双氯芬酸钠的合成工艺的制作方法
1.本发明属于药物合成技术领域,具体涉及一种双氯芬酸钠的合成工艺或方法。
背景技术:
2.双氯芬酸钠,又称双氯灭痛,是一种非甾体强效消炎镇痛药,其对前列腺素合成酶进行抑制,具有清热、消炎及止痛的功效,用于各类风湿性关节炎、红斑狼疮、神经炎及癌症、术后疼痛及发热等,因其疗效好且副作用少,是世界畅销药物之一。
3.双氯芬酸钠最早是由萨尔曼等人合成出来的,国内外研究学者为了提高双氯芬酸产率,在改进优化其合成方法和减少其生产成方面进行了大量的研究工作。目前,双氯芬酸钠的合成路线主要有以下几种:
4.(1)以邻氯(或溴、碘)苯甲酸为起始原料经乌尔曼缩合、脱羧制得关键中间体2,6
‑
二氯二苯胺,然后经酰化、关环生成1
‑
(2,6
‑
二氯苯基)二氢吲哚
‑2‑
酮,水解后得到终产物。本方法是最早用于生产的工艺化路线,其特点是原料易得但反应步骤长。
5.(2)以2,6
‑
二氯二苯胺和氯乙酰氯为主要原料,经酰化、分子内傅克烷基化反应,得到1
‑
(2,6
‑
二氯苯基)二氢吲哚
‑2‑
酮,水解开环后得到产物。
6.(3)以邻卤代苯乙酸、酯或酰胺等为起始原料,与2,6
‑
二氯苯胺进行乌尔曼反应,再经水解或成盐制备得到终产物。
7.(4)环己酮氯化后与邻硝基苯乙酸缩合后,再经芳构化及成盐得到终产物。
8.(5)将n
‑
(2,6
‑
二氯苯)苯乙酰胺氯化后,关环制备得到1
‑
(2,6
‑
二氯苯基)二氢吲哚
‑2‑
酮,水解得到终产物。
9.现有的双氯芬酸钠生产路线绝大多数都存在原料成本高、操作复杂、收率较低及使用危险、剧毒原料等问题。即使是目前工业化的合成路线,也存在着催化剂不够高效、溶剂毒性大、反应时间长、总收率低和耗能高等诸多问题。因此,亟待对现有的双氯芬酸钠生成工艺进行优化,研究出可生产化的制备工艺,从而降低双氯芬酸钠的制备成本,扩展其应用前景。
技术实现要素:
10.为了解决上述问题,本发明提供了一种双氯芬酸钠的合成工艺或方法,以邻氨基苯乙酸酯为原料,与2,6
‑
二氯苯氧酸酯酰化反应,或先与氯代酰氯酰化,再与2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠进行亲核取代,均可得到中间体,并在碱性条件下,重排、氨解、水解,从而反应得到双氯芬酸钠。通过本发明,合成步骤得到有效减少,制备工艺简单易行,反应条件温和,提高双氯芬酸钠的收率,对其推广应用具有重大意义。
11.本发明的目的之一在于提供一种双氯芬酸钠的合成工艺,以邻氨基苯乙酸酯为原料进行制备。
12.由邻氨基苯乙酸酯制备含有2,6
‑
二氯苯氧基团和邻乙酸酯基苯酰氨基团的化合物,然后进行重排反应、氨解,最后水解得到双氯芬酸钠。
13.所述邻氨基苯乙酸酯由苯乙酸酯硝化后,氢化还原制备得到。
14.邻氨基苯乙酸酯与2,6
‑
二氯苯氧酸酯进行酰化反应,重排后进行氨解和/或水解;
15.所述酰化反应在催化剂存在下进行,所述催化剂选自钯催化剂、锌盐、醇钾或醇钠,催化剂与邻氨基苯乙酸酯的摩尔比为1:(1.5
‑
3.5),优选为1:(2
‑
2.5)。
16.所述邻氨基苯乙酸酯为邻氨基苯乙酸烷基酯、邻氨基苯乙酸苯基酯或氨基苯乙酸取代苯基酯,优选为邻氨基苯乙酸烷基酯。
17.所述2,6
