,1h),8.12(d,j=8.1hz,1h),7.86–7.70(m,2h),7.63(q,j=9.1,8.2hz,2h),7.52(t,j=7.6hz,1h).hrms(esi):calcd.forc14h8brfn2os[m+h]+=350.9598found.350.9596.tr=4.172min,hplcpurity:97.49%。
实施例20
2-氟-n-(6-氟苯并[d]异噻唑-3-基)苯甲酰胺(20)
按照实施例11的方法,用2-氯-4-氟苯甲腈替换2-氯苯甲腈,用2-氟苯甲酸替换呋喃-2-羧酸,得目标产物为淡黄色固体(149.6mg,76.9%)。mp202.1-203.1℃.1h-nmr(300mhz,dmso-d6)δ11.31(s,1h),8.20–8.05(m,2h),7.78(td,j=7.4,1.8hz,1h),7.70–7.57(m,1h),7.49–7.38(m,1h),7.44–7.31(m,2h).hrms(esi):calcd.forc14h9f2n2os[m+h]+=291.0398found.291.0398.tr=3.573min,hplcpurity:100%。
实施例21
4-溴-n-(5-(三氟甲基)苯并[d]异噻唑-3-基)苯甲酰胺(21)
按照实施例11的方法,用2-氯-4-三氟甲基苯甲腈替换2-氯苯甲腈,用对溴苯甲酸替换呋喃-2-羧酸,得目标产物为黄色固体(232.5mg,86.5%)。mp170.9-171℃.1h-nmr(400mhz,dmso-d6)δ11.45(s,1h),8.47(d,j=9.3hz,2h),8.01(d,j=8.2hz,2h),7.98–7.90(m,1h),7.79(d,j=8.2hz,2h).hrms(esi):calcd.forc15h8brf3n2os[m+h]+=402.2087found.402.2085.tr=5.234min,hplcpurity:96.65%。
实施例22
n-(6-氯苯并[d]异噻唑-3-基)-2-氟苯甲酰胺(22)
按照实施例12的方法,用2,4-二氯苯甲腈替换2-氯苯甲腈,用2-氟苯甲酸替换呋喃-2-羧酸,得目标产物为黄色固体(166.9mg,81.7%)。mp171-171.1℃.1h-nmr(300mhz,dmso-d6)δ11.34(s,1h),8.42–8.33(m,1h),8.12(dd,j=8.7,0.6hz,1h),7.78(td,j=7.4,1.8hz,1h),7.71–7.53(m,2h),7.46–7.31(m,2h).hrms(esi):calcd.forc14h8clfn2osna[m+na]+=328.9922,found.328.9929.tr=4.923min,hplcpurity:97.32%。
实施例23
n-(苯并[d]异噻唑-3-基)-4-溴-2-甲基苯甲酰胺(23)
按照实施例12的方法,用2-甲基-4-溴苯甲酸替换呋喃-2-羧酸,得目标产物为白色固体(204.5mg,87.9%),mp236-238.7℃.1h-nmr(400mhz,dmso-d6)δ11.19(s,1h),8.20(dt,j=8.3,0.9hz,1h),8.12(dt,j=8.2,1.0hz,1h),7.64(ddd,j=8.1,6.9,1.1hz,1h),7.61(dt,j=1.6,0.8hz,1h),7.56–7.53(m,2h),7.54–7.49(m,1h),2.46(s,3h).hrms(esi):calcd.forc15h11brn2os[m+h]+=346.9868,found.346.9869.tr=7.325min,hplcpurity:100%。
实施例24
n-(6-溴苯并[d]异噻唑-3-基)-2-氟苯甲酰胺(24)
