一种莪术醇酯化物、制备方法及其在治疗结直肠癌药物中的应用
1.本发明属于天然药物及药物化学领域,具体涉及一种莪术醇酯化物及其制备方法,以及在制备 治疗结直肠癌药物中的应用。
背景技术:
2.结肠直肠癌(crc)是当今最常见的疾病之一,每年全球有约120万名患者被确诊为结直肠癌, 而有超过60万名患者直接或间接死于结直肠癌。此外,结直肠癌的发病率会随着年龄的增大而增加, 比如发达国家的结直肠癌发病中位年龄为70岁。虽然遗传因素是结直肠癌的的危险因素,但大部分 结直肠癌都是散发的,并在几年内以腺瘤
‑
肿瘤的形式发生。当前结直肠癌最主要的治疗手段是外科 手术、新辅助放射治疗(患者是直肠癌)以及辅助化疗(患者为iii、iv期或高风险的ii期结肠癌)。 在生存期方面,i期患者的5年生存率可达90%以上,而iv期患者只有略大于10%的生存率。随着城市 现代化程度提高,人民生活水平的提高,生活方式及饮食结构的改变,结直肠肿瘤高发的问题将越 来越突出,值得引起我们的关注。
3.我国有着丰富的中药资源,近年来,研究者对具有肿瘤活性的中药进行了大量的筛选,表明抗 肿瘤中药有调节机体免疫功能、抑制肿瘤微血管生成、直接杀伤肿瘤细胞、诱导肿瘤细胞凋亡、诱 导肿瘤细胞分化、逆转癌细胞的多重耐药、调节细胞信号传导、抑制端粒酶活性等多种作用机制, 且中药具有多靶点、多环节等西药难以具备的特性,药理作用广泛,同一种中药常常从整体调节, 提高机体自身抗病能力,通过多种机制达到抗肿瘤的作用。随着对中药抗肿瘤作用实验和临床研究 的不断深入,中药抗肿瘤作用越来越受到国际社会的认可。
4.莪术醇作为一种愈创木烷型倍半萜天然产物,在抗肿瘤研究方面表现出确切的疗效和潜在的应 用前景,备受药物化学家们的青睐。研究表明,莪术醇可以通过调节细胞基因的表达、抑制核酸代 谢、抑制细胞增殖、诱导细胞分化以及抑制肿瘤细胞的转移扩散、增强免疫等机制发挥作用。目前 已经鉴定出多种与莪术醇及其衍生物相关的潜在的信号传导途径,如激活pten/pi3k/akt途径、抑 制akt/gsk3β/cyclin d1途径等。(参阅文献[1]wei w,azhar rasul a s,sarfraz i,et al.curcumol:from plant roots to cancer roots[j].international journal of biological sciences,2019,15(8):1600.[2]sheema hashem,sabah nisar,geetanjali sageena,et al.therapeutic effects of curcumol in several diseases;an overview[j].nutrition and cancer,2020,73(2),181
‑
195)
[0005]
尽管现有研究已表明莪术醇具有一定的抗肿瘤生物活性,且具有较好的安全性,但是目前研究 仍存在一些问题:(1)莪术醇的生物利用度较低,其较弱的药理活性及选择性阻碍了进一步的药理 学研究,也未能清楚阐述莪术醇衍生物的构效关系;(2)莪术醇水溶性差,导致药代动力学不易开 展;许多研究组通过对莪术醇c
‑
14位环外双键或骨架结构的改造,得到了多个活性较好的莪术醇衍 生物。通过结构优化,引入活性片段后的莪术醇
衍生物,可能会增加与靶点结合的可能。
技术实现要素:
[0006]
本发明目的在于提供一种莪术醇酯化物及其制备方法,以及在制备治疗结直肠癌药物中的应用。 该莪术醇酯化物在有效剂量下对人结直肠癌细胞株sw620、hct116细胞株具有很好的抑制效果。
[0007]
本发明的技术方案如下:
[0008]
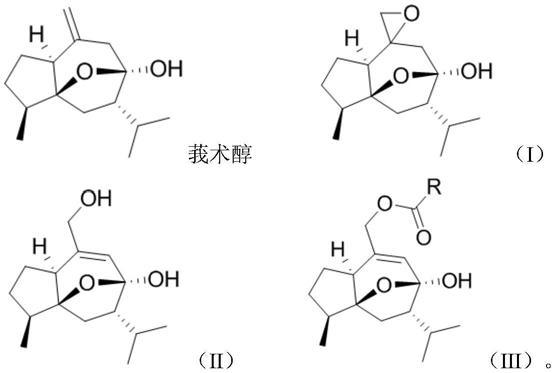
