一种用于氟离子检测的荧光探针、其用途及检测待测样品中氟离子的方法
本发明属于分析化学技术领域,具体涉及一种用于氟离子检测的荧光探针、其用途及检测待测样品中氟离子的方法。
背景技术:
氟是人体必需的微量元素之一,其在生物体内与多种生物过程有关,研究表明生物体内的氟离子浓度失调会引发多种疾病,如口腔疾病等。此外,氟化物还可作为酶抑制剂和药物。因此,在生物医学领域中,对氟离子的的检测具有重要的意义。
荧光探针法检测氟离子的原理是利用氟离子与荧光探针分子中的分子键作用,改变荧光探针分子的结构后,使荧光探针发出荧光。通过检测荧光来判断是否存在氟离子,并通过荧光强度来计算氟离子的含量。荧光探针法具有灵敏度高、选择性好和生物兼容性好等优点。
中国发明专利申请“cn110372742a一种检测氟离子的荧光探针”提供了一种用于检测氟离子的荧光探针,结构如下:
该荧光探针具有响应灵敏、选择性和抗干扰性好等优点。然而,该荧光探针分子的水溶性差,必须溶于dmso等有机溶剂使用,这限制了该荧光探针分子在生物体内的使用。
技术实现要素:
针对现有技术的困难,本发明提供了一种用于氟离子检测的荧光探针,通过对荧光探针的分子结构的改进,使得该荧光探针具有良好的水溶性、选择性高、响应速度快、检测限低、稳定性好且适用ph范围宽。
式ⅰ所示化合物、或其光学异构体、或其盐:
其中,
r选自特异性识别氟离子的基团;
l1和l2分别独立选自取代或未取代的c1-c10亚烷基,所述取代基选自c1-c10烷基、氰基、羟基、羧基、卤素或硝基。
优选的,所述r选自otips或otbdps。
优选的,所述l1和l2选自c3亚烷基。
优选的,所述式i所示化合物具体为:
本发明还提供上述化合物、或其光学异构体、或其盐作为氟离子检测荧光探针的用途。
优选的,所述化合物、或其光学异构体、或其盐在水溶液体系中使用。
优选的,所述化合物、或其光学异构体、或其盐用pbsbuffer配制成溶液使用。
本发明还提供一种检测待测样品中氟离子的方法,它是以上述化合物、或其光学异构体、或其盐作为氟离子检测荧光探针进行检测。
优选的,步骤如下:
1)使所述化合物、或其光学异构体、或其盐与待测样品或待测体系反应;
2)检测所述待测样品或待测体系的荧光强度,根据荧光强度计算待测样品中氟离子的含量。
优选的,所述化合物、或其光学异构体、或其盐在水溶液体系中使用;
和/或,所述待测样品为水溶液样品;
和/或,所述待测体系为生物体,使所述化合物、或其光学异构体、或其盐与待测体系反应的方法为:将所述化合物、或其光学异构体、或其盐注射至所述生物体内;
和/或,步骤1)中,所述反应的时间为大于等于3min。
本发明提供了的用于氟离子检测的荧光探针具有如下有益效果:
1、本发明的荧光探针水溶性好,易溶于pbsbuffer中,能够配置成10mm浓储溶液。这是的荧光分子更加易于在生物体中使用。
2、本发明的荧光探针响应快速,对氟离子的响应在3min即可完成并且趋于稳定。
3、本发明的荧光探针选择性高、稳定性好且适用的ph条件宽泛。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
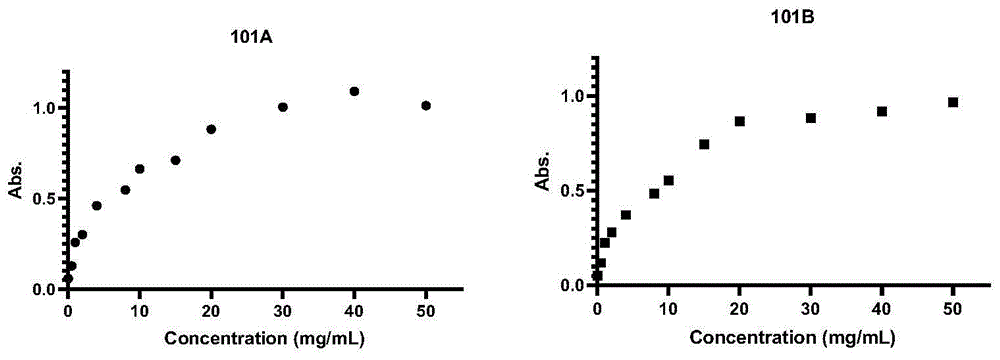
图1为探针101a、101b在pbs中的溶解性实验结果;
图2为探针101a、101b(10μm)与不同浓度naf共同孵育5min的荧光光谱图;
图3为探针101a、101b(10μm)与不同浓度naf共同孵育5min,最大发射波长下的荧光强度曲线图;
图4为探针101a、101b(10μm)与不同浓度naf共同孵育5min;
图5为探针101a、101b(10μm)和naf(100μm)响应不同时间的荧光强度光谱图;
图6为探针101a、101b(10μm)与naf(100μm)响应不同时间,最大发射波长下荧光强度曲线图;
