非同源双链寡聚核苷酸片段在基因敲除系统中的应用的制作方法
1.本发明属于基因编辑技术领域,具体涉及非同源双链寡聚核苷酸片段在基因敲除系统中的应用。
背景技术:
2.随着对基因功能研究的深入,临床把抗病毒治疗方向转向个体化基因编辑领域。然而,现有基因敲除系统(crispr/cas核酸酶系统)对靶基因的敲除效率不高,限制crispr/cas核酸酶系统的发展和应用;同时为了检测脱靶效应,需事先假定脱靶位点与靶标位点相似,但是由于许多脱靶突变发生在与目标位点差异很大的地方,因此脱靶断点的数量和位置是很难预测的。如何提高基因敲除系统的靶基因敲除效率并同时监测脱靶效应成为研究的重点。例如利用crispr/cas9靶向敲除hpv18 e7基因,存在编辑效率有待进一步提高,并且脱靶情况不明等问题,限制了其在临床上的进一步转化应用。
3.现有技术为了获得更高的编辑效率,通常采用寻找新的核酸酶、或采用筛选靶活性高的sgrna、或添加外源增效蛋白、或敲入外源基因片段的同源供体、或对sgrna的改造等。例如专利cn111718418a公布了一种增强基因编辑的融合蛋白;专利cn112601812a公开了在造血干细胞和/或祖细胞基因疗法中使用的促进同源定向dna修复的试剂;专利cn107532162a公开了利用编辑寡核苷酸修饰细胞内的基因组序列;专利cn109295060a公开了一种用于基因编辑的配对sgrna及应用,根据靶标基因或靶标基因组位点的特定编辑需求设计多条sgrna,选择两条特定间距、对应pam特定位置组合的sgrna作为配对sgrna,配对cas9
‑
sgrna可以提高基因编辑效率。更换cas9蛋白,例如sacas9,存在切割效率不足、pam序列要求更严格的局限性;通过同时添加外源性的小分子化合物提高编辑效率,存在化合物不便于同时递送到细胞内的问题;而对sgrna的改造仅针对特定的靶基因获得较好的编辑效率,普适性不高。
技术实现要素:
4.本发明的目的是提供非同源双链寡聚核苷酸片段在基因敲除系统中的应用,非同源双链寡聚核苷酸片段能够增加基因敲除系统对靶基因的切割断点并能够整合到切割断点处,从而增强靶基因的敲除效果并能够准确判定脱靶位点。
5.为达到上述目的,本发明采用的技术方案是:
6.本发明第一方面提供非同源双链寡聚核苷酸片段在基因敲除系统中的应用,所述非同源双链寡聚核苷酸片段能够提高基因敲除系统对靶基因的切割效率并能够整合到切割断点处。
7.具体地,所述非同源双链寡聚核苷酸片段能够激发细胞的非同源末端连接修复,引入更多插入或缺失。
8.具体地,通过将用于敲除靶基因的crispr系统和非同源双链寡聚核苷酸片段共转染细胞。
9.优选地,将所述非同源双链寡聚核苷酸片段还可作为标记物用于监测基因敲除系统的脱靶效应。
10.具体地,将用于敲除靶基因的crispr系统和非同源双链寡聚核苷酸片段共转染细胞后,采用guide
‑
seq方法分析非同源双链寡聚核苷酸片段的整合位置判定脱靶位点。
11.基于现有基因敲除系统的敲除效果不好且脱靶位点难以准确定位的问题,发明人发现仅通过筛选出靶活性高的sgrna,以此sgrna构建的crispr系统的切割效果依旧不够理想,发明人偶然发现将非同源双链寡聚核苷酸片段(非同源dsodn)与基因敲除系统相结合后用于基因敲除,非同源双链寡聚核苷酸片段能够激发细胞的非同源末端连接修复,引入更多插入或缺失(indel),同时将非同源双链寡聚核苷酸片段插入切割断点处还可以其作为标记物准确定位脱靶位点,在显著增强靶基因的敲除效率的同时监测脱靶效应,可谓一举两得。
12.以上应用中,所述crispr系统优选为crispr/cas9系统。目前crispr/cas9系统的研究比较全面,但是其靶基因敲除效率较低且依然存在严重的脱靶效应,将非同源双链寡聚核苷酸片段与之结合能够显著提高crispr/cas9系统的敲除效果并准确定位脱靶位点。
13.所述非同源双链寡聚核苷酸片段的序列优选如seq id no.6所示,或与其具有至少80%及以上的同源性的序列,优选具有至少85%及以上的同源性,进一步优选具有至少90%及以上的同源性,更进一步优选具有至少95%及以上的同源性,再进一步优选具有至少98%及以上的同源性。