一种莪术醇酯化物,结构如式(iii)所示:
[0009][0010]
式(iii)中,r为c1~c4烷基、c2~c4烯基、卤素取代的苯基、苯乙烯基或c4~c6杂环基;优选 甲基、乙烯基、丙烯基、苯乙烯基、4
‑
氟苯基、吡啶基或噻唑基。
[0011]
式(iii)所示的莪术醇酯化物的合成方法为:
[0012]
(1)将天然产物莪术醇溶于有机溶剂中,在
‑
5~0℃下加入间氯过氧苯甲酸(m
‑
cpba),接着 于0~50℃反应1~6h,之后反应液经后处理,得到化合物(i);
[0013]
所述莪术醇与间氯过氧苯甲酸的物质的量之比为1:2~5,优选1:2~3;
[0014]
所述有机溶剂为四氢呋喃、二氯甲烷、氯仿、1,2
‑
二氯乙烷、甲苯、乙腈或1,4
‑
二氧六环,优选 四氢呋喃或二氯甲烷;所述有机溶剂的体积用量以莪术醇的质量计为10~50ml/g;
[0015]
所述后处理的方法为:反应结束后,反应液加水,乙酸乙酯萃取,合并有机相,用饱和氯化钠 溶液洗涤,浓缩,柱层析(石油醚:乙酸乙酯=4:1,体积比)分离纯化,得到化合物(i);
[0016]
(2)将化合物(i)溶于有机溶剂中,在30~100℃、搅拌条件下,加入碱性物质反应1~10h, 之后反应液经后处理,得到化合物(ii);
[0017]
所述化合物(i)与碱性物质的物质的量之比为1:0.2~1,优选1:0.5~0.8;
[0018]
所述碱性物质为氢化钠、甲醇钠、乙醇钠、吡啶、叔丁醇钾、氢氧化钠或氢氧化钾,优选氢氧 化钠或氢氧化钾;
[0019]
所述有机溶剂为甲苯、四氢呋喃、乙醇、甲醇或1,4
‑
二氧六环,优选乙醇或甲醇;所述有机溶剂 的体积用量以化合物(i)的质量计为10~50ml/g;
[0020]
优选反应温度为70~80℃,反应时间为5~8h;
[0021]
所述后处理的方法为:反应结束后,冷却至室温,将反应液倒入冰水中,静置1~3h,析出白色 固体,过滤得到化合物(ii);
[0022]
(3)化合物(ii)与酯化试剂发生酯化反应,得到产物(iii);
[0023]
所述酯化试剂为酰氯化合物rcocl或羧酸化合物rcooh;rcocl或rcooh中,r的定义与式 (iii)中相同;
[0024]
进一步:
[0025]
当r为甲基或乙烯基时,酯化反应的操作方法如下:
[0026]
将化合物(ii)溶于有机溶剂中,加入酰氯化合物rcocl,在0~70℃下搅拌反应2~10h,之后 反应液经后处理,得到产物(iii);
[0027]
所述化合物(ii)与酰氯化合物的物质的量之比为1:1~1.5,优选1:1~1.1;
[0028]
所述有机溶剂为四氢呋喃、二氯甲烷、氯仿、1,2
‑
二氯乙烷、甲苯、乙酸乙酯、乙腈或1,4
‑
二氧 六环,优选二氯甲烷或四氢呋喃;所述有机溶剂的体积用量以化合物(ii)的质量计为50~100ml/g;
[0029]
优选反应温度为10~25℃,反应时间为2~4h;
[0030]
所述后处理的方法为:反应结束后,反应液加水,乙酸乙酯萃取,合并有机相,用无水硫酸钠 干燥,浓缩,柱层析(石油醚:乙酸乙酯=8:1,体积比)分离纯化,得到产物(iii);
[0031]
当r为丙烯基、苯乙烯基、4
‑
氟苯基、吡啶基或噻唑基时,酯化反应的操作方法如下:
[0032]
将化合物(ii)溶于有机溶剂中,加入二环己基碳二酰亚胺(dcc,缩合剂)和4
‑
二甲氨基吡啶 (dmap,碱),再加入羧酸化合物rcooh,在0~70℃下搅拌反应2~10h,之后反应液经后处理, 得到产物(iii);
[0033]
所述化合物(ii)与羧酸化合物的物质的量之比为1:1~1.5,优选1:1~1.1;
[0034]
所述化合物(ii)与二环己基碳二酰亚胺、4