图7为探针101a、101b(10μm)和不同阴离子(100μm)共同孵育5min的荧光强度光谱图;
图8为探针101a、101b(10μm)和不同阴离子(100μm)共同孵育5min,在最大发射波长下荧光强度柱状图;
图9为探针101b(10μm)在不同ph(5.5、7.0、7.4、8.5)pbs缓冲液中对naf(100μm)响应强度变化的光谱图;
图10为探针101b(10μm)在最大发射波长下与不同ph(5.5、7.0、7.4、8.5)下对naf(100μm)响应强度变化的折线图。
图11为探针101b(10μm)在pbs(ph7.4)缓冲液中10h内的荧光强度光谱图;
图12为探针101b(10μm)在最大发射波长下在pbs(ph7.4)缓冲液中10h内的荧光强度的折线图。
具体实施方式
以下实施例和实验例中所用的试剂和材料均为市售。
实施例1化合物101a的合成
本实施例合成化合物101a,结构式如下:
合成反应式如下:
具体步骤为:
(1)化合物2的合成
室温下向化合物1(10.0g,81.89mmol,1eq)和甲醛(6.2ml,81.89mmol,1eq)的hcl(浓,70ml)溶液中,通入hcl(g)3小时,产生黄色沉淀。过滤,并用水(50ml×3)洗涤。将滤饼溶于乙醚中,用na2so4干燥,过滤浓缩除去大部分有机溶液,然后冷却至室温,加入石油醚(60℃),产生白色针状晶体。过滤混合物并用石油醚洗涤,真空干燥,得到化合物2(6.0g,35.0mmol,收率42.7%).1hnmr(400mhz,cdcl3)δ11.08(s,1h),9.90(s,1h),7.65–7.42(m,2h),7.01(d,j=8.4hz,1h),4.60(s,2h);13cnmr(100mhz,cdcl3)δ196.2,161.66,137.37,133.66,129.23,120.37,118.36,45.25.
(2)化合物3的合成
将化合物2(14.5g,84.47mmol,1eq)和六亚甲基四胺(15.5g,109.48mmol,1.3eq)在acoh(85ml,50%)中的混合,115℃下搅拌1h,加入hcl(浓)38ml,在相同温度下搅拌5分钟。反应溶液用冰浴冷却至0℃,产生浅黄色沉淀。过滤混合物,滤饼用冰水(50ml×2)洗涤,然后将滤饼溶于dcm(50ml)中,用na2so4干燥,过滤浓缩,得到浅黄色固体化合物3(6.0g,40.0mmol,收率47.3%).1hnmr(400mhz,cdcl3)δ11.57(s,1h),9.90(s,1h),8.00-8.20(m,1h),7.14(d,j=8.0hz,1h);13cnmr(100mhz,cdcl3)δ196.3,189.4,166.3,137.2,136.6,129.2120.3,118.9.
(3)化合物5的合成
将化合物4(3g,18.9mmol,1eq),1-溴丙烷(2.8g,22.6mmol,1eq)在甲苯(50ml)中混合,无光照的氮气氛下130℃搅拌24小时。产生深紫色沉淀。tlc(二氯甲烷:甲醇=8:1)监控反应完成。过滤反应混合物,用乙醚(40ml×3)洗涤。滤饼用甲基叔丁基醚和乙醚研磨,过滤并真空干燥,得到紫色粉末的化合物5(4g,14.3mmol,收率75.4%).1hnmr(400mhz,dmso-d6)δ8.06(dd,j=6.0,2.8hz,1h),7.83(dd,j=6.0,2.8hz,1h),7.66–7.59(m,2h),4.66(t,j=8.0hz,2h),2.84(s,3h),2.63(t,j=6.4hz,2h),2.16(dt,j=14.4,7.2hz,2h),1.53(s,6h);13cnmr(100mhz,dmso-d6)δ196.52,141.91,141.18,129.30,128.92,123.42,115.42,54.10,47.34,46.55,23.72,22.02,13.77.
