氟雷拉纳晶型及其制备方法与应用与流程
1.本发明涉及兽用原料药氟雷拉纳的晶型及晶型的制备方法,属于化药领域。
背景技术:
2.氟雷拉纳(fluralaner)属于异噁唑啉类化合物,2014年获欧盟和fda批准上市,用于宠物犬、猫的蜱和跳蚤感染,2019年4月,氟雷拉纳咀嚼片获准在国内上市(商品名贝卫多),用于治疗犬体表的跳蚤和蜱感染,及因跳蚤引起的过敏性皮炎的辅助治疗。
3.氟雷拉纳通过干预γ-氨基丁酸(gaba)门控氯离子通道发挥其作用。其制剂咀嚼片犬、猫口服后吸收迅速,2小时起效,4小时杀虫率80%以上,8小时杀虫率95%以上,1天内达到最大血药浓度,且食物可增强其吸收;分布广泛;半衰期约12~15天,12周给药一次。
4.氟雷拉纳溶液还用于预防和治疗红螨,但仅在欧盟获批上市,未进入国内。国内亟需易规模化生产且稳定性良好的氟雷拉纳用于治疗红螨。
5.化合物由于结晶方式的不同,可能存在多种晶型,同一药物的不同晶型可能在稳定性、溶解性等方面产生差异,进而影响药物的临床吸收与生理活性。异噁唑啉类化合物的存在多晶型现象,专利申请cn105121416a提供了一种异噁唑啉类结构化合物的多晶型现象及制备方法。
6.专利申请cn102149695a公开了氟雷拉纳存在多晶型现象,并提供了3种晶型和一种无定型形式的制备方法。cn102149695a提供了氟雷拉纳化合物的制备方法和晶型的制备方法,其中氟雷拉纳晶型ⅰ、ⅱ、ⅲ的制备方法和对应的粉末x射线衍射图谱特征如下:
7.晶型ⅰ的制备是用乙酸乙酯和甲苯溶解氟雷拉纳,滤去不溶物,然后减压蒸馏浓缩至一定体积,再加入乙酸乙酯和甲苯,升温至90℃,缓慢降温至60℃,析出后降温至0℃,搅拌1h,过滤、干燥得到;或通过晶型ⅲ在甲苯中搅拌7天,过滤,自然干燥得到;或者用无定型减压干燥得到,或用无定型样品在室温下混悬在甲苯中,搅拌7天得到;其x射线粉末衍射特征图谱如图1。
8.晶型ⅱ的制备通过氟雷拉纳溶解在四氢呋喃和水的混合溶液中敞口放置,自然挥发26天以上,所得固体过滤、干燥得到;或者用晶型ⅲ在甲醇中搅拌7天,过滤,自然干燥得到;其x射线粉末衍射特征图谱如图2。
9.晶型ⅲ的制备通过乙酸乙酯溶解氟雷拉纳,5℃下滴加到氮气保护的预冷正己烷中,5℃下搅拌30分钟,过滤,干燥得到;其x射线粉末衍射特征图谱如图3。
10.无定型非晶体通过将氟雷拉纳的dmso溶液加入到水中,搅拌,过滤,湿品的x射线粉末衍射特征图谱如图4。
11.上述晶型ⅰ、ⅱ、ⅲ制备时间长,过程繁琐;无定型产品为湿品,干燥后即发生晶型改变,不易储存,不利于氟雷拉纳的后续制剂使用
12.因此,生产上亟需提供晶型一致、稳定性良好氟雷拉纳晶型、且要求其制备方法工艺简单,操作简便。
技术实现要素:
13.本发明提供了4-[5-(3,5-二氯苯基)-5-三氟甲基-4h-1,2-噁唑-3-基]-2-甲基-n-[2-氧代-2-(2,2,2-三氟乙胺基)]乙基苯甲酰胺(氟雷拉纳)新晶型和对应的制备方法,制备得到的氟雷拉纳纯度高,结晶用时短、操作简单,便于实现生产。在确定的制备方法下,所得晶型一致、稳定;进行的稳定性试验表明,所得到的氟雷拉纳新晶型稳定性良好。
[0014]
本发明涉及一种氟雷拉纳的晶型a,所述晶型a的x射线粉末衍射的反射角2θ在4.32
°±
0.2
°
,8.68
°±
0.2
°
,18.75
°±
0.2
°
,21.12
°±
0.2
°
,21.85
