一种N端带有表位标签M的表达载体及其构建方法与应用
一种n端带有表位标签m的表达载体及其构建方法与应用
技术领域
1.本发明属于生物技术领域,涉及一种n端带有表位标签m的表达载体及其制备方法与应用。
背景技术:
2.1984年,1984年munro和pelham发表的文章第一次将抗原表位标签通过重组dna的方法应用于检测果蝇hsp70蛋白的突变体。为了单独观察突变后的蛋白质在细胞中的活动,设计融合一个高敏感性和独特性的表位标签于蛋白末端,用该表位标签所对应的抗体进行检测。由次以后,表位标记技术可以用来检测没有特异抗体的重组蛋白,同时方便科研人员检测比较转基因的产物和内源基因的产物的异同。相较于传统内源性蛋白耗时耗力以及不可预测性,选用为标记蛋白的标签抗体更容易获得,灵敏度更高,可以通过各种生化和细胞生物学技术检测,包括流式细胞术、蛋白免疫印迹和免疫荧光染色,抗体针对每个标记具有特异性。标记系统也广泛应用于亲和纯化和免疫沉淀。
3.能够用作标签的氨基酸序列有一定的要求。一,表位标签蛋白大小一般为6
‑
30氨基酸,不会影响融合蛋白的生物活性。二,很重要的是具有能够特异性识别该表位标签的高灵敏度抗体。提高灵敏度的一种方法是串联重复标签序列,比如常用的3倍flag标签,串联后的标签对于抗体的结合力更强。三,大多数标签为亲水性,含有更多的带电氨基酸,比如9个氨基酸长度的ha标签含有两个天冬氨酸,10个氨基酸长度的c
‑
myc标签含有三个谷氨酸,一个赖氨酸,一个天冬氨酸。表位标签的位置一般位于c末端或n末端,标签位于末端应该能够比插入目的蛋白内部具有更大可能性使目的蛋白保持自身生物活性。有文献报道,大致有63%的融合蛋白对于表位标签有良好的耐受性。同时由于大多数蛋白折叠后末端会暴露于空间结构外部,标签位于末端能够更有利于标签抗体的识别。目前常见的表位标签有如下几种:
4.(1)c
‑
myc,氨基酸长度为10,序列为eqkliseedl,免疫原为人类c
‑
myc基因408
‑
439位产物,市面上对应提供9e10抗体。
5.(2)flag,氨基酸长度为8,序列为dykddddk,免疫原为合成的含有肠激酶裂解位点的段肽,市面上对应提供m1,m2,m3抗体。
6.(3)v5,氨基酸长度为14,序列为gkpipnpllglds,免疫原为猿病毒5(sv5)副粘病毒p蛋白和v蛋白中分离得到,市面上对应提供v5抗体。
7.(4)his6,氨基酸长度为6,序列为hhhhhh,免疫原为重组的带有his标签的融合蛋白,市面上对应提供6
‑
his,6
×
his,his
‑
11抗体。
8.(5)t7,基酸长度为11,序列为masmtggqqmg,免疫原为噬菌体t7主要衣壳蛋白的先导肽,市面上对应提供抗体。
9.(6)hsv,基酸长度为11,序列为qpelapedped,免疫原为来自hsv糖蛋白d的合成肽,市面上对应提供抗体。
10.由于标签各有优缺点,所以不断开发新的标签很有必要,因此,亟需开发一种新新
的表位标签,以能够基本实现对克隆基因在哺乳动物细胞中的对应蛋白表达的监测。
技术实现要素:
11.为了解决所述技术问题,本发明提供了一种n端带有表位标签m的表达载体及其应用,能够用于检测蛋白表达。
12.在本发明的第一方面,提供了一种片段m在真核生物中作为表位标签在外源过表达基因检测上的应用,所述片段m的氨基酸序列如seq id no:1所示,核苷酸序列如seq id no:2所示。
13.进一步地,所述应用包括在蛋白免疫印迹实验、免疫沉淀实验、免疫荧光实验和流式细胞术中的应用。
14.进一步地,所述应用包括:
15.构建n端带有表位标签m的真核表达载体;
16.将所述n端融合有所述表位标签的真核表达载体与编码目标蛋白的核苷酸序列进行重组,获得重组表达载体;
17.将所述重组表达载体在所述真核生物中进行表达;或/和蛋白免疫印迹实验、或/和免疫沉淀实验或/和、免疫荧光实验或/和流式细胞术实验。
18.进一步地,所述片段m串联重复1次或3次作为表位标签m融合在表达载体的n端。
19.在本发明的第二方面,提供了一种n端带有表位标签m的表达载体的构建方法,所述方法包括:
20.用限制性核酸内切酶xhoi和noti酶切空载pqfn
‑
flag3,获得带有xhoi和noti酶切端口的pqfn
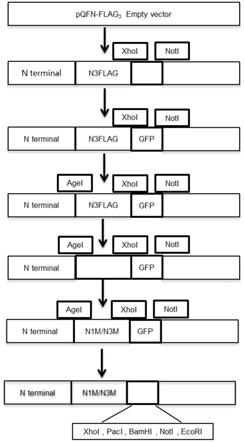
‑
flag3;将含gfp序列载体为模板采用如seq id no:3
‑
seq id no:4所示的引物对进行pcr,获得gfp基因片段;后将所述pqfn
‑
flag3和所述gfp基因片段分别通过xhoi和noti双酶切后酶连,获得pqfn
‑
flag3‑
gfp;
21.将所述pqfn
‑
flag3‑
gfp分别采用如seq id no:5
‑
seq id no:6所示的引物对和如seq id no:7
‑
seq id no:8所示的引物对进行第二pcr,获得长片段和短片段,后将所述长片段和短片段分别通过agei和noti双酶切后并酶连,获得标签前带有agei的pqfn
‑
agei
‑
flag3‑
gfp质粒;
22.将所述标签前带有agei的pqfn
‑
agei
‑
flag3‑
gfp质粒采用agei和xhoi双酶切,获得带有agei和xhoi酶切端口的pqf
‑
gfp;
23.将含表位标签m序列载体为模板采用如seq id no:9
‑