(4)化合物7的合成
将化合物6(5.0g,41.0mmol,1eq)、氯三异丙基硅烷(11.8g,61.5mmol,1.5eq)和咪唑(4.2g,61.5mmol,1.5eq)在mecn(60ml)中混合,室温下搅拌2小时。tlc(石油醚-etoac=4:1)监控反应完成。过滤并浓缩反应混合物。将残余物溶解在etoac(50ml)中,并用水(50ml*3)、盐水(50ml*1)洗涤,用na2so4干燥,过滤并浓缩得到黄色油状化合物7(12g)。无进一步纯化,粗品直接用于下一步。1hnmr(400mhz,cdcl3)δ9.88(s,1h),7.79(d,j=8.4hz,2h),6.98(d,j=8.4hz,2h),1.33-1.24(m,3h),1.11(d,j=7.2hz,18h);13cnmr(100mhz,cdcl3)δ190.89,161.94,131.96,130.16,120.32,17.84,12.69.
(5)化合物8的合成
向化合物7(5.0g,18.0mmol,1eq)在etoh(60ml)中的溶液中添加nabh4(815.0mg,21.4mmol,1.2eq),室温下搅拌过夜。tlc(石油醚:etoac=4:1)监控反应完成。浓缩反应混合物,用水(50ml)稀释残渣,etoac(60ml*3)萃取,盐水(100ml)洗涤合并的有机相,na2so4干燥,过滤浓缩。残渣通过快速柱层析法纯化,石油醚:etoac从0%到30%洗脱,得到油状化合物8(4.5g,16.1mmol,收率84.6%).1hnmr(400mhz,cdcl3)δ7.20(d,j=8.4hz,2h),6.85(d,j=8.4hz,2h),4.57(s,2h),1.29–1.20(m,3h),1.10(d,j=7.2hz,18h);13cnmr(100mhz,cdcl3)δ155.67,133.44,128.53,119.90,65.03,17.93,12.67.
(6)化合物9的合成
将化合物8(4.5g,16.1mmol,1eq)和py(1.8ml,22.5mmol,1.4eq)在乙醚(100ml)中的溶液中混合,逐滴添加pbr3(4.4g,16.1mmol,1eq)的乙醚(20ml)溶液,在室温下搅拌反应过夜。tlc(石油醚:etoac=5:1)监控反应完成。用水(50ml)稀释反应混合物,乙醚(80ml*2)萃取,用水(100ml*2)、盐水(100ml)洗涤合并有机相,用na2so4干燥,过滤并浓缩。残渣通过快速柱层析法纯化,石油醚:etoac从2%到10%洗脱,得到油状化合物9。(2.6g,7.6mmol,收率47.2%).1hnmr(400mhz,cdcl3)δ7.18(d,j=8.8hz,2h),6.78(d,j=5.6hz,2h),4.59(s,2h),1.28–1.33(m,3h),0.94(d,j=6.4hz,18h);13cnmr(100mhz,dmso-d6)δ156.13,130.24,128.56,122.44,31.16,24.28,15.46.
(7)化合物10的合成
在无光照、室温、氮气气氛下,在化合物3(800mg、5.3mmol、1eq)、化合物9(2.0g、5.8mmol、1.1eq)和k2co3(1.5g,10.7mmol,2eq)在dmf(20ml)中的混合物进行搅拌。薄层色谱(石油醚:etoac=1:1)监控反应完成。反应混合物用水(40ml)稀释,etoac(40ml*3)萃取,用水(80ml*4)、盐水(60ml*1)洗涤,na2so4干燥,过滤浓缩。用闪蒸柱色谱纯化,其中石油醚:ac从5%到30%洗脱,得白色固体化合物10(1.6g,3.9mmol,收率73.6%).1hnmr(400mhz,cdcl3)δ10.52(s,1h),9.94(s,1h),8.34(d,j=2.4hz,1h),8.10(dd,j=8.8,2.4hz,1h),7.30(d,j=8.4hz,2h),7.21(d,j=8.8hz,1h),6.92(d,j=8.4hz,2h),5.21(s,2h),1.29-1.23(m,3h),1.12-1.09(m,18h);13cnmr(100mhz,cdcl3)δ190.23,188.72,165.20,156.60,135.56,131.91,129.69,129.14,127.25,125.09,120.28,113.69,71.08,17.91,12.66.