°±
0.2
°
,22.28
°±
0.2
°
,22.93
°±
0.2
°
,24.91
°±
0.2
°
,26.29
°±
0.2
°
,27.19
°±
0.2
°
处具有特征峰。
[0015]
本发明的氟雷拉纳的晶型a,是氟雷拉纳的新晶型,具有稳定结晶形态,可用于氟雷拉纳的生产、存储及后续使用。
[0016]
本发明还涉及一种制备所述氟雷拉纳的晶型a的方法,其中,所述方法包括:步骤(1)将氟雷拉纳加入低级醇,加热回流;步骤(2)将所述步骤(1)中回流的氟雷拉纳溶液过滤,除去不溶物后,在室温重结晶;以及步骤(3)将所述步骤(2)重结晶的氟雷拉纳晶体过滤,干燥,得到所述氟雷拉纳的晶型a。
[0017]
本发明氟雷拉纳的晶型a的制备方法,操作简便,重结晶用时短,且能稳定、一致地得到具有稳定结晶形态的晶型a。
[0018]
作为本发明的一种实施方式,所述步骤(1)中低级醇为甲醇、异丙醇、或乙醇;所述回流温度为60℃~90℃;氟雷拉纳与所述溶剂的重量体积比为1∶2~1∶10;所述回流时间为10min-2h;优选地,所述回流时间为30min。
[0019]
作为本发明的一种实施方式,所述步骤(2)的重结晶条件为搅拌1~4小时。
[0020]
本发明的重结晶条件温和,便于操作,且用时短。
[0021]
作为本发明的一种优选实施方式,本发明氟雷拉纳的晶型a的制备采用重结晶法得到,将氟雷拉纳加入到2-10倍量的异丙醇、甲醇或乙醇等溶剂中搅拌,升温至回流(回流温度即为溶剂的沸点,例如异丙醇为溶剂时,回流温度为80
±
3℃),保持10min-2h后,为基本澄清的溶液,趁热过滤除去不溶性杂质,自然降温至室温,降温过程持续30min-1h,可见有大量固体析出,室温下继续搅拌1-4h,过滤,滤饼在50℃下鼓风干燥4-12h,得到白色固体粉末,即为氟雷拉纳晶型a。
[0022]
本发明还涉及所述的氟雷拉纳的晶型a在制备治疗红螨的药物中的应用。
[0023]
本发明的氟雷拉纳的晶型a稳定性良好,有利于后续加工生产、存储及使用。
[0024]
本发明还涉及一种氟雷拉纳的晶型b,所述晶型b的x射线粉末衍射的反射角2θ在4.32
°±
0.2
°
,8.68
°±
0.2
°
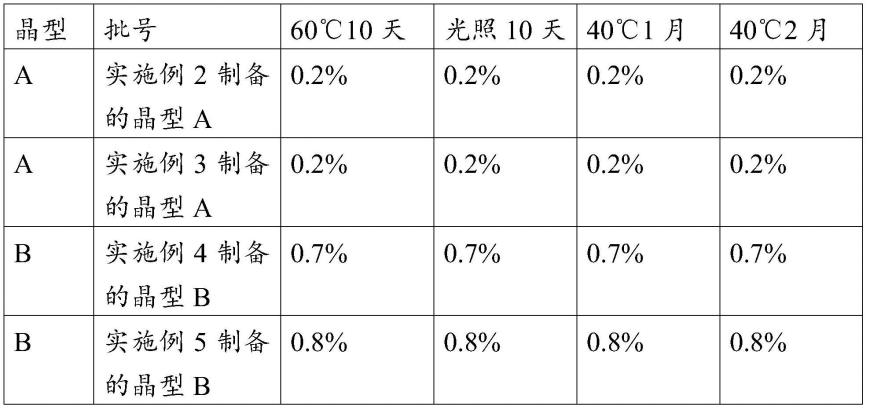
,14.44
°±
0.2
°
,16.30
°±
0.2
°
,16.84
°±
0.2
°
,19.36
°±
0.2
°
,21.18
°±
0.2
°
,21.86
°±
0.2
°
,22.32
°±
0.2
°
,23.84
°±
0.2
°
,24.43
°±
0.2
°
,26.19
°±
0.2
°
处具有特征峰。
[0025]
本发明的氟雷拉纳的晶型a,是氟雷拉纳的新晶型,具有稳定结晶形态,可用于氟雷拉纳的生产、存储及后续使用。
[0026]
本发明还涉及一种制备所述氟雷拉纳的晶型b的方法,其中,所述方法包括:步骤(1)将氟雷拉纳加入低级醇和低级烷烃的混合溶液,搅拌打浆;以及步骤(2)将所述步骤(1)中重结晶的氟雷拉纳抽滤,干燥,到所述氟雷拉纳的晶型b。
[0027]
本发明氟雷拉纳的晶型b的制备方法,操作简便,重结晶条件温和,且能稳定、一致
地得到具有稳定结晶形态的晶型b。
[0028]