seq id no:10所示的引物对进行第一退火,获得带有agei和xhoi酶切端口的m1产物;将所述m1产物与所述带有agei和xhoi酶切端口的pqf
‑
gfp质粒酶连,获得pqfn
‑
m
‑
gfp;
24.或者将含表位标签m序列载体为模板采用如seq id no:11
‑
seq id no:12所示的引物对和如seq id no:13
‑
seq id no:14所示的引物对进行第二退火,获得带有agei和xhoi酶切端口的m3产物;将所述m3产物与所述带有agei和xhoi酶切端口的pqf
‑
gfp质粒酶连,获得pqfn
‑
m3‑
gfp;
25.用xhoi和ecori酶切所述pqfn
‑
m
‑
gfp或者所述pqfn
‑
m3‑
gfp,获得具有xhoi和ecori酶切端口的线性载体pqfn
‑
m或者pqfn
‑
m3;
26.将如seq id no:15
‑
seq id no:16所示的退火引物退火后分别连接到所述具有
xhoi和ecori酶切端口的线性载体pqfn
‑
m或者pqfn
‑
m3,得到具有xhoi、paci、bamhi、noti和ecori五个酶切位点的空载pqfn
‑
m或者pqfn
‑
m3。
27.进一步地,所述pcr的退火温度为60℃,所述第一退火和所述第二退火的条件均包括:37℃30min,95℃5min,然后自然冷却至25℃。
28.在本发明的第三方面,提供了一种采用所述方法构建获得的一种n端带有表位标签m的表达载体。
29.在本发明的第四方面,提供了一种所述的n端带有表位标签m的表达载体的应用,所述应用包括:
30.在ncbi上查找出需要研究的目的基因x的序列,根据基因x序列设计并获得带有xhoi和noti多克隆位点处酶切位点的上下游引物对,采用所述上下游引物对扩增带有xhoi和noti酶切位点的x基因,获得x基因片段;
31.对所述x基因片段进行xhoi和noti双酶切并进行酶切产物回收,获得带有xhoi和noti酶切端口的x基因片段;
32.将所述的n端带有表位标签m的表达载体采用xhoi和noti酶切,电泳回收,获得带有xhoi和noti酶切端口的pqfn
‑
m和pqfn
‑
m3;
33.将所述带有xhoi和noti酶切端口的pqfn
‑
m和pqfn
‑
m3分别与所述带有xhoi和noti酶切端口的x基因片段进行酶切连接,获得位标签序列的重组质粒pqfn
‑
m
‑
x和pqfn
‑
m3‑
x。
34.进一步,所述应用还包括:
35.将所述重组质粒pqfn
‑
m
‑
x和pqfn
‑
m3‑
x分别转染至细胞,利用标签m通过蛋白免疫印迹技术检测x基因编码蛋白的表达;
36.和/或利用标签m对所述x基因进行免疫沉淀实验;
37.和/或将所述重组质粒pqfn
‑
m
‑
x和pqfn
‑
m3‑
x分别转染至哺乳动物细胞,后经过固定、打孔、封闭、单克隆抗体m一抗孵育、荧光二抗孵育、dapi染色和封片,利用标签m通过免疫荧光技术监测x基因对应编码蛋白的定位情况。
38.本发明实施例中的一个或多个技术方案,至少具有如下技术效果或优点:
39.1、本发明提供的一种n端带有表位标签m的表达载体及其制备方法与应用,本发明构建的可以融合目标蛋白n端的表达载体pqfn
‑
m,pqfn
‑
m3,通过蛋白免疫印迹检测发现,m标签序列能够被特异性识别来表征融合蛋白的表达。构建的带有m标签的表达载体为哺乳动物蛋白表达领域提供了一种新型的通用融合标签载体资源,丰富了国内的表位标签资源;
40.2、方便更换基因。多克隆位点处提供5个酶切位点用于选择。其中xhoi和noti两个酶切位点为大多数基因中所没有的,少数含有xhoi位点的基因上游可以设计带有xhoi的同尾酶sali的引物来替代。通过本发明构建成功的表达载体pqfn
‑
m,pqfn
‑
m3,只需要简单进行xhoi和noti双酶切后就可以通过酶切连接将gfp基因替换成所要研究的基因;
41.3、表位标签m所对应的单克隆鼠单抗3b9具有特异性好,灵敏度高,能够用于检测蛋白表达;
42.4、本发明构建成功的表达载体pqfn
‑
m,pqfn
‑
m3可用于免疫沉淀和免疫共沉淀实验;可尝试用于蛋白亲和纯化实验;可尝试用于蛋白质组学实验。
附图说明
43.为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其它的附图。
44.图1为构建带有m表位标签的表达载体流程示意图;包括构建pqfn
‑
m/m3‑
gfp和pqfn
‑
m/m3空载;
45.图2为pqfn
‑
m3表达载体模式图及多克隆位点处的序列;
46.图3为应用例1中n端融合的m标签在免疫印迹实验中的应用;a为n端带有三个m标签序列的蛋白表达鉴定,b为n端带有三个flag标签序列的蛋白表达鉴定;
47.图4为应用例2中使用n端融合的flag标签和m标签检测sqstm1蛋白的亚细胞定位。
48.图5为应用例3中n端融合的m标签在流式细胞术中的应用。
具体实施方式
49.下文将结合具体实施方式和实施例,具体阐述本发明,本发明的优点和各种效果将由此更加清楚地呈现。本领域技术人员应理解,这些具体实施方式和实施例是用于说明本发明,而非限制本发明。
50.在整个说明书中,除非另有特别说明,本文使用的术语应理解为如本领域中通常所使用的含义。因此,除非另有定义,本文使用的所有技术和科学术语具有与本发明所属领域技术人员的一般理解相同的含义。若存在矛盾,本说明书优先。