(8)化合物11(101a)的合成
化合物10(100.0mg,0.24mmol,1eq)、化合物12(136.0mg,0.49mmol,2eq)和naoac(40.0mg,0.49mmol,2eq)在ac2o(2ml)中的混合物在80℃、氮气气氛下、避光搅拌30min,tlc(二氯甲烷:甲醇=6:1)监控反应完成。将反应混合物浓缩。残渣通过快速柱层析纯化,二氯甲烷:甲醇从0%到11%洗脱,得到深黄色固体。etoac研磨黄色固体,过滤并在真空中干燥,得到橙色固体化合物11(160mg,0.17mmol,收率70.3%).1hnmr(400mhz,dmso-d6))δ8.98(s,2h),8.20(s,2h),7.44-7.31(m,5h),7.06(d,j=5.6hz,1h),6.88–6.67(m,7h),5.72(d,j=10.0hz,1h),5.48(d,j=9.6hz,1h),5.18(s,2h),4.12(t,j=6.8hz,4h),2.56(t,j=8.4hz,4h),1.92–1.88(m,4h),1.46–1.40(m,15h),1.11–1.02(m,18h).13cnmr(100mhz,dmso-d6)δ178.16,158.24,155.34,142.58,142.14,138.22,130.64,128.52,126.28,123.16,120.10,117.42,114.86,113.66,113.85,111.73,56.71,54.37,51.24,26.08,24.56,20.09,19.90,14.06.
实施例2化合物101b的合成
本实施例合成化合物101b,结构式如下:
合成反应式如下:
具体步骤如下:
(1)化合物12的合成
0℃下向化合物4(2.0g,16.4mmol,1eq)的dcm溶液中添加咪唑(5.6g,40.9mmol,2.5eq),室温搅拌1h。加入叔丁基氯二苯基硅烷(4.9g,18.0mmol,1.1eq),室温搅拌2h。tlc(石油醚:etoac=4:1)监控反应完成。用水(50ml*3)、盐水(50ml*1)洗涤反应混合物,用na2so4干燥,过滤并浓缩。残渣用快速柱层析法石油醚:etoac从0%到20%洗脱,得到黄色油状化合物12(5g,13.9mmol,收率84.6%).1hnmr(400mhz,cdcl3)δ9.80(s,1h),7.75-7.67(m,4h),7.64(d,j=8.4hz,2h),7.48-7.34(m,6h),6.86(d,j=8.4hz,2h),1.11(s,9h);13cnmr(100mhz,cdcl3)δ190.94,161.22,135.40,131.92,131.74,130.28,130.22,128.01,120.31,26.38,19.48.
(2)化合物13的合成
向化合物12(2.0g,5.6mmol,1eq)的etoh(20ml)溶液中添加nabh4(420.0mg,6.7mmol,1.2eq),室温下搅拌过夜。tlc(石油醚:etoac=2:1)监控反应完成。浓缩反应混合物,用水(30ml)稀释残渣,用etoac(40ml*3)萃取,盐水(60ml)洗涤合并的有机相,用na2so4干燥,过滤并浓缩。残渣通过快速柱层析法纯化,石油醚:etoac从0%到30%洗脱,得到油状化合物13(1.3g,3.6mmol,收率64.1%).1hnmr(400mhz,cdcl3)δ7.71(dd,j=8.0,1.4hz,4h),7.44-7.28(m,6h),7.06(d,j=8.4hz,2h),6.74(d,j=8.4hz,2h),4.49(s,2h),1.09(s,9h);13cnmr(100mhz,cdcl3)δ155.22,135.53,133.42,132.86,129.95,128.43,127.83,119.75,65.05,26.54,19.49.