14.进一步优选地,所述非同源双链寡聚核苷酸片段的5
′
端进行磷酸化修饰、5
′
端的三个核苷酸中相连两个核苷酸之间进行硫甙修饰、3
′
的三个核苷酸中相连两个核苷酸之间进行硫甙修饰。
[0015]5′
端进行磷酸化修饰有利于非同源双链寡聚核苷酸片段与切割端点整合;硫甙修饰有利于提高非同源双链寡聚核苷酸片段在细胞内存在时的稳定性而不易被降解。
[0016]
本发明第二方面提供一种基因敲除系统,其包括crispr系统和非同源双链寡聚核苷酸片段。
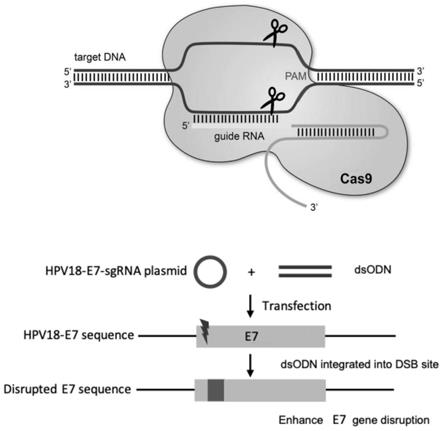
[0017]
优选地,所述crispr系统为crispr/cas9系统。
[0018]
优选地,用于敲除靶基因的crispr系统和非同源双链寡聚核苷酸片段共转染细胞。
[0019]
优选地,所述非同源dsodn的序列如seq id no.6所示,或与其具有至少80%及以上的同源性的序列,优选具有至少85%及以上的同源性,进一步优选具有至少90%及以上的同源性,更进一步优选具有至少95%及以上的同源性,再进一步优选具有至少98%及以上的同源性。
[0020]
进一步优选地,所述非同源双链寡聚核苷酸片段的5
′
端进行磷酸化修饰、5
′
端的三个核苷酸中相连两个核苷酸之间进行硫甙修饰、3
′
的三个核苷酸中相连两个核苷酸之间进行硫甙修饰。5
′
端进行磷酸化修饰有利于非同源双链寡聚核苷酸片段与切割端点整合;硫甙修饰有利于提高非同源双链寡聚核苷酸片段插入切割端点后的稳定性。
[0021]
优选地,所述基因敲除系统针对人乳头瘤病毒18型e7基因的敲除。
[0022]
根据一些具体实施方式,所述的crispr系统的sgrna的序列如seq id no.1所示,或与其具有至少80%及以上的同源性的序列,优选具有至少85%及以上的同源性,进一步
优选具有至少90%及以上的同源性,更进一步优选具有至少95%及以上的同源性,再进一步优选具有至少98%及以上的同源性;
[0023]
或者所述的sgrna的序列如seq id no.2所示,或与其具有至少80%及以上的同源性的序列,优选具有至少85%及以上的同源性,进一步优选具有至少90%及以上的同源性,更进一步优选具有至少95%及以上的同源性,再进一步优选具有至少98%及以上的同源性。
[0024]
具体地,将能够与人乳头瘤病毒18型e7基因相结合的sgrna、cas9克隆表达载体px330得到一种靶向敲除人乳头瘤病毒18型e7基因的质粒;
[0025]
将用于靶向敲除人乳头瘤病毒e7基因的质粒和非同源dsodn共转染细胞。
[0026]
利用构建的靶向高危型hpv e7基因的crispr/cas9表达质粒,通过将表达质粒和非同源dsodn共同转染到细胞中,特异性的诱导相应hpv亚型阳性细胞的hpv致癌元件移码突变,失去致癌特性,甚至直接由于dsb过多导致细胞凋亡。有效增强了crispr/cas9系统特异性敲除高危型hpv e7基因的效率,从而达到降低病毒负荷、清除病毒及病变细胞、逆转癌变的目的,具有重要的临床应用价值。
[0027]
优选地,所述系统用于靶向敲除人乳头瘤病毒18型e7基因,所述非同源dsodn的序列如seq id no.6所示,或与其具有至少80%及以上的同源性的序列,优选具有至少85%及以上的同源性,进一步优选具有至少90%及以上的同源性,更进一步优选具有至少95%及以上的同源性,再进一步优选具有至少98%及以上的同源性。
[0028]
由于上述技术方案运用,本发明与现有技术相比具有下列优点:
[0029]
基于现有基因敲除系统的靶基因敲除效率不高,且脱靶情况不明朗,脱靶位点难以准确定位等问题,首次将非同源双链寡聚核苷酸片段与基因敲除系统相结合用于基因敲除,非同源双链寡聚核苷酸片段能够激发细胞的非同源末端连接修复,引入更多插入/缺失,同时非同源双链寡聚核苷酸片段可插入切割断点处,可作为标记物采用guide
‑
seq方法准确定位脱靶位点,有助于增强靶基因的敲除效率并同时监测脱靶效应。
附图说明
[0030]