‑
二氯苯氧酸酯通过2,6
‑
二氯苯酚与氯烷基酸酯亲核取代反应进行制备,所述氯烷基酸酯为氯烷基酸烷基酯、氯烷基酸苯基酯和氯烷基酸取代苯基酯中的一种,优选为氯烷基酸烷基酯。
18.所述亲核取代反应在溶剂中进行,所述溶剂选自醇类、酯类和醚类溶剂中的一种或几种,优选为甲醇、异丙醇、石油醚和乙酸乙酯中的一种或几种,优选为乙酸乙酯。
19.邻氨基苯乙酸酯与氯代酰氯(优选ω
‑
氯代烷基酰氯)在溶剂存在下进行酰化,反应温度为100
‑
160℃,优选为110
‑
140℃,得到n
‑
氯酰基苯胺。
20.n
‑
氯酰基苯胺与2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠在碱性条件下,优选在相转移催化剂如聚乙二醇存在下,在80
‑
150℃,优选100
‑
120℃温度下进行亲核取代。再加入无机强碱,保温反应,氨解和/或水解,得到双氯芬酸钠。
21.本发明的再一目的在于提供一种根据以上工艺或方法制得的双氯芬酸钠。
22.本发明提供的双氯芬酸钠的合成工艺或方法具有以下有益效果:
23.(1)本发明中以邻氨基苯乙酸酯为反应原料,与2,6
‑
二氯苯氧酸酯酰化反应过程中,先后进行重排及氨解,反应进行迅速,收率高,条件温和,再经碱性条件进行酯基水解,可直接得到双氯芬酸钠,产物收率得到有效提高。
24.(2)本发明中以邻氨基苯乙酸酯为反应原料,还可以与氯代酰氯先进行酰化,再与2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠进行亲核取代,在碱性条件下,同样先后进行重排及氨解,酯基水解,得到双氯芬酸,反应收率高,容易进行。
25.(3)本发明中,在制备过程中产生中间物ⅰ后,后续重排和氨解反应容易进行,能够顺利得到2
‑
[(2,6
‑
二氯苯基)氨基]
‑
苯乙酸酯,再经酯基水解,可直接得到双氯芬酸钠。无需进行后续2,6
‑
二氯二苯胺的酰胺化、成环和水解,生产工艺得以简化,收率大幅提高。
[0026]
(4)所述合成工艺或方法中,无需使用高温高压条件,所用溶剂均可回收套用,降低三废,提高双氯芬酸钠质量。
附图说明
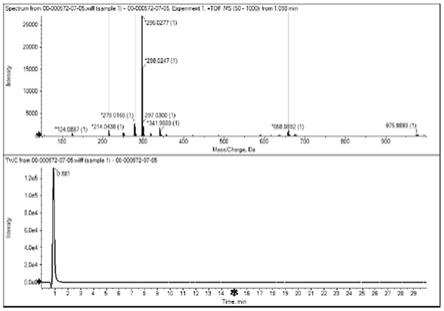
[0027]
图1示出实施例3制得的双氯芬酸钠的高分辨质谱
‑
液相色谱图;
[0028]
图2示出实施例3制得的双氯芬酸钠的高效液相色谱图。
具体实施方式
[0029]
下面通过具体实施方式对本发明进行详细说明,本发明的特点和优点将随着这些说明而变得更为清楚、明确。
[0030]
本发明提供的双氯芬酸钠的合成工艺或方法,以邻氨基苯乙酸酯为原料,在与2,6
‑
二氯苯氧酸酯酰化反应后,直接进行重排及氨解,另外,还可通过与氯代酰氯进行酰化,
与2,6
‑
二氯苯酚反应,实现重排和氨解,最终在碱性条件下酯基水解,直接得到双氯芬酸钠。合成工艺中,制备步骤缩短,使后处理过程得到有效简化,条件温和,有利于推广及应用。
[0031]
根据本发明,双氯芬酸钠的合成工艺以邻氨基苯乙酸酯为原料进行制备。
[0032]
所述工艺中,由邻氨基苯乙酸酯制备中间物ⅰ,经重排反应、氨解、水解得到双氯芬酸钠。
[0033]
本发明中,所述中间物ⅰ为含有2,6