按照实施例11的方法,用2-氯-4-溴苯甲腈替换2-氯苯甲腈,用2-氟苯甲酸替换呋喃-2-羧酸,得目标产物为白色固体(184.8mg,76.8%)。mp166.5-167.1℃.1h-nmr(400mhz,dmso-d6)δ11.33(s,1h),8.54(d,j=1.7hz,1h),8.05(d,j=8.7hz,1h),7.77(td,j=7.4,1.8hz,1h),7.70(dd,j=8.7,1.8hz,1h),7.66–7.60(m,1h),7.40–7.33(m,2h).hrms(esi):calcd.forc14h7brfn2osna[m+na]+=372.9417,found.372.9421.tr=5.613min,hplcpurity:99.18%。
实施例25
n-(5-氯苯并[d]异噻唑-3-基)-2-氟苯甲酰胺(25)
按照实施例11的方法,用2,5-二氯苯甲腈替换2-氯苯甲腈,用2-氟苯甲酸替换呋喃-2-羧酸,得目标产物为白色固体(157.8mg,76.8%)。mp>300℃.1h-nmr(300mhz,dmso-d6)δ11.33(s,1h),8.33–8.18(m,2h),7.80(d,j=1.9hz,1h),7.69(dd,j=8.7,2.0hz,2h),7.44–7.33(m,2h).hrms(esi):calcd.forc14h9clfn2os[m+h]+=307.0103,found.307.0109.tr=5.017min,hplcpurity:100.0%。
实施例26
n-(5-(三氟甲基)苯并[d]异噻唑-3-基)苯甲酰胺(26)
按照实施例11的方法,用2-氯-5-(三氟甲基)苯甲腈替换2-氯苯甲腈,用对溴苯甲酸替换呋喃-2-羧酸,得目标产物为黄色固体(211.9mg,78.9%)。mp261.6-263.6℃.1h-nmr(300mhz,dmso-d6)δ11.40(s,1h),8.52–8.43(m,2h),8.12–8.04(m,2h),7.96(s,1h),7.63–7.55(m,2h).hrms(esi):calcd.forc15h9brf3n2os[m+h]+=400.9566,found.400.9568.tr=4.672min,hplcpurity:96.48%。
实施例27
2-氟-n-(5-(三氟甲基)苯并[d]异噻唑-3-基)苯甲酰胺(27)
按照实施例11的方法,用2-氯-5-(三氟甲基)苯甲腈替换2-苯甲苄腈,用2-氟苯甲酸替换呋喃-2-羧酸,得目标产物为白色固体(175.1mg,76.8%)。m.p.>300℃.1h-nmr(300mhz,dmso-d6)δ11.48(s,1h),8.62(s,1h),8.48(d,j=8.6hz,1h),7.96(dd,j=8.5,1.7hz,1h),7.80(d,j=1.9hz,1h),7.70–7.60(m,1h),7.39(d,j=6.9hz,2h).hrms(esi):calcd.forc15h9f4n2os[m+h]+=341.0366,found.341.0368.tr=5.613min,hplcpurity:99.06%。
实施例28
n-(5,6-二氟苯并[d]异噻唑-3-基)苯甲酰胺(28)
按照实施例11的方法,用3,4-二氟-5-氯-苯甲腈替换2-氯苯甲腈,用苯甲酸替换呋喃-2-羧酸,得目标产物为白色固体(150.0mg,77.1%)。mp>300℃.hrms(esi):calcd.forc14h8brf2n2os[m+h]+=368.9503,found.368.9506.tr=7.324min,hplcpurity:98.1%。
实施例29
n-(苯并[d]异噻唑-3-基)-4-溴-2-甲氧基苯甲酰胺(29)
按照实施例12的方法,用2-甲氧基-4-溴苯甲酸替换吡咯-2-羧酸,得目标产物为白色固体(162.8mg,66.9%)。mp>300℃.1h-nmr(400mhz,dmso-d6)δ10.87(s,1h),8.19(s,1h),8.07(s,1h),7.69–7.27(m,5h),3.91(s,3h).hrms(esi):calcd.forc15h11brn2o2s[m+h]+=362.9797,found.362.9798.tr=5.532min,hplcpurity:98.46%。
实施例30