51.除非另有特别说明,本发明中用到的各种原材料、试剂、仪器和设备等,均可通过市场购买得到或者可通过现有方法制备得到。
52.本发明实施例提供的技术方案为解决上述技术问题,总体思路如下:
53.片段m为最小抗原表位为6个氨基酸长度的ppyddd序列,比常用的8个氨基酸长度的flag序列更短,可以考虑是否可以用来作为标签融合表达没有特异性抗体的蛋白。新型表位标签m的氨基酸序列ppyddd(seq id no:1),碱基序列为:cctccgtatgacgatgac(seq id no:2)。
54.根据本发明实施例一种典型的实施方式,提供一种n端带有表位标签m的表达载体的构建方法,所述方法包括:
55.用限制性核酸内切酶xhoi和noti酶切空载pqfn
‑
flag3,获得带有xhoi和noti酶切端口的pqfn
‑
flag3;将含gfp序列载体为模板采用如seq id no:3
‑
seq id no:4所示的引物对进行pcr,获得gfp基因片段;后将所述pqfn
‑
flag3和所述gfp基因片段分别通过xhoi和noti双酶切后酶连,获得pqfn
‑
flag3‑
gfp;
56.将所述pqfn
‑
flag3‑
gfp分别采用如seq id no:5
‑
seq id no:6所示的引物对和如seq id no:7
‑
seq id no:8所示的引物对进行第二pcr,获得长片段和短片段,后将所述长片段和短片段分别通过agei和noti双酶切后并酶连,获得标签前带有agei的pqfn
‑
agei
‑
flag3‑
gfp质粒;
57.将所述标签前带有agei的pqfn
‑
agei
‑
flag3‑
gfp质粒采用agei和xhoi双酶切,获得带有agei和xhoi酶切端口的pqf
‑
gfp;
58.将含表位标签m序列载体为模板采用如seq id no:9
‑
seq id no:10所示的引物对进行第一退火,获得带有agei和xhoi酶切端口的m1产物;将所述m1产物与所述带有agei和xhoi酶切端口的pqf
‑
gfp质粒酶连,获得pqfn
‑
m
‑
gfp;
59.或者将含表位标签m序列载体为模板采用如seq id no:11
‑
seq id no:12所示的引物对和如seq id no:13
‑
seq id no:14所示的引物对进行第二退火,获得带有agei和xhoi酶切端口的m3产物;将所述m3产物与所述带有agei和xhoi酶切端口的pqf
‑
gfp质粒酶连,获得pqfn
‑
m3‑
gfp;
60.用xhoi和ecori酶切所述pqfn
‑
m
‑
gfp或者所述pqfn
‑
m3‑
gfp,获得具有xhoi和ecori酶切端口的线性载体pqfn
‑
m或者pqfn
‑
m3;
61.将如seq id no:15
‑
seq id no:16所示的退火引物退火后分别连接到所述具有xhoi和ecori酶切端口的线性载体pqfn
‑
m或者pqfn
‑
m3,得到具有xhoi、paci、bamhi、noti和ecori五个酶切位点的空载pqfn
‑
m或者pqfn
‑
m3。
62.本发明实施例以pqcxip为表达载体骨架进行改造,对其复制后命名为pqf,以氨基酸ppyddd为标签序列m,构建可以融合目标蛋白n端的表达载体pqfn
‑
m,pqfn
‑
m3,并在n端融合有m标签的多克隆位点处设计xhoi、paci、bamhi、noti和ecori5个酶切位点,多个酶切位点的存在可以适用包容更多的基因插入。同时为方便后期载体更换标签使用,在n端融合的m标签前加入agei酶切位点,将gfp和sqstm1基因融入该表达载体,通过蛋白免疫印迹检测发现,m标签序列能够被特异性识别来表征融合蛋白的表达。构建的带有m标签的表达载体为哺乳动物蛋白表达领域提供了一种新型的通用融合标签载体资源。
63.作为可选的实施方式,所述pcr的退火温度为60℃,所述第一退火和所述第二退火的条件均包括:37℃30min,95℃5min,然后自然冷却至25℃。
64.根据本发明另一种典型的实施方式,提供了一种所述的n端带有表位标签m的表达载体的应用,所述应用包括:
65.在ncbi上查找出需要研究的目的基因x的序列,根据基因x序列设计并获得带有xhoi和noti多克隆位点处酶切位点的上下游引物对,采用所述上下游引物对扩增带有xhoi和noti酶切位点的x基因,获得x基因片段;
66.对所述x基因片段进行xhoi和noti双酶切并进行酶切产物回收,获得带有xhoi和noti酶切端口的x基因片段;
67.将所述的n端带有表位标签m的表达载体采用xhoi和noti酶切,电泳回收,获得带有xhoi和noti酶切端口的pqfn
‑
m和pqfn
‑
m3;
68.将所述带有xhoi和noti酶切端口的pqfn
‑
m和pqfn
‑
m3分别与所述带有xhoi和noti酶切端口的x基因片段进行酶切连接,获得位标签序列的重组质粒pqfn
‑
m
‑
x和pqfn
‑
m3‑
x。
69.将所述重组质粒pqfn
‑
m
‑
x和pqfn
‑
m3‑
x分别转染至细胞,利用标签m通过蛋白免疫印迹技术检测x基因编码蛋白的表达;
70.和/或利用标签m对所述x基因进行免疫沉淀实验;
71.和/或将所述重组质粒pqfn
‑
m
‑
x和pqfn
‑
m3‑
x分别转染至哺乳动物细胞,后经过固定、打孔、封闭、单克隆抗体m一抗孵育、荧光二抗孵育、dapi染色和封片,利用标签m通过免疫荧光技术监测x基因对应编码蛋白的定位情况。