作为本发明的一种实施方式,所述步骤(1)中的低级醇为异丙醇、或甲醇,所述低级烷烃为正己烷;所述低级醇和所述低级烷烃的体积比为3∶7~1∶1;氟雷拉纳与所述混合溶液的重量体积比为1∶10;所述搅拌打浆时间为24h以上。
[0029]
作为本发明的一种优选实施方式,本发明氟雷拉纳的晶型b的制备采用打浆的方法制备得到,将氟雷拉纳加入到10倍体积的正己烷和异丙醇的混合溶剂中,正己烷和异丙醇的体积比例范围为7:3-1:1,搅拌打浆24h以上,过滤,50℃下鼓风干燥4-12h,得到白色固体粉末,即为晶型b。
[0030]
本发明还涉及所述的氟雷拉纳的晶型b在制备治疗红螨的药物中的应用。
附图说明
[0031]
图1为专利cn102149695a氟雷拉纳晶型ⅰ的x射线粉末衍射图;
[0032]
图2为专利cn102149695a氟雷拉纳晶型ⅱ的x射线粉末衍射图;
[0033]
图3为专利cn102149695a氟雷拉纳晶型ⅲ的x射线粉末衍射图;
[0034]
图4为专利cn102149695a氟雷拉纳无定型形态的x射线粉末衍射图;
[0035]
图5为本发明实施例2制备的氟雷拉纳晶型a的x射线粉末衍射图;
[0036]
图6为本发明实施例3制备的另一氟雷拉纳晶型a的x射线粉末衍射图;
[0037]
图7为本发明实施例4制备的氟雷拉纳晶型b的x射线粉末衍射图;
[0038]
图8为本发明实施例5制备的另一氟雷拉纳晶型b的x射线粉末衍射图;
[0039]
图9为氟雷拉纳市售片的x射线粉末衍射图。
具体实施方式
[0040]
以下,对本发明的实施方式进行说明。
[0041]
定义
[0042]
术语“晶型”用来描述固体化合物的存在状态,描述晶体内部的离子、原子或分子组成、对称性质与周期排列规律的多种参量集合体。
[0043]
术语“熔点”(melting point)是固体将其物态由固态转变(熔化)为液态的温度,缩写为m.p.。
[0044]
术语“x-射线粉末衍射(xrpd)”是指在x-射线粉末衍射图中,纵坐标为用计数(counts)表示的衍射强度,横坐标为用度(
°
)表示的衍射角2θ。当提及图谱和/或图中数据,术语“衍射峰”是指本领域的技术人员不会归属于背景噪音的一个特征峰。
[0045]
术语“2θ或2θ角度(angle)”是指光通过晶体产生的衍射角,θ为布拉格角,单位为
°
或度,2θ的误差范围为
±
0.3或
±
0.2或
±
0.1或
±
0.05。
[0046]
术语“晶面间距(d-value,d值)”是指空间点阵选择3个不相平行的连结相邻两个点阵点的单位矢量a,b,c,它们将点阵划分成并置的平行六面体单位,称为晶面间距。
[0047]
术语“相对强度i%”是指将归属于某一晶型的一组衍射峰中的第一强峰的强度定义为100%时,其它峰的强度与第一强峰的强度的比值。
[0048]
术语“溶解度”,是指在一定的温度下,某固体物质在100克溶剂里达到饱和状态时所能溶解的质量,中国药典明确规定,溶解度是药品的一种物理性质,各品种项下选用的部
分溶剂及其在该溶剂中的溶解性能,可供精制或制备溶液时参考;对在特定溶剂中的溶解性能需作质量控制时,在该品种(检查)项下作具体规定药品的近似溶解度以下名词术语表示:
[0049]
极易溶解系指溶质1g(ml)能在溶剂不到1ml中溶解;易溶系指溶质1g(ml)能在溶剂1~不到10ml中溶解;溶解系指溶质1g(ml)能在溶剂10~不到30ml中溶解;略溶系指溶质1g(ml)能在溶剂30~不到100ml中溶解;微溶系指溶质1g(ml)能在溶剂100~不到1000ml中溶解;极微溶解系指溶质1g(ml)能在溶剂1000~不到10000ml中溶解;几乎不溶或不溶系指溶质1g(ml)在溶剂10000中不能完全溶解。