n-(苯并[d]异噻唑-3-基)-4-溴-2,6-二氟苯甲酰胺(30)
按照实施例12的方法,用用2,5-二氟-4-溴苯甲酸替换吡咯-2-羧酸,得目标产物为白色固体(120.2mg,48.6%)。mp>300℃.1h-nmr(400mhz,chloroform-d)δ9.12(s,1h),8.16(d,j=8.2hz,1h),7.90(dd,j=8.2,0.9hz,1h),7.58(d,j=7.0hz,1h),7.50(t,j=7.1hz,1h),7.25(d,j=7.4hz,2h).hrms(esi):calcd.forc14h7brf2n2os[m+h]+=368.9503,found.368.9505.tr=8.485min,hplcpurity:96.53%。
下面是本发明化合物的部分药效学实验数据:
(1)荧光素酶报告基因实验测试化合物对hif-2的激动活性
实验方法:将含有缺氧诱导元件和荧光素酶基因序列的商用病毒(cls-007l,qiagen)转染至786-o细胞中,得到稳定转染的786-o-hre工具细胞(j.med.chem.61,9691-9721(2018))。将786-o-hre细胞均匀地接种于96孔白板中。待细胞贴壁,加药组加入相应化合物的二甲亚砜(dmso)溶液,空白对照组加入相应浓度的dmso。在37℃、5%co2培养条件的培养箱中孵育24小时后,在白板中根据试剂盒说明书加入10μl的荧光素酶底物(rg055s,beyotime),立即检测发光值。加药孔发光值对比空白对照组发光值得到化合物的激动效应e。同时使用graphpadprism8对化合物不同浓度的发光值进行非线性回归分析计算ec50值。
e=加药组发光值/空白对照组发光值×100%
表1、本发明部分化合物在20μm浓度下对hif-2转录活性的激动效应
表1数据为本发明部分化合物在20μm浓度下对hif-2转录活性的激活效应。活性数据表明,本发明中的化合物均有一定程度的hif-2激动活性且优于阳性化合物m1002(141.6±8.2)。其中部分化合物的hif-2激动活性显著优于阳性药化合物m1002,如化合物2、4、8、10、14、16和18等。
本发明化合物的活性数据表明化合物异噻唑环中的磺酰胺结构的两个s=o双键并不是hif-2激动活性的药效基团,可去除s=o双键得到新的母体结构。本发明化合物的异噻唑环的硫原子上仅含一个s=o双键或不含s=o双键取代,化合物活性普遍优于阳性化合物m1002。
同时,本发明部分化合物在低浓度下即能激活hif-2转录活性,其在荧光素酶报告基因实验中的ec50数据如表2所示。其中,化合物2、18、19、21在纳摩尔浓度下即能有效激活hif-2。而阳性化合物m1002在多浓度荧光素酶报告基因实验中,高浓度荧光数值反常下降,这可能是由于其对786-o-hre细胞的细胞毒性原因。荧光数值反常导致其ec50数值无法拟合,其活性弱于本发明化合物。
表2、本发明部分化合物在荧光素酶报告基因实验中的ec50
(2)细胞计数(cellcountingkit-8,cck-8)法测试本发明化合物对肾上皮细胞株hek293细胞毒性
为了初步评价本发明化合物的安全性,采用cck-8法测试肾上皮细胞株hek293的细胞毒性。
实验方法:将hek293细胞接种在96孔板中,细胞贴壁后,给药组加入相应化合物的dmso溶液,以保证每个化合物终浓度为2、20μm。空白对照组加入给药组中最高浓度的dmso。同时设置3个复孔。处理24小时后,根据试剂盒说明书将cellcountingkit-8(c0037,beyotime)添加到每个孔中。在37℃条件下将96孔板避光孵育2小时,使用酶标仪检测每孔450nm波长的吸光度。计算给药组吸光度值与空白对照组吸光度的比值以确定抑制率i,定义i值高于10%为有细胞毒性。
i=(1-加药组吸光度/空白组吸光度)×100%
以下是部分化合物的细胞毒性实验数据(表3):
cck-8法测试的实验结果表明,本发明化合物在20μm高浓度条件下均无显著细胞毒性。而阳性化合物m1002在2μm浓度下即展现出细胞毒性。本发明化合物在细胞水平安全性方面优于阳性化合物m1002。