图1为实施例中使用的非同源双链寡聚核苷酸片段(dsodn)的序列示意图;
[0031]
图2为crispr/cas9共转非同源dsodn敲除高危型hpv e7基因作用示意图;
[0032]
图3为实施例中应用靶向hpv18 e7的crispr/cas9共转非同源dsodn,靶基因断点处检测到odn整合。图中“blank”代表未处理组断点pcr结果,
“‑”
代表仅转染靶向hpv18e7的crispr/cas9质粒组断点pcr结果,“ss”代表共转靶向hpv18 e7的crispr/cas9质粒和非同源单链寡聚核苷酸片段(ssodn)组断点pcr结果,“ds”代表共转靶向hpv18e7的crispr/cas9质粒和非同源双链寡聚核苷酸片段(dsodn)组断点pcr结果;
[0033]
图4为实施例中应用靶向hpv18 e7的crispr/cas9共转非同源dsodn组断点pcr产物sanger测序结果;
[0034]
图5为实施例中应用靶向hpv18 e7的crispr/cas9质粒和非同源dsodn共转hela细胞后hpv18 e7 mrna表达水平。图中“blank”代表未处理组hpv18 e7 mrna表达水平,
“‑”
代表仅转染靶向hpv18 e7的crispr/cas9质粒组hpv18 e7 mrna表达水平,“ss”代表共转靶向hpv18 e7的crispr/cas9质粒和非同源ssodn组hpv18 e7 mrna表达水平,“ds”代表共转靶
向hpv18e7的crispr/cas9质粒和非同源dsodn组hpv18 e7 mrna表达水平;
[0035]
图6为实施例中应用靶向hpv18 e7的crispr/cas9质粒和非同源dsodn共转hela细胞后hpv18
‑
e7基因敲除效率(indel%)。图中
“‑”
代表仅转染靶向hpv18 e7的crispr/cas9质粒组,“ss”代表共转靶向hpv18 e7的crispr/cas9质粒和非同源ssodn组,“ds”代表共转靶向hpv18 e7的crispr/cas9质粒和非同源dsodn组。non
‑
odn%代表断点处有插入或缺失(indel)但无odn插入的比例,odn%代表断点处有odn插入的比例;
[0036]
图7为guide
‑
seq建库流程示意图;
[0037]
图8为实施例中共转染的dsodn作为标记物监测的脱靶情况。“20bp target site”指的是靶向的目标基因的dna序列,pam为前间区序列临近基序。“·”代表监测到的位点的碱基和目标dna序列匹配一致,全部为“·”表示这个位点是在靶位点,有不一致的表明这个位点是脱靶位点。
具体实施方式
[0038]
为更好地说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。
[0039]
实施例中使用的引物由苏州金唯智生物科技有限公司合成;qrt
‑
pcr试剂采用premix ex taq
tm
,购自宝生物工程(大连)有限公司(货号为code no.rr420a)。
[0040]
实施例1、非同源odn序列(seq id no.6)以及获得方式。
[0041]
(1)ssodn donor的获得:
[0042]
ssodn donor的序列为:
[0043]5′‑
p
‑
g*t*ttaattgagttgtcatatgttaataacggt*a*t
‑3′
[0044]
其中“p”表示5
′
磷酸化,“*”表示2个核苷酸之间的硫甙修饰。序列合成和修饰均由苏州金唯智生物公司完成。合成的干粉加入无菌的去离子水,稀释成50μm的浓度用于后续转染。
[0045]
(2)dsodn donor的获得:
[0046]
首先合成2条反向互补配对的ssodn,序列如下:
[0047]
oligo 1:5
′‑
p
‑
g*t*ttaattgagttgtcatatgttaataacggt*a*t
‑3′
[0048]
oligo 2:5
′‑
p
‑
a*t*accgttattaacatatgacaactcaattaa*a*c
‑3′
[0049]
同样的,其中“p”表示5