‑
二甲氨基吡啶的物质的量之比为1:1:1;
[0035]
所述有机溶剂为四氢呋喃、二氯甲烷、氯仿、1,2
‑
二氯乙烷、甲苯、乙腈或1,4
‑
二氧六环,优选 四氢呋喃或二氯甲烷;所述有机溶剂的体积用量以化合物(ii)的质量计为50~100ml/g;
[0036]
优选反应温度为10~25℃,反应时间为2~4h;
[0037]
所述后处理的方法为:反应结束后,反应液加水,乙酸乙酯萃取,合并有机相,用无水硫酸钠 干燥,浓缩,柱层析(石油醚:乙酸乙酯=8:1,体积比)分离纯化,得到产物(iii);
[0038][0039]
本发明式(iii)所示的莪术醇酯化物具有抑制人结直肠癌细胞株sw620和hct116活性的作用, 可用于制备治疗结直肠癌的药物。
[0040]
与现有技术相比,本发明具有以下有益效果:
[0041]
(1)本发明通过对莪术醇c
‑
14位环外双键进行结构修饰,通过形成酯键引入不同
的片段,体外 细胞实验显示,该类莪术醇衍生物对结肠癌细胞sw620和hct116细胞株表现出良好的生物活性,可 用于预防或/和治疗结直肠癌,在医药领域具有一定的应用前景。
[0042]
(2)本发明公开的c
‑
14位改造的莪术醇衍生物的合成方法简便、反应条件温和、易于操作,且 合成过程中原料易得、生产成本较低,适于工业化生产应用。
附图说明
[0043]
图1为实施例1得到的化合物3
‑
1的核磁谱图。
[0044]
图2为实施例2得到的化合物3
‑
2的核磁谱图。
[0045]
图3为实施例3得到的化合物3
‑
3的核磁谱图。
[0046]
图4为实施例4得到的化合物3
‑
4的核磁谱图。
[0047]
图5为实施例5得到的化合物3
‑
5的核磁谱图。
[0048]
图6为实施例6得到的化合物3
‑
6的核磁谱图。
[0049]
图7为实施例7得到的化合物3
‑
7的核磁谱图。
具体实施方式
[0050]
下面通过具体实施例进一步描述本发明,但本发明的保护范围并不仅限于此。
[0051]
以下实施例中原料莪术醇厂家:江西吉安中香天然植物有限公司,白色固体,纯度98%以上。
[0052]
实施例1:c
‑
14位改造的莪术醇衍生物3
‑
1的合成
[0053][0054]
(1)取莪术醇(5.0g,21.15mmol),溶于50ml二氯甲烷中,冰水浴条件下,分批加入m
‑
cpba (间氯过氧苯甲酸)(7.30g,42.31mmol),30min加毕,转移至室温25℃下搅拌3h,通过tlc 检测反应,直到反应完全,浓缩反应液,加入饱和碳酸氢钠除去残余的间氯过氧苯甲酸,用乙酸乙 酯萃取,合并乙酸乙酯相,饱和氯化钠水溶液洗涤乙酸乙酯层三次,无水硫酸钠干燥,减压浓缩, 得化合物1
‑
1(4.87g),为淡黄色油状产物1
‑
1(无需纯化直接用于下一步反应),收率为91.22%。
[0055]
(2)取化合物1
‑
1(2.0g,7.93mmol),溶于20ml乙醇中,加热至70℃搅拌,加入氢氧化 钠(0.17g,4.25mmol),加热回流2h,自然冷却至室温,通过tlc检测直到反应完全,加入至 100ml冰水浴中,静置析出白色固体,得化合物2
‑
1(456.80mg),收率78.84%。1h nmr
(500mhz, cd3od)δ5.80(d,j=1.2hz,1h),4.01(d,j=1.4hz,2h),2.21(dd,j=12.6,10.9hz,1h),2.10(t,j= 8.7hz,1h),1.96(m,3h),1.87(m,1h),1.62(m,3h),1.56(dd,j=12.7,7.4hz,1h),1.01(d,j=6.5hz, 3h),1.29(d,j=6.5hz,3h),0.91(d,j=6.6hz,3h).