[0050]
试验法:除另有规定外,称取研成细粉的供试品或量取液体供试品,于25℃
±
2℃一定容量的溶剂中,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,如无目视可见的溶质颗粒或液滴时,即视为完全溶解。
[0051]
下面结合具体实施例来进一步描述本发明,本发明的优点和特点将会随着描述更为清楚。但这些实施例仅是范例性的,并不对本发明的范围构成任何限制。本领域技术人员应该理解的是,在不偏离本发明的精神和范围下可以对本发明技术方案的细节和形式进行修改或替换,但这些修改和替换均落入本发明的保护范围内。
[0052]
本发明中所用的化学试剂均为分析纯,购自国药集团。本发明所述的实验方法,若无特殊说明,均为常规方法;所述的生物材料,若无特殊说明,均可从商业途径获得。
[0053]
实施例1氟雷拉纳的制备
[0054]
氟雷拉纳的制备方法简述如下:在n2保护下,4-溴-2-甲基苯甲酸甲酯、三正丁基(1-乙氧基乙烯)锡和四(三苯基膦)钯在1,4-二氧六环中回流反应,反应结束后向反应体系中加入稀盐酸,萃取、浓缩,所得浓缩液不经纯化,直接在dmso体系中,nah作用下与3,5-二氯-2,2,2-三氟苯乙酮缩合,萃取、浓缩、析晶纯化,得到的固体在碱性条件下与盐酸羟胺成环,水解,再与2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐缩合得到目标物氟雷拉纳。
[0055]
氟雷拉纳也可以通过商售得到。
[0056]
实施例2本发明氟雷拉纳晶型a的制备和鉴定
[0057]
向实施例1制备的10g氟雷拉纳中加入70ml异丙醇,搅拌下升温至82.5℃回流,回流30分钟后,过滤除去不溶物,移至室温下继续搅拌,可见晶体缓慢析出,继续搅拌1h,过滤,滤饼50℃鼓风干燥,即得氟雷拉纳晶型a7.2g,纯度99.8%。
[0058]
通过上述方法制备的氟雷拉纳晶型a的x射线粉末衍射图用x-射线粉末衍射法测定其晶面间距d、布拉格2θ角和相对强度i(以相对于最强射线的百分数表示)如图5所示。
[0059]
其特征衍射数据见表1:
[0060]
表1本发明氟雷拉纳晶型a的x-射线粉末衍射法检测结果
[0061]
2-θ晶面间距(d-value)相对强度(i%)4.3220.42100.08.6810.1850.618.754.7311.121.124.2032.221.854.0749.922.283.9914.5
22.933.8812.924.913.5723.426.293.3933.827.193.2810.4
[0062]
上述本发明提供的氟雷拉纳新晶型a的制备方法,其结晶溶剂可选甲醇、异丙醇、乙醇等低级醇类,均可得到与晶型a实质上相同的产物。
[0063]
实施例3本发明另一氟雷拉纳晶型a的制备和鉴定
[0064]
向10g实施例1制备的氟雷拉纳中加入50ml甲醇,搅拌下升温至64.7℃回流,回流30分钟,过滤除去不溶物,移至室温下继续搅拌,晶体缓慢析出,继续搅拌1h,过滤,滤饼50℃鼓风干燥,即得氟雷拉纳5.8g,纯度99.6%。
[0065]