72.下面将结合实施例及实验数据对本技术的效果进行详细说明。下列实施例中未注
明的具体实验条件和方法,通常按照常规条件如:j.萨姆布鲁克等主编,科学出版社,1992,分子克隆实验指南(第三版);d.l.斯佩克特等,科学出版社,2001,细胞实验指南等书中所述的条件,或按照制造厂商所建议的条件。
73.实施例1、n端带有表位标签m的表达载体的构建方法
74.1、n端融合m标签:pqfn
‑
m/m3‑
gfp的构建。
75.(1)用限制性核酸内切酶xhoi和noti酶切空载pqfn
‑
flag3,得到带有xhoi和noti酶切端口的pqfn
‑
flag3。
76.(2)设计上游带有xhoi,下游带有noti酶切位点的gfp基因引物,通过pcr(pcr体系如表2所示,pcr条件如表3所示),酶切连接等方法将gfp基因片段连接到pqfn
‑
flag3载体上得到pqfn
‑
flag3‑
gfp。
77.表1
[0078][0079]
表2
[0080][0081][0082]
表3
[0083][0084]
(3)将pqfn
‑
flag3‑
gfp分为两段,一段为短片段nflag3+gfp,另一段为长片段剩余载体序列。在nflag3序列前设计带有agei酶切位点的引物进行pcr,回收后酶切连接得到在标签前带有agei的pqfn
‑
agei
‑
flag3‑
gfp质粒。
[0085]
表4
[0086][0087]
将所述pqfn
‑
flag3‑
gfp分别采用如seq id no:5
‑
seq id no:6所示的引物对和如seq id no:7
‑
seq id no:8所示的引物对进行第二pcr,获得长片段和短片段,后将所述长片段和短片段分别通过agei和noti双酶切后并酶连,获得标签前带有agei的pqfn
‑
agei
‑
flag3‑
gfp质粒;
[0088]
长片段pcr体系为如表5。
[0089]
表5
[0090][0091]
长片段pcr条件为如表6。
[0092]
表6
[0093][0094]
短片段pcr体系如表7。
[0095]
表7
[0096][0097]
短片段pcr条件如表8。
[0098]
表8
[0099][0100][0101]
(4)用agei和xhoi酶切pqfn
‑
agei
‑
flag3‑
gfp得到带有agei和xhoi酶切端口的pqf
‑
gfp。
[0102]
(5)分别设计n1m和n3m的退火引物,然后将其连接于pqf
‑
gfp,得到pqfn
‑
m/m3‑
gfp。
[0103]
表9
[0104][0105]
具体地,将含表位标签m序列载体为模板采用如seq id no:9
‑
seq id no:10所示的引物对进行第一退火,获得带有agei和xhoi酶切端口的m1产物;将所述m1产物与所述带有agei和xhoi酶切端口的pqf
‑
gfp质粒酶连,获得pqfn
‑
m
‑
gfp;
[0106]
或者将含表位标签m序列载体为模板采用如seq id no:11
‑
seq id no:12所示的引物对和如seq id no:13
‑
seq id no:14所示的引物对进行第二退火,获得带有agei和xhoi酶切端口的m3产物;将所述m3产物与所述带有agei和xhoi酶切端口的pqf
‑
gfp质粒酶连,获得pqfn
‑
m3‑
gfp;
[0107]
退火体系如表10所示,退火条件为:37℃30min,95℃5min,然后自然冷却至25℃。
[0108]
表10
[0109][0110][0111]
2.n端融合m标签:pqfn
‑
m/m3的构建。
[0112]
(1)用xhoi和ecori酶切pqfn
‑
m/m3‑
gfp得到具有xhoi和ecori酶切端口的线性载体pqfn
‑
m/m3。
[0113]
(2)设计具有xhoi、paci、bamhi、noti和ecori5个酶切位点的退火引物,退火后将其连接到pqfn
‑
m/m3(xhoi,ecori)得到具有五个酶切位点的空载pqfn
‑
m/m3(xhoi、paci、bamhi、noti和ecori)。退火体系如表12所示,退火条件为:37℃30min,95℃5min,然后自然冷却至25℃。
[0114]
表11
[0115][0116]
表12
[0117][0118]
实施例2、重组质粒pqfn
‑
m3‑
sqstm1的构建
[0119]
1.引物的设计合成
[0120]
根据ncbi中提供的人源sqstm1基因,gene id 8878得到sqstm1 isoform 1的编码序列,设计合成一对特异性引物,扩增sqstm1基因。引物由上海生工生物工程技术有限公司合成,引物的序列如下:
[0121]
sqstm1
‑
f:ccctcgagatgtcggaagcgggcgaggag(seq id no:17);
[0122]
sqstm1
‑
r:atttgcggccgcgtatggcttgtagttattctgatg(seq id no:18);
[0123]
2.hek293细胞总rna提取及cdna的合成
[0124]
在24孔板中培养hek293细胞,待其长满一个孔之后,吸去培养基,用总rna提取试剂盒(plus mini kit,公司产品),按试剂盒操作说明书介绍的方法提
取细胞总rna。提取的rna使用逆转录试剂盒(monscript
tm rtⅲsuper mix with dsdnase(two
‑