′
磷酸化,“*”表示2个核苷酸之间的硫甙修饰连接。2条互补的ssodn序列合成和修饰均由苏州金唯智生物公司完成。合成的干粉加入无菌的去离子水,稀释成125μm的浓度用于后续的退火步骤。
[0050]
2条单链ssodn退火形成双链dsodn donor
[0051]
首先,配置10x的oligoduplex annealing buffer(ste)作为退火所需要的缓冲液:
no.1)或e7
‑
sgrna2(seq id no.2)。
[0069]
(2)crispr/cas9
‑
e7的表达载体构建方法参考引文中的构建方法完成构建(cong,l.et al.multiplex genome engineering using crispr/cas systems.science 339,819
‑
823,doi:10.1126/science.1231143(2013).):
[0070]
将e7
‑
sgrna1或e7
‑
sgrna2和cas9序列克隆到表达载体px330上,构建针对hpv18 e7的真核表达载体crispr/cas9
‑
e7质粒,构建完成后,通过常规测序比对确定构建载体序列正确无突变,挑选出完全正确的克隆进行扩增并提取质粒。
[0071]
实施例3、crispr/cas9
‑
e7质粒及非同源dsodn共转后dsodn能整合至dsb处的验证
[0072]
crispr/cas9
‑
e7质粒转染到细胞后表达出来的靶向hpv18 e7的crispr/cas9
‑
e7系统,可以迅速识别hpv18 e7序列,发挥切割作用。切割后细胞主要通过不受细胞周期限制的nhej途径快速修复,从而引入小片段的插入或缺失(indel),导致移码突变,最终使得e7功能丧失,hpv18 e7癌蛋白的表达受到抑制。共转dsodn后,一方面dsodn能够整合至断点处,另一方面能够激发细胞nhej途径,引入更多indel,从而提高靶基因的敲除效率。
[0073]
具体操作方法是:
[0074]
(1)细胞培养
[0075]
hpv18阳性宫颈癌细胞系hela用含有10%血清的dmem完全培养基在37℃、5%co2培养箱里培养。待细胞融合度达到90%时用0.25%的胰酶消化后,用dmem完全培养基终止消化,接种到6孔板中,继续培养24小时。
[0076]
(2)crispr/cas9
‑
e7质粒和非同源dsodn共同转染
[0077]
24小时后,确认细胞贴壁良好,细胞融合度达到80%,即可进行转染。每孔转染2μgcrispr/cas9
‑
e7质粒和2μl非同源dsodn,使用roche公司的x
‑
tremegene hp dna transfection reagent转染试剂按照说明书要求进行转染,与未处理的细胞组、转染等量crispr/cas9
‑
e7质粒的细胞组、转染等量crispr/cas9
‑
e7质粒和非同源ssodn的细胞组对照。转染后的细胞继续在37℃、5%co2培养箱中培养。
[0078]
(3)基因组dna提取
[0079]
转染48小时后,常规0.25%的胰酶消化,用dmem完全培养基终止消化,收集细胞到离心管中,300g离心5分钟,弃除培养基,pbs洗涤一次,再次300g离心5分钟,弃除pbs,获得细胞渣,使用细胞基因组提取试剂盒(全式金生物技术有限公司,货号:ee101
‑
01)提取细胞基因组dna,测量dna浓度。
[0080]
(4)引物的设计
[0081]
根据hpv18 e7基因序列及非同源dsodn设计引物,引物一端在dsodn上,另一端位于靶基因上。本实施例中对应的引物序列为:
[0082]
odn
‑
f:ttgagttgtcatatgttaataacggt(seq id no.7)。
[0083]
hpv18
‑
e7
‑
r:gttgcttactgctgggatgc(seq id no.8)。
[0084]
(5)pcr反应
[0085]
以上述提取的基因组dna为模版,用上述引物进行pcr反应。本实验使用的高保真dna聚合酶为北京全式金生物科技有限公司的pcr supermix(货号:as111