通过上述方法制备的氟雷拉纳晶型a的x射线粉末衍射图如图6所示。
[0066]
其特征衍射数据见表2:
[0067]
表2
[0068]
2-θ晶面间距(d-value)相对强度(i%)4.3420.35100.08.6910.1738.018.774.729.319.384.5819.121.164.2079.921.874.0632.322.303.9822.022.973.8712.523.863.7320.924.963.5632.826.273.3951.227.233.2714.5
[0069]
因此,本发明的制备方法能在产业化上应用、程序简单方便,且能稳定得到氟雷拉纳新晶型a。
[0070]
与专利申请cn102149695a提供的氟雷拉纳晶型x射线粉末衍射图(参见图1~图4)对比可知,本发明的氟雷拉纳晶型a与cn102149695a的晶型均不一致。对市售氟雷拉纳片进行x-射线粉末衍射分析,所得图谱如图9所示,比较图9与本发明氟雷拉纳晶型a的x-射线粉末衍射分析,证明本发明氟雷拉纳晶型b与市售氟雷拉纳片晶型也不一致。本发明的氟雷拉纳晶型a为氟雷拉纳的新晶型。
[0071]
将实施例3得到的氟雷拉纳晶型a使用熔点仪测定,熔点为174
±
3℃。
[0072]
实施例4本发明氟雷拉纳晶型b的制备和鉴定
[0073]
向100g实施例1制备的氟雷拉纳中加入500ml异丙醇和500ml正己烷的混合溶剂中,搅拌打浆24h,抽滤,滤饼在50℃鼓风干燥,即得晶型b的氟雷拉纳92g,纯度99.3%。
[0074]
通过上述方法制备的氟雷拉纳晶型b的x射线粉末衍射图如图7所示。
[0075]
其特征衍射数据见表3:
[0076]
表3
[0077]
2-θ晶面间距(d-value)相对强度(i%)4.3220.4240.48.6810.1816.114.446.1322.616.305.4313.316.845.2637.819.364.5836.521.184.19100.021.864.0613.122.323.9827.923.843.7336.924.433.6418.126.193.4040.9
[0078]
将实施例4得到的氟雷拉纳晶型b使用熔点仪测定,熔点为174
±
3℃。
[0079]
实施例5本发明另一氟雷拉纳晶型b的制备和鉴定
[0080]
向100g实施例1制备的氟雷拉纳中加入300ml异丙醇和700ml正己烷的混合溶剂,搅拌打浆24h,抽滤,滤饼在50℃鼓风干燥,即得晶型b的氟雷.拉纳94g,纯度99.1%。
[0081]
通过上述方法制备的氟雷拉纳晶型b的x射线粉末衍射图如图8所示。
[0082]
其特征衍射数据见表4:本发明氟雷拉纳a型和b型的衍射峰强度有较大差异,晶型a在2θ值4.34
±
0.2处有衍射最强峰;而晶型b最强峰在2θ值21.19
±
0.2处。
[0083]
其特征衍射数据见表4:
[0084]
表4
[0085]
2-θ晶面间距(d-value)相对强度(i%)4.3420.3442.68.7010.1515.814.466.1222.316.325.438.916.855.2636.919.364.5835.721.194.19100.021.874.0614.522.343.9828.323.863.7339.324.453.6419.826.193.4044.6
[0086]