step),ref:mr05201,莫纳生物公司产品),按试剂盒操作说明书介绍的方法进行逆转录,获得hek293细胞基因组cdna,冻存备用。
[0125]
3.pcr扩增及产物回收
[0126]
以步骤2合成的hek293细胞基因组cdna为模板,pcr扩增sqstm1基因,扩增反应体系和具体反应条件如下:
[0127][0128]
用移液器将上述组分混匀,在pcr热循环仪上进行以下反应:
[0129][0130]
110v,40min琼脂糖凝胶电泳,胶回收试剂盒(d2500
‑
02,omega公司产品,下同)回收dna片段(按试剂盒说明书操作)。
[0131]
4.酶切载体和基因片段并回收
[0132]
(1)用限制性核酸内切酶xhoi和noti双酶(neb公司产品,下同)消化pqfn
‑
m3空载或pqfn
‑
m3‑
gfp质粒,110v,80min琼脂糖凝胶电泳,胶回收试剂盒回收约7.2kbp dna片段pqfn
‑
m3。
[0133]
(2)用限制性核酸内切酶xhoi和noti双酶消化步骤3中sqstm1胶回收产物,直接使用胶回收试剂盒回收酶切产物。
[0134]
5.载体和基因片段的连接转化及阳性克隆筛选
[0135]
(1)用载体pqfn
‑
m3:基因片段sqstm1=1:3的摩尔比例,使用连接酶(t4 dna ligase,m0202l,neb公司产品,下同)进行连接(按说明书操作)。
[0136]
(2)连接产物转化大肠杆菌,挑起重组子阳性克隆接种5毫升含有50μg/ml氨苄抗性lb培养基中37℃培养过夜,小提质粒纯化提取质粒dna,经核苷酸测序验证,获得重组质粒pqfn
‑
m3‑
sqstm1。
[0137]
实施例3、重组质粒pqfn
‑
m3‑
rig
‑
i的构建
[0138]
1.引物的设计合成
[0139]
根据ncbi中提供的人源rig
‑
i基因,gene id 23586的编码序列,设计合成一对特
异性引物,扩增rig
‑
i基因。引物由上海生工生物工程技术有限公司合成,引物的序列如下:
[0140]
rig
‑
i
‑
f(xho1):ccctcgaggccaccatgaccaccgagcagcgac(seq id no:19);
[0141]
rig
‑
i
‑
r(not1):atttgcggccgctttggacatttctgctggatc(seq id no:20);
[0142]
2.hek293细胞总rna提取及cdna的合成
[0143]
在24孔板中培养hek293细胞,待其长满一个孔之后,吸去培养基,用总rna提取试剂盒(plus mini kit,公司产品),按试剂盒操作说明书介绍的方法提取细胞总rna。提取的rna使用逆转录试剂盒(monscript
tm rtⅲsuper mix with dsdnase(two
‑
step),ref:mr05201,莫纳生物公司产品),按试剂盒操作说明书介绍的方法进行逆转录,获得hek293细胞基因组cdna,冻存备用。
[0144]
3.pcr扩增及产物回收
[0145]
以步骤2合成的hek293细胞基因组cdna为模板,pcr扩增rig
‑
i基因,扩增反应体系和具体反应条件如下:
[0146][0147]
用移液器将上述组分混匀,在pcr热循环仪上进行以下反应:
[0148][0149]
110v,40min琼脂糖凝胶电泳,胶回收试剂盒(d2500
‑
02,omega公司产品,下同)回收dna片段(按试剂盒说明书操作)。
[0150]
4.酶切载体和基因片段并回收
[0151]
(1)用限制性核酸内切酶xhoi和noti双酶(neb公司产品,下同)消化pqfn
‑
m3空载或pqfn
‑
m3‑
gfp质粒,110v,80min琼脂糖凝胶电泳,胶回收试剂盒回收约7.2kbp dna片段pqfn
‑
m3。
[0152]
(2)用限制性核酸内切酶xhoi和noti双酶消化步骤3中rig胶回收产物,直接使用胶回收试剂盒回收酶切产物。
[0153]
5.载体和基因片段的连接转化及阳性克隆筛选
[0154]
(1)用载体pqfn
‑
m3:基因片段rig=1:3的摩尔比例,使用连接酶(t4 dna ligase,m0202l,neb公司产品,下同)进行连接(按说明书操作)。
[0155]
(2)连接产物转化大肠杆菌,挑起重组子阳性克隆接种5毫升含有50μg/ml氨苄抗性lb培养基中37℃培养过夜,小提质粒纯化提取质粒dna,经核苷酸测序验证,获得重组质粒pqfn
‑
m3‑
rig
‑
i。
[0156]
实施例4、重组质粒pqfn
‑
m3‑
sting的构建
[0157]
1.引物的设计合成
[0158]
根据ncbi中提供的人源sting基因,gene id 340061的编码序列,设计合成一对特异性引物,扩增sting基因。引物由上海生工生物工程技术有限公司合成,引物的序列如下:
[0159]
sting
‑
f(xho1):ccctcgaggccaccatgccccactccagcctgcatc(seq id no:21);
[0160]
sting
‑
r(not1):atttgcggccgcagagaaatccgtgcggagag(seq id no:22);
[0161]
2.hek293细胞总rna提取及cdna的合成
[0162]
在24孔板中培养hek293细胞,待其长满一个孔之后,吸去培养基,用总rna提取试剂盒(plus mini kit,公司产品),按试剂盒操作说明书介绍的方法提取细胞总rna。提取的rna使用逆转录试剂盒(monscript