[0056]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml干燥的dcm中,在室温条件下,向反应 瓶中缓慢滴加乙酰氯(80mg,1.03mmol),室温下搅拌3h。tlc检测(pe:ea=4:1)反应结束, 加水淬灭反应,乙酸乙酯萃取(3
×
100ml),无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状 粗品,用硅胶柱层析纯化产物(pe:ea=8:1),以88.30%的收率得到黄色油状物化合物3
‑
1。yellow oil.yield:88.30%.1h nmr(400mhz,chloroform
‑
d)δ5.65(s,1h),4.58(s,1h),4.45
‑
4.24(m,2h), 2.08
‑
1.98(m,1h),1.91(s,3h),1.89
‑
1.80(m,1h),1.78
‑
1.52(m,4h),1.46
‑
1.24(m,3h),1.07(m,1h), 0.83(t,j=6.8hz,6h),0.71(d,j=6.4hz,3h).
13
c nmr(101mhz,chloroform
‑
d)δ170.73,138.32, 138.29,127.16,127.14,103.22,87.17,65.48,58.87,58.84,49.65,40.16,36.25,31.08,30.64,27.26,22.57, 21.29,20.82,11.64.hrms m/z(esi):calcd for c
17
h
26
o4na[m+na]
+
:317.1723,found:317.1728.
[0057]
实施例2:c
‑
14位改造的莪术醇衍生物3
‑
2的合成
[0058][0059]
(1)与实施例1步骤(1)相同。
[0060]
(2)与实施例1步骤(1)相同。
[0061]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml干燥的dcm中,在室温条件下,向反应 瓶中缓慢滴加丙烯酰氯(80mg,0.87mmol),室温下搅拌3h。tlc检测(pe:ea=4:1)反应结束, 加水淬灭反应,乙酸乙酯萃取(3
×
100ml),无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状 粗品,用硅胶柱层析纯化产物(pe:ea=8:1),以74.90%的收率得到黄色油状物化合物3
‑
2。yellow oil.yield:74.90%.1h nmr(500mhz,chloroform
‑
d)δ6.47
‑
6.34(m,1h),6.21
‑
6.05(m,1h),5.91
‑
5.79 (m,2h),4.69
‑
4.38(m,2h),2.20(dd,j=12.6,11.0hz,1h),2.00(t,j=8.3hz,1h),1.87
‑
1.93(m,2h), 1.86
‑
1.78(m,1h),1.63
‑
1.71(m,1h),1.61
‑
1.53(m,2h),1.50
‑
1.41(m,1h),1.23(dd,j=12.7,7.4hz, 1h),0.99(dd,j=11.8,6.6hz,6h),0.87(d,j=6.6hz,3h).
13
c nmr(126mhz,chloroform
‑
d)δ 165.75,138.62,131.00,128.20,126.99,103.23,87.19,65.51,59.36,49.80,40.19,36.33,31.14,30.65, 27.45,22.57,21.33,11.63ppm.hrms m/z(esi):calcd for c
18
h
26
o4na[m+na]
+
:329.1723,found: 329.1722.