(3)化合物14的合成
向化合物13(1.3g,3.6mmol,1eq)和py(397mg,5.0mmol,1.4eq)的乙醚(40ml)溶液中逐滴添加pbr3(970mg,3.6mmol,1eq),在室温下搅拌过夜。tlc(石油醚:etoac=4:1)监控反应完成。用水(40ml)稀释反应混合物,乙醚(40ml*2)萃取混合物,用水(60ml*2)、盐水(50ml)洗涤合并的有机相,用na2so4干燥,过滤并浓缩。残渣通过快速柱层析法纯化,石油醚:etoac从2%到15%洗脱,得到透明油状化合物14(1.3g,3.1mmol,收率85.2%).1hnmr(400mhz,cdcl3)δ7.72–7.68(m,4h),7.44–7.34(m,6h),7.11(d,j=8.4hz,2h),6.71(d,j=8.4hz,2h),4.41(s,2h),1.29-1.23(m,3h),1.09(s,9h);13cnmr(100mhz,cdcl3)δ155.77,135.49,132.62,130.25,130.01,127.85,119.96,34.04,26.47,19.46.
(4)化合物15的合成
将化合物3(450mg,3.0mmol,1eq)、化合物14(1.4g,3.3mmol,1.1eq)和k2co3(828mg,6.0mmol,2eq)在dmf(20ml)中的混合物在室温、无光、氮气气氛下搅拌2小时。tlc(石油醚:etoac=4:1)监控反应完成。用水(40ml)稀释反应混合物,etoac(40ml*3)萃取混合物,用水(60ml*4)、盐水(60ml*1)洗涤合并的有机相,用na2so4干燥,过滤并浓缩。残渣通过快速柱层析法纯化,石油醚:etoac从2%到40%洗脱,得到淡黄色固体化合物15(1.0g,2.0mmol,收率66.7%).1hnmr(400mhz,cdcl3)δ10.47(s,1h),9.93(s,1h),8.32(d,j=2.0hz,1h),8.08(dd,j=8.8,2.0hz,1h),7.73-7.69(m,4h),7.46-7.41(m,2h),7.39-7.34(m,4h),7.20-7.13(m,3h),6.80(d,j=8.4hz,2h),5.14(s,2h),1.11(s,9h);13cnmr(100mhz,cdcl3)δ190.22,188.67,165.14,156.07,135.51,132.60,131.92,130.04,129.68,128.96,127.86,127.32,125.07,120.12,113.67,71.01,26.49,19.47.
(5)化合物16(101b)的合成
将化合物15(200.0mg,0.4mmol,1eq)、化合物5(228.0mg,0.8mmol,2eq)和naoac(66.0mg,0.8mmol,2eq)的ac2o(4ml)中的混合物在80℃、氮气气氛、避光搅拌30min,tlc(二氯甲烷:甲醇=8:1)监控反应完成。浓缩反应混合物,通过快速柱层析法纯化残余物,二氯甲烷:甲醇从0%到12%洗脱,得到黄色固体。然后,用etoac研磨黄色固体,过滤并在真空中干燥,得到黄色固体化合物16(110mg,0.106mmol,收率26.3%).1hnmr(400mhz,dmso-d6)δ9.62(s,1h),8.73-8.50(m,3h),8.18(d,j=16.4hz,1h),7.97-8.06(m,3h),7.96-7.91(m,1h),7.89-7.84(m,1h),7.75-7.68(m,6h),7.67–7.63(m,2h),7.58(d,j=9.2hz,1h),7.52-7.41(m,8h),6.85(d,j=8.4hz,2h),5.36(s,2h),4.90(t,j=6.8hz,2h),4.73(t,j=6.8hz,2h),1.94-1.80(m,10h),1.70(s,6h),1.10-1.04(m,12h),0.94(t,j=7.2hz,3h);13cnmr(100mhz,dmso-d6)δ182.18,181.99,162.07,155.23,152.70,147.17,143.94,143.69,140.76,137.64,135.03,133.99,131.92,130.30,129.85,129.73,129.71,129.37,129.24,129.08,128.56,128.09,127.90,123.51,123.12,119.45,115.68,115.35,114.61,114.36,52.27,52.20,48.31,47.87,26.03,25.91,21.96,21.85,18.94,14.06,40.73,10.64.