tm rtⅲsuper mix with dsdnase(two
‑
step),ref:mr05201,莫纳生物公司产品),按试剂盒操作说明书介绍的方法进行逆转录,获得hek293细胞基因组cdna,冻存备用。
[0163]
3.pcr扩增及产物回收
[0164]
以步骤2合成的hek293细胞基因组cdna为模板,pcr扩增sting基因,扩增反应体系和具体反应条件如下:
[0165][0166]
用移液器将上述组分混匀,在pcr热循环仪上进行以下反应:
[0167][0168]
110v,40min琼脂糖凝胶电泳,胶回收试剂盒(d2500
‑
02,omega公司产品,下同)回收dna片段(按试剂盒说明书操作)。
[0169]
4.酶切载体和基因片段并回收
[0170]
(1)用限制性核酸内切酶xhoi和noti双酶(neb公司产品,下同)消化pqfn
‑
m3空载或pqfn
‑
m3‑
gfp质粒,110v,80min琼脂糖凝胶电泳,胶回收试剂盒回收约7.2kbp dna片段
pqfn
‑
m3。
[0171]
(2)用限制性核酸内切酶xhoi和noti双酶消化步骤3中sting胶回收产物,直接使用胶回收试剂盒回收酶切产物。
[0172]
5.载体和基因片段的连接转化及阳性克隆筛选
[0173]
(1)用载体pqfn
‑
m3:基因片段sting=1:3的摩尔比例,使用连接酶(t4 dna ligase,m0202l,neb公司产品,下同)进行连接(按说明书操作)。
[0174]
(2)连接产物转化大肠杆菌,挑起重组子阳性克隆接种5毫升含有50μg/ml氨苄抗性lb培养基中37℃培养过夜,小提质粒纯化提取质粒dna,经核苷酸测序验证,获得重组质粒pqfn
‑
m3‑
sting。
[0175]
实施例5、重组质粒pqfn
‑
m3‑
tbk1的构建
[0176]
1.引物的设计合成
[0177]
根据ncbi中提供的人源tbk1基因,gene id 29110的编码序列,设计合成一对特异性引物,扩增tbk1基因。引物由上海生工生物工程技术有限公司合成,引物的序列如下:
[0178]
tbk1
‑
f(xho1):ccctcgaggccaccatgcagagcacttctaatcatc(seq id no:23);
[0179]
tbk1
‑
r(not1):atttgcggccgcaagacagtcaacgttgcgaag(seq id no:24);
[0180]
2.hek293细胞总rna提取及cdna的合成
[0181]
在24孔板中培养hek293细胞,待其长满一个孔之后,吸去培养基,用总rna提取试剂盒(plus mini kit,公司产品),按试剂盒操作说明书介绍的方法提取细胞总rna。提取的rna使用逆转录试剂盒(monscript
tm rtⅲsuper mix with dsdnase(two
‑
step),ref:mr05201,莫纳生物公司产品),按试剂盒操作说明书介绍的方法进行逆转录,获得hek293细胞基因组cdna,冻存备用。
[0182]
3.pcr扩增及产物回收
[0183]
以步骤2合成的hek293细胞基因组cdna为模板,pcr扩增tbk1基因,扩增反应体系和具体反应条件如下:
[0184][0185]
用移液器将上述组分混匀,在pcr热循环仪上进行以下反应:
[0186]
[0187][0188]
110v,40min琼脂糖凝胶电泳,胶回收试剂盒(d2500
‑
02,omega公司产品,下同)回收dna片段(按试剂盒说明书操作)。
[0189]
4.酶切载体和基因片段并回收
[0190]
(1)用限制性核酸内切酶xhoi和noti双酶(neb公司产品,下同)消化pqfn
‑
m3空载或pqfn
‑
m3‑
gfp质粒,110v,80min琼脂糖凝胶电泳,胶回收试剂盒回收约7.2kbp dna片段pqfn
‑
m3。
[0191]
(2)用限制性核酸内切酶xhoi和noti双酶消化步骤3中tbk1胶回收产物,直接使用胶回收试剂盒回收酶切产物。
[0192]
5.载体和基因片段的连接转化及阳性克隆筛选
[0193]
(1)用载体pqfn
‑
m3:基因片段tbk1=1:3的摩尔比例,使用连接酶(t4 dna ligase,m0202l,neb公司产品,下同)进行连接(按说明书操作)。
[0194]
(2)连接产物转化大肠杆菌,挑起重组子阳性克隆接种5毫升含有50μg/ml氨苄抗性lb培养基中37℃培养过夜,小提质粒纯化提取质粒dna,经核苷酸测序验证,获得重组质粒pqfn
‑
m3‑
tbk1。
[0195]
实施例6、重组质粒pqfn
‑
m3‑
irf3的构建
[0196]
1.引物的设计合成
[0197]
根据ncbi中提供的人源irf3基因,gene id 3661的编码序列,设计合成一对特异性引物,扩增irf3基因。引物由上海生工生物工程技术有限公司合成,引物的序列如下:
[0198]
irf3
‑
f(xho1):ccctcgaggccaccatgggaaccccaaagccacg(seq id no:25);
[0199]
irf3
‑
r(not1):atttgcggccgcgctctccccagggccctg(seq id no:26);
[0200]
2.hek293细胞总rna提取及cdna的合成