[0062]
实施例3:c
‑
14位改造的莪术醇衍生物3
‑
3的合成
[0063][0064]
(1)与实施例1步骤(1)相同。
[0065]
(2)与实施例1步骤(2)相同。
[0066]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml dcm中,向反应瓶中缓慢加入dcc(96mg, 0.79mmol)和dmap(154mg,0.79mmol),室温下搅拌5min后,加入巴豆酸(75mg,0.88mmol) 室温下继续反应8h。tlc检测(pe:ea=4:1)反应结束,加水淬灭反应,乙酸乙酯萃取(3
×
30ml), 无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状粗品,用硅胶柱层析纯化产物(pe:ea=8:1), 以62.70%的收率得到黄色油状液体3
‑
3。yellow oil.yield:62.70%.1h nmr(500mhz,chloroform
‑
d) δ7.05
‑
6.84(m,1h),5.89
‑
5.68(m,2h),4.64
‑
4.39(m,2h),3.78(s,1h),2.17(dd,j=12.7,10.9hz,1h), 1.97(t,j=8.6hz,1h),1.87(dd,j=6.9,1.8hz,5h),1.83
‑
1.76(m,1h),1.62
‑
1.71(m,1h),1.61
‑
1.50(m, 2h),1.39
‑
1.47(m,1h),1.24
‑
1.17(m,1h),0.97(dd,j=11.2,6.6hz,7h),0.84(d,j=6.5hz,3h).
13
c nmr(126mhz,chloroform
‑
d)δ165.96,144.97,138.59,126.85,122.30,103.16,87.11,65.10,59.00, 49.70,40.10,36.22,31.05,30.54,27.31,22.49,21.23,17.85,11.55ppm.hrms m/z(esi):calcd for c
19
h
28
o4na[m+na]
+
:320.1880,found:343.1883.
[0067]
实施例4:c
‑
14位改造的莪术醇衍生物3
‑
4的合成
[0068]
[0069]
(1)与实施例1步骤(1)相同。
[0070]
(2)与实施例1步骤(2)相同。
[0071]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml dcm中,向反应瓶中缓慢加入dcc(96mg, 0.79mmol)和dmap(154mg,0.79mmol),室温下搅拌5min后,加入肉桂酸(75mg,0.88mmol) 室温下继续反应8h。tlc检测(pe:ea=4:1)反应结束,加水淬灭反应,乙酸乙酯萃取(3
×
30ml), 无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状粗品,用硅胶柱层析纯化产物(pe:ea=8:1), 以65.90%的收率得到黄色油状液体3
‑
4。yellow oil.yield:65.90%.1h nmr(500mhz,chloroform
‑
d)δ 7.71(d,j=16.0hz,1h),7.53(dd,j=6.6,3.1hz,2h),7.42
‑
7.34(m,3h),6.48(d,j=16.0hz,1h),5.91 (d,j=1.4hz,1h),4.78
‑
4.50(m,2h),3.87
‑
3.68(m,1h),2.21(dd,j=12.8,10.8hz,1h),2.04(dd,j= 10.3,5.8hz,1h),1.98
‑
1.80(m,3h),1.68
‑
1.76(m,1h),1.61(m,2h),1.45
‑
1.55(m,1h),1.26(dd,j= 12.7,7.5hz,2h),1.02(t,j=6.4hz,6h),0.89(d,j=6.6hz,3h).
13
c nmr(126mhz,chloroform
‑
d)δ 166.42,145.06,138.55,138.54,134.18,130.23,128.74,127.99,127.08,127.06,117.63,103.19,87.14, 65.45,59.05,59.03,49.72,40.11,36.22,31.07,30.57,27.36,22.51,21.28,11.58ppm.hrms m/z(esi): calcd for c
24
h
30
o4na[m+na]
+
:405.2036,found:405.2034.