‑
二氯苯氧基团和邻乙酸酯基苯酰氨基团的化合物,优选地,所述中间物ⅰ具有以下结构:
[0034][0035]
其中,
[0036]
r为脂族或芳族基团,优选为未取代或取代的烷基、烯基、炔基,未取代或取代的芳基或杂芳基,如未取代或取代的苯基、萘基、蒽基、吡啶基或哌啶基,更优选为低级烷基,如c1‑
c5烷基、苯基或低级烷基(如c1‑
c5烷基)取代的苯基;
[0037]
n为整数,优选为1
‑
15,更优选为1
‑
8,如1
‑
3。
[0038]
本发明中,所述邻氨基苯乙酸酯为已知化合物,可由商业途径购买或自制得到。
[0039]
根据本发明优选的实施方式,所述邻氨基苯乙酸酯由苯乙酸酯硝化后,氢化还原制备得到。优选地,在溶剂中,所述苯乙酸酯在硝化反应剂存在下进行硝化,得到邻氨基苯乙酸酯。
[0040]
根据本发明,所述硝化反应剂为浓硝酸或发烟硝酸。所述溶剂选自卤代烷烃,优选为二氯甲烷。
[0041]
在本发明优选的实施方式中,所述硝化反应剂在低温下进行滴加,滴加结束后在15
‑
25℃下进行反应,得到邻硝基苯乙酸酯。
[0042]
根据本发明,所述氢化还原在钯催化剂催化下进行,常压下通入氢气,使硝基还原为氨基。
[0043]
在本发明的一种实施方式中,所述邻氨基苯乙酸酯与2,6
‑
二氯苯氧酸酯酰化反应,经重排后,氨解、水解得到双氯芬酸钠。
[0044]
所述邻氨基苯乙酸酯为邻氨基苯乙酸烷基酯、邻氨基苯乙酸苯基酯或氨基苯乙酸取代苯基酯,优选为邻氨基苯乙酸烷基酯。所述烷基选自c1‑
c5烷基,优选为c1‑
c3烷基;所述取代苯基优选为c1‑
c5烷基取代苯基。
[0045]
本发明中,所述2,6
‑
二氯苯氧酸酯优选具有以下结构:
[0046][0047]
其中,
[0048]
r1为脂族或芳族基团,优选为未取代或取代的烷基、烯基、炔基,未取代或取代的芳基或杂芳基,如未取代或取代的苯基、萘基、蒽基、吡啶基或哌啶基,更优选为低级烷基,如c1‑
c5烷基、苯基或低级烷基(如c1‑
c5烷基)取代苯基;
[0049]
n为整数,优选为1
‑
15,更优选为1
‑
8,如1
‑
3。
[0050]
例如,所述2,6
‑
二氯苯氧酸酯可以为2,6
‑
二氯苯氧酸烷基酯、2,6
‑
二氯苯氧酸苯基酯或2,6
‑
二氯苯氧酸取代苯基酯,优选为2,6
‑
二氯苯氧酸烷基酯。所述烷基选自c1‑
c5烷基,优选为c1‑
c3烷基;所述取代苯基优选为c1‑
c5烷基取代苯基。
[0051]
本发明中,所述2,6
‑
二氯苯氧酸酯为由2,6
‑
二氯苯酚与氯烷基酸酯亲核取代反应制备得到。
[0052]
根据本发明,所述氯烷基酸酯为氯烷基酸烷基酯、氯烷基酸苯基酯和氯烷基酸取代苯基酯中的一种,优选为氯烷基酸烷基酯,如氯乙酸乙酯。所述烷基选自c1‑
c5烷基,优选为c1‑
c3烷基;所述取代苯基优选为c1‑
c5烷基取代苯基。
[0053]
本发明优选的实施方式中,所述亲核取代反应在溶剂中进行,所述溶剂选自醇类、酯类和醚类溶剂中的一种或几种,优选为甲醇、异丙醇、石油醚和乙酸乙酯中的一种或几种,优选为乙酸乙酯。所述氯烷基酸酯与溶剂的摩尔体积比为1:(150
‑
260),优选为1:(180
‑
210)。
[0054]