因此,本发明的制备方法能在产业化上应用、程序简单方便,且能稳定得到氟雷拉纳新晶型b。
[0087]
实施例6本发明氟雷拉纳晶型a、晶型b的稳定性实验
[0088]
选择实施例中制备得到的样品,对所得晶型a与晶型b样品进行影响因素、加速试验判断稳定性差异,加速条件为40℃
±
2℃、rh75%
±
5%;hplc法测得的有关物质结果见表5:
[0089]
表5本发明氟雷拉纳晶型a、晶型b的稳定性实验结果
[0090][0091]
结果可知,两种晶型稳定性良好。而无定型形态的氟雷拉纳,随着放置时间的延长,水分明显降低,无法保持,稳定性差。
[0092]
本发明的氟雷拉纳新晶型具有稳定的结晶形态,有利于氟雷拉纳的生产、存储及后续使用。
[0093]
实施例7本发明氟雷拉纳晶型a、晶型b的溶解度实验
[0094]
选择实施例2、4中制备得到的样品,对所得晶型a与晶型b样品进行溶解度试验,结果见表6。
[0095]
表6本发明氟雷拉纳晶型a、晶型b的溶解度实验
[0096]
[0097][0098]
从表6结果可知,本发明氟雷拉纳晶型a在水中几乎不溶,在乙醇、异丙醇中微溶,在甲醇、乙腈中略溶,在二氧六环中溶解,在dmso、dmf中易溶;氟雷拉纳晶型b在水中几乎不溶,在乙腈、乙醇、异丙醇中微溶,在甲醇、dmf中略溶,在dmso、二氧六环中易溶。
[0099]
实施例8本发明氟雷拉纳晶型a、晶型b流动性检测
[0100]
流动性检测方法:以注入法测定物料的休止角对比粉末的流动性。在固定高度下,粉体经过两个漏斗缓慢匀速流出,落在备好的平皿上,测量其粉料堆积体的高和底面直径,计算得出休止角。
[0101]
经检测,本发明氟雷拉纳晶型a、晶型b两种晶型粉体休止角θ<30
°
;因此本发明晶型a、晶型b在生产中易于操作、储存和转移,有利于制备成各种不同的药物制剂形式,如与药学上可接受的辅料混合溶解,制成口服液体制剂;或与各种辅料混合制成口服固体制剂;或制备成固体分散体;或药学上可接受的盐,进而制成各种固体制剂。
[0102]
实施例9本发明氟雷拉纳晶型a、晶型b用于治疗红螨
[0103]
本发明氟雷拉纳晶型a或晶型b与适量的溶剂混合,加入吐温、抗氧剂搅拌分散成均一澄清溶液,加水保持澄清,饮水用于鸡的红螨的治疗。
[0104]
试验于某禽业公司一栋种鸡舍进行试验,以0.5mg/kg剂量给药1次,给药前红螨多不可数,给药后粪便传输带上布满脱落的螨虫。用瓦楞板放进50ml的ep管中,固定于鸡笼中进行螨虫的收集,给药前捕获器中螨虫重量为1.212g、0771g、0.771g,给药2天捕获器中螨虫重量为0.109g、0.136g、0.172g,给药4天捕获器螨虫重量为0.030g、0.025g、0.032g。以螨虫减少率评价更为客观准确,螨虫减少率(%)=(试验前活螨虫数-试验后活螨虫数)/试验前活螨虫数*100,给药4天螨虫减少率最低为98.1%,最高为99.6%。一次给药,螨虫减少率超过90%。当第2次给药4天后,螨虫减少率100%,达到完全灭杀的效果。
[0105]
以上所述仅是本发明的优选实施例而已,并非对本发明做任何形式上的限制,虽然本发明已以优选实施例揭露如上,然而并非用以限定本发明,任何熟悉本专业的技术人员,在不脱离本发明技术方案的范围内,当可利用上述揭示的技术内容作出些许更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1