[0072]
实施例5:c
‑
14位改造的莪术醇衍生物3
‑
5的合成
[0073][0074]
(1)与实施例1步骤(1)相同。
[0075]
(2)与实施例1步骤(2)相同。
[0076]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml dcm中,向反应瓶中缓慢加入dcc(96mg, 0.79mmol)和dmap(154mg,0.79mmol),室温下搅拌5min后,加入4
‑
氟苯甲酸(122mg,0.88mmol) 室温下继续反应8h。tlc检测(pe:ea=4:1)反应结束,加水淬灭反应,乙酸乙酯萃取(3
×
30ml), 无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状粗品,用硅胶柱层析纯化产物(pe:ea=8:1), 以81.20%的收率得到黄色油状液体3
‑
5。yellow oil.yield:81.20%.1h nmr(500mhz,chloroform
‑
d)δ 8.15
‑
7.89(m,2h),7.02
‑
7.10(m,2h),5.90(s,1h),4.82
‑
4.59(m,2h),4.20(s,1h),2.17(dd,j=12.7, 10.8hz,1h),2.05
‑
1.96(m,1h),1.92
‑
1.74(m,3h),1.65
‑
1.74(m,1h),1.50
‑
1.62(m,2h),1.49
‑
1.39(m, 1h),1.21(dd,j=
12.7,7.3hz,2h),1.01
‑
0.92(m,6h),0.83(d,j=6.5hz,3h).
13
c nmr(126mhz, chloroform
‑
d)δ166.50,164.90,164.48,138.00,131.91(j
c
‑
f
=9.4hz),127.46,125.99(j
c
‑
f
=3.0hz), 115.35(j
c
‑
f
=21.8hz),103.06,87.03,65.95,58.65,49.66,39.93,36.06,30.91,30.47,27.28,22.32,21.08, 11.41ppm.hrms m/z(esi):calcd for c
22
h
27
o4fna[m+na]
+
:397.1786,found:397.1787.
[0077]
实施例6:c
‑
14位改造的莪术醇衍生物3
‑
6的合成
[0078][0079]
(1)与实施例1步骤(1)相同。
[0080]
(2)与实施例1步骤(2)相同。
[0081]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml dcm中,向反应瓶中缓慢加入dcc(96mg, 0.79mmol)和dmap(154mg,0.79mmol),室温下搅拌5min后,加入2
‑
吡啶甲酸(107mg,0.88mmol) 室温下继续反应8h。tlc检测(pe:ea=4:1)反应结束,加水淬灭反应,乙酸乙酯萃取(3
×
30ml), 无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状粗品,用硅胶柱层析纯化产物(pe:ea=8:1), 以80.70%的收率得到黄色油状液体3
‑
6。yellow oil.yield:80.70%.1h nmr(500mhz,chloroform
‑
d)δ 8.67(d,j=4.7hz,1h),7.98(d,j=7.3hz,1h),7.75(t,j=7.7hz,1h),7.39(dd,j=7.5,4.8hz,1h), 5.87(s,1h),4.86
‑
4.65(m,2h),4.25(s,1h),2.08(t,j=11.8hz,1h),2.00(s,1h),1.68
‑
1.89(m,4h), 1.57
‑
1.66(m,1h),1.45
‑
1.55(m,2h),1.35
‑
1.43(m,1h),1.15(dd,j=12.8,7.4hz,1h),0.89(dd,j= 11.3,6.5hz,7h),0.75(d,j=6.6hz,3h).
13
c nmr(126mhz,chloroform
‑
d)δ164.36,149.69,147.49, 137.99,136.85,127.53,127.51,126.73,124.85,103.02,86.87,66.41,58.80,49.49,39.93,36.03,30.91, 30.38,27.24,22.38,21.12,11.47.hrms m/z(esi):calcd for c
21
h
27
no4na[m+na]
+
:380.1832,found: 380.1830.