‑
02)。
[0086]
pcr反应体系:
[0087][0088]
pcr反应条件:
[0089][0090]
pcr反应完成后,取少量pcr产物进行琼脂糖凝胶电泳,根据电泳结果初步判定pcr产物的浓度以及条带大小是否正确等。
[0091]
(6)目标条带sanger测序
[0092]
图3、图4试验结果显示琼脂糖凝胶电泳有目标条带,条带测序结果比对正确,说明共转crispr/cas9
‑
e7质粒和非同源dsodn的hela细胞组已将dsodn整合至dsb处。未处理的细胞组、转染等量crispr/cas9
‑
e7质粒的细胞组、转染等量crispr/cas9
‑
e7质粒和ssodn的细胞组均无目标条带显示。
[0093]
实施例4、qrt
‑
pcr明确共转crispr/cas9
‑
e7质粒和dsodn对hpv 18 e7 mrna转录水平的影响
[0094]
(1)样品rna的抽提
[0095]
①
取冻存已裂解的细胞,室温放置5分钟使其完全溶解。
[0096]
②
两相分离:每1ml的trizol试剂裂解的样品中加入0.2ml的氯仿,盖紧管盖。手动剧烈振荡管体15秒后,15到30℃孵育2到3分钟。4℃下12000rpm离心15分钟。离心后混合液体将分为下层的红色酚氯仿相,中间层以及无色水相上层。rna全部被分配于水相中。水相上层的体积大约是匀浆时加入的trizol试剂的60%。
[0097]
③
rna沉淀:将水相上层转移到一干净无rna酶的离心管中。加等体积异丙醇混合以沉淀其中的rna,混匀后15到30℃孵育10分钟后,于4℃下12000rpm离心10分钟。此时离心前不可见的rna沉淀将在管底部和侧壁上形成胶状沉淀块。
[0098]
④
rna清洗:移去上清液,每1mltrizol试剂裂解的样品中加入至少1ml的75%乙醇(75%乙醇用depch2o配制),清洗rna沉淀。混匀后,4℃下7000rpm离心5分钟。
[0099]
⑤
rna干燥:小心吸去大部分乙醇溶液,使rna沉淀在室温空气中干燥5
‑
10分钟。
[0100]
⑥
溶解rna沉淀:溶解rna时,先加入无rna酶的水40μl用枪反复吹打几次,使其完全溶解,获得的rna溶液保存于
‑
80℃待用。
[0101]
(2)按下列组份配制pcr反应液(反应液配制均在冰上进行)
[0102]
试剂使用量终浓度sybr premix ex taq(tli rnaseh plus)(2
×
)10μl1
×
pcr forward primer(10μm)0.4μl02μm
*1
pcr reverse primer(10μm)0.4μl0.2μm
*1
dna模板(<100ng)
*2
2μl灭菌水7.2μl total20ul
[0103]
应用applied biosystems 7500 fast real
‑
time pcr system进行real time pcr反应,反应程序采用两步法pcr扩增标准程序:
[0104]
stage 1:预变性
[0105]
reps:1
[0106]
95℃30秒
[0107]
stage 2:pcr反应
[0108]
reps:40
[0109]
95℃5秒
[0110]
60℃30~34秒
[0111]
图5显示,共转crispr/cas9
‑
e7质粒和dsodn的hela细胞组hpv18 e7 mrna转录水平明显降低,说明其靶向敲除hpv18 e7基因的效果优于转染等量crispr/cas9
‑
e7质粒的细胞组、转染等量crispr/cas9
‑
e7质粒和共转非同源ssodn的细胞组。
[0112]
实施例5、扩增子测序进一步评估共转crispr/cas9
‑
e7质粒和非同源dsodn对靶基因的编辑效率
[0113]
以实施例3中提取的基因组dna为模版,根据靶点设计引物并进行pcr反应。本实验使用试剂为kapabiosystems公司的kapahifi hotstart readymix(货号kk2602)。
[0114]
(1)引物的设计
[0115]
根据靶点设计引物,引物两端分别位于靶点两侧。本实施例中对应的引物序列为:
[0116]
e7
‑
ngs
‑
f:
[0117]
acactctttccctacacgacgctcttccgatcttgcatggacctaaggcaaca(seq id no.9)。
[0118]
e7
‑
ngs
‑
r:gtgactggagttcagacgtgtgctcttccgatctgctcaattctggcttcacact(seq id no.10)。
[0119]
(2)第一轮pcr
[0120]
反应体系:
[0121]
nuclease
‑
free h2o补齐至25ulkapa hifi hotstart readymix12.5ule7
‑
ngs
‑
f(10μm)0.75ule7
‑
ngs
‑
r(10μm)0.75ulgenome dna1ugtotal25ul
[0122]
反应条件:98℃for 3min;25cycles of(98℃for 20s,65℃for 15s,72℃for 15s),72℃for 1min,4℃for∞
[0123]
(3)第二轮pcr
[0124]
反应体系:
[0125]
nuclease
‑
free h2o补齐至25ul
kapahifi hotstart readymix12.5ulnebnext i5 primer(10μm)2.5ulnebnext i7 primer(10μm)2.5uldna from pcr12ultotal25ul
[0126]
反应条件:98℃for 3min;11cycles of(98℃for 20s,65℃for 15s,72℃for 15s),72℃for 1min,4℃for∞
[0127]
(4)第二轮pcr扩增产物切胶回收,定量,进行测序及数据分析。
[0128]
图6实验结果显示,共转crispr/cas9
‑
e7质粒和非同源dsodn的hela细胞组indel%明显高于转染等量crispr/cas9
‑
e7质粒的细胞组、转染等量crispr/cas9
‑
e7质粒和非同源ssodn的细胞组,其中一部分是由于odn的整合插入。
[0129]
当sgrna为e7
‑
sgrna1时,共转crispr/cas9
‑
e7质粒和非同源dsodn的hela细胞组indel%可达40%,相比转染等量crispr/cas9
‑
e7质粒的细胞组、转染等量crispr/cas9
‑
e7质粒和非同源ssodn的细胞组分别提高了25%左右,其中代表断点处有odn插入占总插入缺失(indel)的66%左右;当sgrna为e7
‑
sgrna2时,共转crispr/cas9
‑
e7质粒和非同源dsodn的hela细胞组indel%可达65%左右,相比转染等量crispr/cas9
‑
e7质粒的细胞组、转染等量crispr/cas9
‑
e7质粒和非同源ssodn的细胞组分别提高了20%左右,其中“odn%”代表断点处有odn插入占总插入缺失(indel)的17%左右。crispr/cas9
‑
e7质粒共转非同源dsodn可显著增强靶向敲除人乳头瘤病毒e7基因的效率。
[0130]
实施例6、共转染的dsodn作为标记物监测的脱靶情况
[0131]
1)guide
‑
seq建库流程示意图见图7。
[0132]
2)准备y型接头:
[0133]
y型接头是通过将miseq通用寡核苷酸(miseq common adapter)分别与每个样品条形码接头引物(a##adapter)进行退火而制成的:其中,miseq通用寡核苷酸接头序列为p
‑
gatcggaagagc*c*a(“p”表示磷酸化,“*”表示硫甙修饰),样品条形码接头引物序列为:
[0134]
aatgatacggcgaccaccgagatctacactagatcgcnnwnnwnnacactctttccctacacgacgctcttccgatc*t
[0135]
其中,“nnwnnwnn”为分子index标签,“*”表示硫甙修饰。
[0136]
准备y型接头的退火反应体系为:
[0137][0138]
3)准备dna样品:
[0139]