实验例1101a和101b水溶性实验
分别将不同量的探针101a、101b加入10mmpbs(ph7.4)混合溶解,得到充分溶解后的溶液。检测各组紫外可见吸收,检测条件:激发波长600nm,狭缝3,发射波长630-900nm。
测试结果如图1所示。结果表明,101a最大溶解度在30-40mg/ml,101b最大溶解度在20-30mg/ml,水溶性良好。
实验例2探针与naf浓度依赖性实验
(1)实验方法
分别将探针101a、101b加入10mmpbs(ph7.4)充分溶解,最终配制成100μm的工作液。然后将naf用上述pbs配制成1mm的溶液。
对照组:分别取探针101a、101b与等体积pbs共同孵育5min;实验组:分别取探针101a、101b工作液与等体积不同浓度naf溶液共同孵育。检测各组紫外可见吸收,检测条件:激发波长600nm,狭缝3,发射波长630-900nm。
(2)实验结果
图2是探针101a、101b(10μm)与不同浓度naf共同孵育5min的荧光光谱图;图3是101a、101b(10μm)与不同浓度naf共同孵育5min,最大发射波长下的荧光强度曲线图;图4是101a、101b(10μm)与不同浓度naf共同孵育5min,最大发射波长下的荧光强度-浓度线性关系图。从图中可以看到,荧光强度与naf浓度在0-100μm范围内呈很好的线性关系。
根据公式lod=3σ/s可计算检测限,其中:σ是空白值的标准偏差,s为待测物含量趋近于0时,检测到的信号差值对待测物含量的斜率。经计算,探针101a、101b对氟离子的检测限分别为2.3×10-8m和1.8×10-9m。
实验例3探针与naf反应最佳时间探索实验
(1)实验方法
分别将探针101a、101b加入10mmpbs(ph7.4)充分溶解,最终配制成80μm的浓储液。然后将naf用上述pbs配制成1mm的溶液。
对照组:分别取探针101a、101b与pbs缓冲液等体积混合;实验组:分别取探针101a、101b(10μm)与等体积naf(1000μm)溶液。检测条件:激发波长600nm,狭缝3,发射波长630-900nm。根据探针响应特性不同时间采一次样。
(2)实验结果
图5是探针101a、101b(10μm)和naf(100μm)响应不同时间的荧光强度光谱图;图6是探针101a、101b(10μm)与naf(100μm)响应不同时间,最大发射波长下荧光强度曲线图。从图中可以看到,探针101a和101b对氟离子的响应在3min即可完成并且趋于稳定,响应非常迅速。
实验例4探针的离子选择性
(1)实验方法
将探针101a、101b加入10mmpbs(ph7.4)充分溶解,配制成80μm的浓储液,方法同上,同时分别用10mmpbs(ph7.4)将不同的阴离子(hco3-、h2po4-、hcoo-、oh-、hso4-、i-、so3-、no2-、no3-、cl-、hpo4-、f-)配制成1000μm的溶液。
对照组:分别取探针101a、101b与pbs等体积混合均匀;实验组::分别取探针101a、101b与不同阴离子溶液(1000μm)等体积混合均匀。检测条件同上,孵育5min。
(2)实验结果
图7是探针101a、101b(10μm)和不同阴离子(100μm)共同孵育5min的荧光强度光谱图;图8是探针101a、101b(10μm)和不同阴离子(100μm)共同孵育5min,在最大发射波长下荧光强度柱状图。从图中可以看出,探针101a、101b对氟离子具有很高的选择性,对氟离子的相应强度约为其他干扰离子的16-50倍。
实验例5不同ph下的响应性检测
(1)实验方法
取探针101b分别用pbs-ph4.5、pbs-ph6.0、pbs-ph7.4、pbs-ph10.5溶解配成100μm的工作液,同时与相对应的naf(1000μm)等体积均匀混合,孵育5min。检测条件同上。
(2)实验结果
图9为探针101b(10μm)在不同ph(5.5、7.0、7.4、8.5)pbs缓冲液中对naf(100μm)响应强度变化的光谱图;图10为探针101b(10μm)在最大发射波长下与不同ph(5.5、7.0、7.4、8.5)下对naf(100μm)响应强度变化的折线图。从图中可以看到,ph的变化对荧光强度的影响较小,探针101b的ph适用范围较宽。
实验例6在10mmpbs(ph7.0)中的稳定性实验
(1)实验方法
取探针101b用pbs(ph7.4)配成浓度为10μm的溶液。检测条件同上,每隔1小时测一次。
(2)实验结果
图11为探针101b(10μm)在pbs(ph7.4)缓冲液中10h内的荧光强度光谱图;图12为探针101b(10μm)在最大发射波长下在pbs(ph7.4)缓冲液中10h内的荧光强度的折线图。从图中可以看到,在24小时内,荧光强度并没有发生显著的降低,可见探针101b稳定性很好。
从上述实施例和实验例中可以看到,本申请提供了一种新的用于检测氟离子的荧光探针,该荧光探针具有良好的水溶性、选择性高、响应速度快、检测限低、稳定性好且适用ph范围宽,具有很好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!