表3、本发明部分化合物对hek293细胞的细胞毒性
(3)荧光素酶报告基因实验测试化合物和phd抑制剂的协同作用
此外,本发明测试了部分化合物与phd抑制剂联用对hif-2的活性提升的协同作用。在细胞水平,采用荧光素酶报告基因实验测试化合物和phd抑制剂的协同作用。由于在786-o细胞中vhl蛋白缺失,导致hif-2无法被phd羟基化从而降解。所以本发明选择人肾上皮细胞株hek293细胞株作为工具进行研究。以下是实验方法:将含有缺氧诱导元件和荧光素酶基因序列的商用病毒(cls-007l,qiagen)转染至hek293细胞中,得到稳定转染的hek293-hre工具细胞。将hek293-hre细胞均匀地接种于96孔白板中。待细胞贴壁,联合给药组同时加入相应浓度本发明化合物的dmso溶液以保证其终浓度为0.2、2、20μm,与相应浓度的phd抑制剂的dmso溶液以保证其终浓度为2μm;phd抑制剂对照组只加入相应浓度phd抑制剂的dmso溶液以保证其终浓度为0.2、2、20μm;空白对照组加入给药组中最高浓度的dmso。同时设置三个复孔。在37℃和5%co2培养条件的细胞培养箱中孵育24小时之后,根据说明书在96孔白板中加入荧光素酶底物(rg055s,beyotime),使用酶标仪立即检测其发光值。计算联合用药效应值r,定义本发明化合物在0.2、2、20μm三个浓度下的r值均大于100%则表示两者在提升hif-2转录活性中具有协同作用。
r=联合给药组发光值/(单独给药组发光值+2μmphd抑制剂组发光值)×100%
表4、本发明部分实施例化合物与phd抑制剂联合使用的实验数据
本发明部分化合物与phd抑制剂联合使用在提升hif-2转录活性中展现协同作用,其中,nd表示未测试。实验结果表明,本发明化合物与phd抑制剂联合使用在提升hif-2转录活性中可展现协同作用。同时本发明公布了化合物2与phd抑制剂联用的荧光素酶报告基因实验结果图(附图2)。结果表明化合物2和phd抑制剂在细胞中对hif-2的激活具有良好的协同作用,其联用效果显著优于单独给药组。
(4)体内血浆epo提升活性研究
实验方法:选用18-20克的c56bl/6小鼠,将其随机分为8组:空白对照组、化合物2的低剂量单独给药组、化合物2的中剂量单独给药组、化合物2的高剂量单独给药组组,phd抑制剂单独给药组组、phd抑制剂和低剂量的化合物2联用组、phd抑制剂和中剂量的化合物2联用组、phd抑制剂和高剂量的化合物2联用组,每组5只小鼠。给药组分别灌胃给药相应浓度的化合物,空白对照组灌胃给药与给药组相同体积的生理盐水。4-6个小时后,采用眼眶后静脉丛取血法对各组小鼠取血,将血样在6000r/min转速下离心5min取上层获得血浆。根据试剂盒说明书说明,使用鼠源epo定量elisa检测试剂盒(ab119593,abcam)检测各组小鼠血浆中epo的含量。
本发明公布了化合物2与phd抑制剂akb-6548联用的实验结果(附图3)。结果表明在动物水平,本发明化合物2与phd抑制剂akb-6548联合给药对c57bl/6小鼠体内epo的提升展现出良好的协同作用。联合给药组中的小鼠血浆epo显著高于akb-6548单独使用组,且效果呈化合物2剂量依赖性。相比单独使用phd抑制剂或单独使用本发明化合物,两者联合使用在快速提升epo水平中有着独特优势。
本发明的苯并异噻唑衍生物无论在细胞水平还是在动物水平均具有良好的生物活性。该类化合物能激动缺氧诱导因子2转录活性,增强促红细胞生成素的生成和分泌,从而促进红细胞的生成;本发明所制备的苯并异噻唑类缺氧诱导因子2激动剂类化合物或其药学上可接受的盐可与脯氨酰羟化酶抑制剂联合使用在提升hif-2转录活性中起协同作用,其可用于治疗缺氧诱导因子2相关疾病,如缺血性疾病等。相比单独phd抑制剂,两者联用能够快速提升小鼠血液中epo水平,在肾性贫血、缺血性脑卒等缺血性疾病中有着一定的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!