所述亲核取代反应在碱性条件下进行,在反应液中加入弱碱类物质,优选为碳酸盐,如碳酸钠、碳酸钾、片碱等,所述弱碱类物质与氯烷基酸酯的摩尔比为1:(0.3
‑
0.65),优选为1:(0.4
‑
0.55)。
[0055]
所述反应温度为60
‑
95℃,优选为70
‑
85℃。
[0056]
所述邻氨基苯乙酸酯与2,6
‑
二氯苯氧酸酯酰化反应后,经中间物ⅰ进行查普曼重排。
[0057]
所述酰化反应在催化剂存在下进行,所述催化剂选自钯催化剂、锌盐、醇钾或醇钠,优选选自pbo、钯碳、氯化锌、醋酸锌、甲醇钠或甲醇钾,更优选为甲醇钠。所述催化剂与邻氨基苯乙酸酯的摩尔比为1:(1.5
‑
3.5),优选为1:(2
‑
2.5)。
[0058]
所述酰化反应在溶剂存在下进行,所述溶剂选自醇类溶剂中的一种或几种,优选为正丁醇、异丁醇、丙醇和异丙醇中的一种或几种,更优选为正丁醇。所述溶剂与邻氨基苯乙酸酯的体积摩尔比为(20
‑
80)ml:(0.55
‑
0.65)mol,优选为(30
‑
60)ml:(0.55
‑
0.65)mol。
[0059]
所述酰化反应温度为80
‑
120℃,优选为90
‑
110℃。
[0060]
根据本发明,在酰化反应条件下,所述中间产物ⅰ经查普曼重排得到中间物ⅱ,其具有以下结构:
[0061][0062]
其中,r和n如以上对中间物ⅰ所定义。
[0063]
在酰化反应条件下,中间物ⅱ继续氨解,其中叔胺基转化为仲胺基。
[0064]
然后,后处理得到有机相,加入碱性物质,再经水解将中间物ⅱ中的酯基转化为羧酸盐,得到双氯芬酸钠。所述水解反应温度为60
‑
100℃,优选为70
‑
90℃。
[0065]
在本发明的另一种实施方式中,所述邻氨基苯乙酸酯与氯代酰氯酰化,得到n
‑
氯酰基苯胺,再与2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠进行亲核取代,最后经中间物ⅰ重排后,氨解、水解得到双氯芬酸钠。
[0066]
所述氯代酰氯为ω
‑
氯代烷基酰氯,如氯乙酰氯、氯丁酰氯、2
‑
氯代异丁酰氯、5
‑
氯代戊酰氯、氯代特戊酰氯。
[0067]
所述邻氨基苯乙酸酯与氯代酰氯酰化反应在溶剂存在下进行,所述溶剂选自醇类,如正丁醇、异丁醇、丙醇、异丙醇,芳香烃类,如甲苯、二甲苯,醚类,如石油醚、四氢呋喃,酮类,如2
‑
甲基吡咯烷酮,和酰胺类溶剂,如n,n
‑
二甲基甲酰胺、n,n
‑
二甲基乙酰胺、甲酰胺,其中的一种或几种,优选为正丁醇、异丁醇、丙醇、异丙醇、甲苯、二甲苯、2
‑
甲基吡咯烷酮、n,n
‑
二甲基乙酰胺、甲酰胺中的一种或几种,更优选为正丁醇或二甲苯。所述反应温度为100
‑
160℃,优选为110
‑
140℃。
[0068]
酰化反应结束后,加入2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠,在碱性条件下,加入相转移催化剂,如聚乙二醇,进行亲核取代。所述碱性条件为向反应液中加入碱金属碳酸盐,如碳酸钠、碳酸钾。
[0069]
所述2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠与邻氨基苯乙酸酯的摩尔比为(0.8
‑
1.6):1,优选为(1