电转3天后收细胞,按照前述抽提dna的方法抽提dna,dna采用qubit测定浓度,
a260/280需在1.8
‑
2.0之间为合格,浓度大于20ng/μl、总量大于1μg为合格。
[0140]
使用1xte缓冲液(即10mm tris
‑
hcl,无edta)稀释到最终体积为120μl。
[0141]
根据covaris s2仪器的标准操作方案,将每个样品的dna打断至平均长度为500bp。
[0142]
4)打断的dna样本纯化:
[0143]
将纯化磁珠提前拿至室温,震荡混匀室温孵育30min后再使用。
[0144]
将打断后的样品转入1.5ml专用纯化ep管中,加入120μl磁珠。
[0145]
轻轻吸打混匀6次,室温静置孵育10min,将pcr管置于磁力架上3min使溶液澄清。
[0146]
移除上清,pcr管继续放置在磁力架上,向pcr管内加入200μl新鲜配制的80%乙醇溶液,静置30s。
[0147]
移除上清,再次向pcr管内加入200μl80%乙醇溶液,静置30s后彻底移除上清。
[0148]
室温静置5min,使残留乙醇彻底挥发。
[0149]
加入20μl的1x te缓冲液,轻轻吸打重悬磁珠后移开磁力架,室温静置2min。
[0150]
将pcr管置于磁力架上2min使溶液澄清。
[0151]
用移液器小心吸取15μl上清液(磁珠洗脱后,吸取上清液时不要吸取磁珠),转移到新的pcr管中,标记好样本,用于下一步反应。
[0152]
5)末端修复
[0153]
在200μl pcr管中,添加以下(每个反应),反应体系为:
[0154][0155]
6)加a尾和连接
[0156]
在上一步完成的反应管中,添加以下(每个反应),反应体系为:
[0157][0158]
7)加a尾后的产物纯化
[0159]
将peg/naclsolution预先拿出来恢复到室温。
[0160]
每个样品中加入0.9x(也即是22.95μl)的peg/naclsolution后,充分混匀,转移到1.5ml低吸附管中,室温孵育15min后按照常规纯化步骤进行纯化。
[0161]
每个磁珠样品中加入12μl1xte缓冲液进行洗脱后,转移到标注好的pcr小管。
[0162]
8)第一轮pcr及纯化
[0163]
在200μl的反应管中,添加以下(每个反应),反应体系为:
[0164][0165]
所对应的引物使用如下:
[0166]
p5
‑
1的引物序列为:aatgatacggcgaccaccgagatcta(seq id no.11),由苏州金维智公司合成。
[0167]
gsp1 primer:由gsp1(+)和gsp1(
‑
)混合而成,gsp1(+)引物的序列为:ggatctcgacgctctccctgtttaattgagttgtcatatgttaataac(seq id no.12);gsp1(
‑
)引物的序列为:ggatctcgacgctctccctataccgttattaacatatgaca(seq id no.13)。
[0168]
第一轮pcr的磁珠纯化:
[0169]
将peg/naclsolution预先拿出来恢复到室温。
[0170]
每个样品中加入1.2x(也即是36μl)的peg/naclsolution后,充分混匀,转
移到1.5ml低吸附管中,室温静置孵育15min后按照常规纯化步骤进行纯化。
[0171]
每个磁珠样品中加入15μl的1xte缓冲液进行洗脱。
[0172]
第二轮pcr及纯化
[0173]
所对应的引物使用如下:
[0174]
p5
‑
2的引物序列为:aatgatacggcgaccaccgagatctacac(seq id no.14),由苏州金维智公司合成。
[0175]
gsp2 primer的序列:由gsp2(+)和gsp2(
‑
)混合而成,gsp2(+)引物的序列为:cctctctatgggcagtcggtgatacatatgacaactcaattaaac(seq id no.15);gsp2(
‑
)引物的序列为:cctctctatgggcagtcggtgatttgagttgtcatatgttaataacggta(seq id no.16)。