[0201]
在24孔板中培养hek293细胞,待其长满一个孔之后,吸去培养基,用总rna提取试剂盒(plus mini kit,公司产品),按试剂盒操作说明书介绍的方法提取细胞总rna。提取的rna使用逆转录试剂盒(monscript
tm rtⅲsuper mix with dsdnase(two
‑
step),ref:mr05201,莫纳生物公司产品),按试剂盒操作说明书介绍的方法进行逆转录,获得hek293细胞基因组cdna,冻存备用。
[0202]
3.pcr扩增及产物回收
[0203]
以步骤2合成的hek293细胞基因组cdna为模板,pcr扩增irf3基因,扩增反应体系和具体反应条件如下:
[0204][0205]
用移液器将上述组分混匀,在pcr热循环仪上进行以下反应:
[0206][0207]
110v,40min琼脂糖凝胶电泳,胶回收试剂盒(d2500
‑
02,omega公司产品,下同)回收dna片段(按试剂盒说明书操作)。
[0208]
4.酶切载体和基因片段并回收
[0209]
(1)用限制性核酸内切酶xhoi和noti双酶(neb公司产品,下同)消化pqfn
‑
m3空载或pqfn
‑
m3‑
gfp质粒,110v,80min琼脂糖凝胶电泳,胶回收试剂盒回收约7.2kbp dna片段pqfn
‑
m3。
[0210]
(2)用限制性核酸内切酶xhoi和noti双酶消化步骤3中irf3胶回收产物,直接使用胶回收试剂盒回收酶切产物。
[0211]
5.载体和基因片段的连接转化及阳性克隆筛选
[0212]
(1)用载体pqfn
‑
m3:基因片段irf3=1:3的摩尔比例,使用连接酶(t4 dna ligase,m0202l,neb公司产品,下同)进行连接(按说明书操作)。
[0213]
(2)连接产物转化大肠杆菌,挑起重组子阳性克隆接种5毫升含有50μg/ml氨苄抗性lb培养基中37℃培养过夜,小提质粒纯化提取质粒dna,经核苷酸测序验证,获得重组质粒pqfn
‑
m3‑
irf3。
[0214]
应用例1、n端含m标签载在蛋白免疫印迹实验中的应用
[0215]
将实施例1所得的pqfn
‑
m3‑
gfp、实施例2所得的重组质粒pqfn
‑
m3‑
sqstm1、实施例3所得的重组质粒pqfn
‑
m3‑
rig
‑
i、实施例4所得的重组质粒pqfn
‑
m3‑
sting、实施例5所得的重组质粒pqfn
‑
m3‑
tbk1和实施例6所得重组质粒pqfn
‑
m3‑
irf3分别进行表达进行验证实验。
[0216]
1、细胞传代并铺板:取合适密度的细胞进行铺板。
[0217]
(1)当培养皿中的细胞生长至90%时进行传代。首先吸去培养皿中的旧培养液,加入2ml的pbs轻轻润洗细胞,洗干净培养基残留;加入1.5ml 0.25%的胰蛋白酶消化液,于37℃消化1min;加入6mldmem培养基终止消化,用移液枪吹打重悬细胞。吸去1ml细胞悬浮液转移至新培养皿并加入9ml培养基继续培养。
[0218]
(2)吸取细胞悬液1.5ml于一新的15ml离心管,再用dmem培养基补足12ml,充分混匀后以每孔500μl的细胞密度铺于24孔板进行培养,用于第二日转染。
[0219]
2、磷酸钙转染法进行所述质粒分别进行转染:细胞密度达到60%
‑
70%时转染。
[0220]
细胞数达到60%
‑
70%后转染,配制如下体系:
[0221]
表13
[0222] 10cm dish6well12well24wellmilliq h2o补足1ml补足150ul补足60ul补足30ul2mcacl2124ul20ul10ul5ulsqstm115
‑
20ug2
‑
3.5ug1
‑
1.5ug400
‑
500ng
[0223]
加完各成分后轻柔混匀,缓慢逐滴加入等量的2
×
hbs,轻柔混匀,短离。等待10分钟后至出现细小颗粒沉淀,将混合液加入细胞培养皿中,轻轻混匀。
[0224]
3、细胞裂解与制样:向细胞板中加入适量的裂解液冰上裂解30min后制样。
[0225]
(1)转染后24小时左右将24孔板从培养箱取出,于冰上每孔加入100μl tap裂解液,在4℃摇床裂解30分钟。
[0226]
(2)将裂解后的细胞裂解液用移液枪吸取到1.5ml ep管中,4℃,15000rpm离心10min。吸取45μl离心后的上清于新的1.5ml ep管中,并加入15μl 4
×
loading buffer。于70℃金属浴中煮样15min。
‑
20℃冻存。
[0227]
4.蛋白免疫印迹(wb)
[0228]
(1)配胶:分别配制10%sds page分离胶和5%浓缩胶。
[0229]
(2)电泳:每个孔加8
‑
10μl的样品,180v,50min电泳进行电泳。
[0230]
(3)电转(湿转):按如下顺序放置:透明孔板,黑色海绵,滤纸,pvdf膜,蛋白胶(按镜面对称的方式放置),滤纸(加上这层滤纸后,用移液管赶走其中的气泡),黑色海绵,黑色孔板。80v(电流保持与0.18
‑
0.2a)转膜1小时。
[0231]
(4)封闭:5%脱脂牛奶,水平摇床上缓慢封闭1h。
[0232]
(5)孵一抗:4℃孵育过夜。
[0233]
(6)洗膜:1
×
tbs洗3次,每次5min。
[0234]
(7)孵二抗:室温下孵育1h。
[0235]
(8)洗膜:1
×
tbs洗6次,每次5min
[0236]