[0082]
实施例7:c
‑
14位改造的莪术醇衍生物3
‑
7的合成
[0083][0084]
(1)与实施例1步骤(1)相同。
[0085]
(2)与实施例1步骤(2)相同。
[0086]
(3)将衍生物2
‑
1(200mg,0.79mmol)溶于15ml dcm中,向反应瓶中缓慢加入dcc(96mg, 0.79mmol)和dmap(154mg,0.79mmol),室温下搅拌5min后,加入噻吩
‑2‑
甲酸(112mg,0.88 mmol)室温下继续反应8h。tlc检测(pe:ea=4:1)反应结束,加水淬灭反应,乙酸乙酯萃取(3
×
30 ml),无水硫酸钠固体干燥,减压浓缩,制备得棕黄色油状粗品,用硅胶柱层析纯化产物(pe:ea =8:1),以73.10%的收率得到黄色油状液体3
‑
7。yellow oil.yield:73.10%.1h nmr(500mhz, chloroform
‑
d)δ8.09(dd,j=3.0,1.2hz,1h),7.49(dd,j=5.0,1.2hz,1h),7.28(dd,j=5.1,3.1hz, 1h),5.89(s,1h),4.83
‑
4.58(m,2h),4.09(s,1h),2.23
‑
2.13(m,1h),2.05
‑
1.97(m,1h),1.94
‑
1.76(m, 3h),1.66
‑
175(m,1h),1.63
‑
1.53(m,2h),1.50
‑
1.39(m,1h),1.21(dd,j=12.7,7.4hz,1h),0.98(dd,j= 9.5,6.5hz,6h),0.84(d,j=6.6hz,3h).
13
c nmr(126mhz,chloroform
‑
d)δ162.12,138.23,133.12, 132.74,127.63,127.26,125.96,103.10,87.07,65.57,58.79,49.69,39.99,36.12,30.98,30.50,27.34, 22.43,21.18,11.52ppm.hrms m/z(esi):calcd for c
20
h
26
o4sna[m+na]
+
:385.1444,found:385.1447.
[0087]
实施例8:体外抗肿瘤实验
[0088]
选取上述实施例合成的莪术醇衍生物进行体外抗肿瘤活性实验,分别进行了hct116(人结肠癌 细胞)和sw620(人结肠癌细胞)细胞株的活性筛选。发现该类化合物对sw620细胞有较好的抑制 效果。
[0089]
选用对数生长期的sw620人结肠癌细胞,用胰酶进行消化后,l
‑
15培养基配成6
×
104/ml的细胞 悬液,然后将细胞悬液加入到96孔板中,每孔细胞数为15000个,37℃下,无co2培养24h,将事先 配置好的不同浓度的药物分别加入到96孔板中,浓度梯度为100μm、50μm、20μm、10μm、2μm, 每个浓度梯度设置4个副孔,37℃下,无co2培养72小时,每孔加入10μlmtt,37℃下,无co2培 养3h,弃去上清液,加入150μl dmso,振荡均价后,酶标仪在490nm处测定光密度(od值)
[0090]
选用对数生长期的hct116人结肠癌细胞,用胰酶进行消化后,dmem培养基配成6
×
104/ml的 细胞悬液,然后将细胞悬液加入到96孔板中,每孔细胞数为5000个,37℃下,5%
co2培养24h,将 事先配置好的不同浓度的药物分别加入到96孔板中,浓度梯度为100μm、50μm、20μm、10μm、 2μm,每个浓度梯度设置4个副孔,37℃下,5%co2培养72h,每孔加入10μl mtt(5mg/ml)溶液, 37℃下,5%co2培养3h,弃去上清液,加入150μl dmso,振荡均价后,酶标仪在490nm处测定 光密度(od值)
[0091]
抑制率计算:
[0092]
生长抑制率=(od对照组
‑
od实验组)/(od对照组
‑
od空白组)
[0093]
根据药物浓度
‑
生长抑制率曲线,计算ic
50
,结果如下表1所示:
[0094]
表1
[0095][0096]
注:5
‑
fu表示5
‑
氟尿嘧啶,阳性对照药物
[0097]
由表1可知,本发明所提供化合物均具有较好的抗结肠癌作用,其中3
‑
1、3
‑
2、3
‑
4和3
‑
5化合物 表现出比莪术醇更强的抑制活性,特别是化合物3
‑
2,对两种肿瘤细胞株表现出较好的抑制效果,在 药物化学领域拥有较好的发展前景。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1