[0176]
p7
‑
#引物的序列为:caagcagaagacggcatacgagat(nnnnnnnn)
[0177]
gtgactggagtcctctctatgggcagtcggtga,其中“nnnnnnnn”为8bp的barcode序列,用于区分样品。
[0178]
在200μl的反应管中,添加以下(每个反应),反应体系为:
[0179][0180]
第二轮pcr的磁珠纯化步骤:
[0181]
将peg/naclsolution预先拿出来恢复到室温。
[0182]
每个样品中加入0.7x(也即是21μl)的peg/naclsolution后,充分混匀,转移到1.5ml低吸附管中,室温静置孵育15min后按照常规纯化步骤进行纯化。
[0183]
每个磁珠样品中加入30μl的1xte缓冲液进行洗脱。
[0184]
第二轮pcr纯化后的样品,先使用qubit定量,根据a260/280查看文库dna的纯度,1.8~2.0之间为合格。用于后续的高通量测序上机。
[0185]
10)上机测序:
[0186]
qpcr文库定量:将每个子文库稀释到4nm然后等体积混合成pool。
[0187]
文库平衡:用25%文库比例的phix进行文库平衡:先将phix稀释至4nm(原浓度为10nm),然后以体积比phix文库(4nm):pool文库=1:3进行混合得到25%phix的pooling文库。具体地,就是phix取0.8μl原液(即8nm)加入1.2μl水,phix总的就2μl。phix文库(4nm):pooling文库=2μl:6μl,总共8μl体积。
[0188]
文库变性:从上一步的8μl中取5μl,与5μl 0.2m的naoh混合变性5min(此时文库浓度为2nm)。
[0189]
文库稀释:上一步的文库上机前需稀释成1.2pm终浓度,以得到更好的上机数据质量。稀释方法为:取10μl上步骤得到的变性文库与990μl的ht1缓冲液进行震荡混合(此时文库浓度为20pm)。然后取90μl上混合液与1410μl的ht1缓冲液进行震荡混合得到终浓度为1.2pm的上机文库。
[0190]
提前将
‑
20℃存放的上机试剂(方板)化冻1
‑
2h,化冻后需拍打除掉底部小气泡。并且。提前将flowcell从4℃取出平衡至室温,使其干燥干净。
[0191]
上机引物稀释:上机引物稀释成0.3μm的浓度。
[0192]
选择选择nextseq mid试剂盒,paired end双端测序程序,按测序仪指示步骤进行上机测序。
[0193]
11)数据分析:
[0194]
序列读取的处理和整合:将测序所得序列中,前6个碱基相同的序列,以及具有同样8个碱基的分子index标签的序列归一整合在一起,将其认定为来自同一个pcr处理前的样本。利用bwa
‑
mem软件程序,将整合后的序列比对到人类基因组参考序列(grch38)。
[0195]
分析判定脱靶位点:将比对质量≥50的reads所起始的匹配位置保留并显示,采用10bp的滑动比对窗口将比对区域进行分组。分析出包含有整合的dsodn序列的reads,然后采用基于bin
‑
consensus变异调用算法的分子index和samtools的计算机分析工具,在这些有dsodn序列的位置调用为snps和indels,然后将其比对参考基因组的非靶标序列,从而判断dsodn序列在细胞基因组上整合的具体参考坐标,从而判定为在靶或脱靶位点。
[0196]
共电转的dsodn donor监测crispr
‑
cas9系统在hpv18 e7靶点的在靶与脱靶情况。图8显示当采用single e7
‑
sgrna1靶向切割hpv18 e7基因时,在靶位点可以检测到36505条reads,并且可以检测到1个脱靶位点。当采用single e7
‑
sgrna2靶向切割hpv18 e7基因时,在靶位点可以检测到75596条reads,并且可以检测到2个脱靶位点。
[0197]
上述实施例只为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1