(9)显影:ecl substrate(a\b液等量混匀),3min;暗室显影,放入x光片,1min后放入洗片机。得到曝光出sqstm1蛋白条带的x光片。
[0237]
如图3所示,通过蛋白质免疫印迹法检测hek293细胞裂解物中融合m标签的蛋白(sqstm1、rig
‑
i、sting、tbk1和irf3),此外结果还表明:对于蛋白的大小不同进行比较,3b9抗体对于小分子量蛋白识别能力更强。
[0238]
应用例2、n端含m标签载体在免疫荧光实验中的应用
[0239]
1.细胞爬片培养及转染
[0240]
(1)转染前一天将细胞爬片放入24孔板中,铺入一定数量的hela细胞。
[0241]
(2)将pqfn
‑
m3‑
sqstm1转染至hela细胞中,转染方法同应用例1。
[0242]
2.免疫荧光实验
[0243]
(1)吸去培养基,用pbs润洗细胞1次。
[0244]
(2)使用预冷的无水甲醇固定细胞10min,结束后pbs润洗3次,每次3min。
[0245]
(3)使用0.5%triton x
‑
100破膜打孔15min,结束后pbs润洗3次,每次3min。
[0246]
(4)使用2%的bsa封闭30min。
[0247]
(5)使用1%的bsa配制一抗,将其放到4℃冰箱中孵育过夜,完成后pbs洗三次,每次3min。
[0248]
(6)使用1%的bsa配制带有鼠种属的荧光基团的二抗594
‑
mouse,37℃培养箱内遮光孵育1h,结束后使用pbs洗三次,每次3min。
[0249]
(7)使用dapi染细胞核10min,用pbs洗细胞3次,每次3min。
[0250]
(8)用封片剂进行封片,待其晾干后在荧光显微镜下进行观察拍照。
[0251]
结果显示,天然的人源选择性自噬接头蛋白sequestosome
‑
1(sqstm1)蛋白和标签标记的sqstm1蛋白都主要聚集在细胞质中,并且呈点状分布,说明m标签/3b9单克隆抗体系统能够指示sqstm1的正确定位,即定位于螯合小体中。从图4中可以观察到除了共定位的sqstm1为红色荧光外,没有额外的背景染上红色,证明m标签/3b9单克隆抗体系统在免疫荧光定位中的特异性良好。此外,m标签比flag标签的红色荧光更加明亮一些,证明m标签系统在免疫荧光定位中的应用比flag似乎更灵敏一些。
[0252]
应用例3、n端含m标签载体在流式细胞术实验中的应用
[0253]
(1)转染前一天按实验需要接种一定数量细胞到6孔板中;
[0254]
(2)待细胞密度达到60%
‑
70%左右后,按实验需要转染pqfn
‑
m3‑
gfp质粒;
[0255]
(3)转染48小时后,去除培养基加入适量胰酶进行消化,并用dmem重悬后加入1.5ml离心管收集细胞;
[0256]
(4)4℃,300g,离心5min,去除dmem和胰酶;
[0257]
(5)加入200μl预冷的pbs洗涤1次,4℃,300g,离心5min,去除pbs;
[0258]
(6)加入4%多聚甲醛混匀,室温固定细胞15min;
[0259]
(7)加入200μl预冷的pbs洗涤2次,3min/次,4℃,300g,离心5min,去除固定液;
[0260]
(8)加入0.1%triton x
‑
100于冰上透化处理10min;
[0261]
(9)加入200μl预冷的pbs洗涤2次,3min/次,4℃,300g,离心5min,去除透化液;
[0262]
(10)加入blocking buffer室温封闭处理30min;
[0263]
(11)使用fasc buffer配制抗flag和抗m的一抗在室温孵育1h,孵育完成后pbs洗3次,3min/次;
[0264]
(12)使用fasc buffer配制带有相应荧光基团的二抗室温遮光孵育30min,结束后使用pbs洗3次,3min/次;
[0265]
(13)加入500μl pbs重悬细胞;
[0266]
(14)用尼龙网膜将离心管内的细胞过滤到流式中,做好标记;
[0267]
(15)用流式细胞仪进行检测。
[0268]
为了将m标签/3b9单克隆抗体系统应用于融合蛋白的流式细胞分析,将pqfn
‑
m3‑
gfp转染至hek293细胞。用3b9单克隆抗体染色后,通过流式细胞术分析细胞(图5)。未转染任何质粒的hek293空白细胞作为阴性对照组;由于gfp自身发绿色荧光,用fitc检测重组蛋白细胞比例,作为阳性对照组;由于647荧光二抗染色3b9单克隆抗体标记的gfp蛋白,用apc检测重组蛋白细胞比例,作为实验组。在阴性对照中没有观察到标记,阳性对照组检测到的
pqfn
‑
m3‑
gfp重组蛋白的细胞分别为26.15%,实验组中3b9单克隆抗体检测到的pqfn
‑
m3‑
gfp重组蛋白的细胞为7.69%。
[0269]
最后,还需要说明的是,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
[0270]
尽管已描述了本发明的优选实施例,但本领域内的技术人员一旦得知了基本创造性概念,则可对这些实施例作出另外的变更和修改。所以,所附权利要求意欲解释为包括优选实施例以及落入本发明范围的所有变更和修改。
[0271]
显然,本领域的技术人员可以对本发明进行各种改动和变型而不脱离本发明的精神和范围。这样,倘若本发明的这些修改和变型属于本发明权利要求及其等同技术的范围之内,则本发明也意图包含这些改动和变型在内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1