‑
1.3):1。所述相转移催化剂与邻氨基苯乙酸酯的质量摩尔比为(10
‑
25)g:1mol,优选为(15
‑
20)g:1mol。
[0070]
所述亲核取代反应温度为80
‑
150℃,优选为100
‑
120℃。
[0071]
根据本发明,在亲核反应结束后,加入无机强碱,如氢氧化钠、氢氧化钾,保温反应,进行重排、氨解及酯基水解,得到双氯芬酸钠。所述无机强碱与2,6
‑
二氯苯酚或2,6
‑
二氯苯酚钠的摩尔比为(0.8
‑
1.5):1,优选为(1
‑
1.3):1。
[0072]
本发明中,以邻氨基苯乙酸酯与2,6
‑
二氯苯氧酸酯酰化反应,或者先与氯代酰氯酰化,再与二氯苯酚取代,得到中间产物ⅰ和中间产物ⅱ,氨解、水解后制备得到双氯芬酸钠。缩短合成工艺,制备原料易得,反应条件温和,能够提高产物收率,有利于工业化推广。
[0073]
实施例
[0074]
实施例1
[0075]
将1mol苯乙酸甲酯加入到5l二氯甲烷中,0℃下剧烈搅拌,缓慢滴加2.5kg质量浓度为24%发烟硝酸的二氯甲烷溶液。滴加结束后,升温至20℃,继续搅拌反应1.5小时。反应结束后,用去离子水洗涤至中性,静置分层后,将有机相用质量浓度为10%的硫酸钠溶液洗涤三次,静置分层得到有机相,减压分馏得166.09g邻硝基苯乙酸甲酯,收率约为85.1%。
[0076]
将0.7mol所制得的邻硝基苯乙酸甲酯和1.5g钯碳加入到260ml甲苯中,搅拌混合,在5
‑
7℃下常压通入氢气,氢化反应12h。反应结束后,先在52℃下过滤出钯碳,冷却后,产物析出,用甲苯洗涤,合并滤液和洗液,回收甲苯,析出的固体在真空下干燥,得到113.34g邻氨基苯乙酸甲酯,收率约为98.1%。
[0077]
实施例2
[0078]
将1mol的3,5
‑
二氯
‑4‑
羟基苯甲酸加入到150ml的dmf中,加热至90℃,加入20ml的2,4,6
‑
三甲基吡啶,升温至120℃,搅拌均匀,进行脱羧反应。随后将温度缓慢升温至150℃进行反应,直至没有二氧化碳生成。
[0079]
再将60g的钾碱、200ml的乙酸乙酯和110ml的氯乙酸乙酯加入到上述反应液中,在50℃下混合均匀。升温至80℃时有二氧化碳溢出,继续升温,回流反应2h。反应结束后,加入600ml的水,洗涤,冷却至室温,分离有机相,减压蒸馏回收溶剂,得到201.76g 2,6
‑
二氯苯氧乙酸乙酯,反应收率约为81%。
[0080]
实施例3
[0081]
将0.5mol实施例2所制得的2,6
‑
二氯苯氧乙酸乙酯和0.6mol的实施例1所制得的邻氨基苯乙酸甲酯加入到40ml的正丁醇中,搅拌混合,加热至100℃。将45ml的浓度为5.0m的甲醇钠溶液,缓慢滴加到反应液中,在100℃下,继续搅拌反应0.5h,并持续蒸馏分液。反应结束后,加入1l的水,搅拌混合,然后冷却至60℃,进行相分离,得到有机相。
[0082]
向有机相中加入30ml水和0.5mol的氢氧化钠,再加入60ml正丁醇,80℃反应10h。反应结束后,冷却至室温,分别减压蒸馏回收正丁醇和水,得到双氯芬酸钠粗产品。
[0083]
向所述粗产品中加入水150ml,活性炭脱色,冷却结晶得到122.64g双氯芬酸钠,为类白色固体,收率约为77.1%,图1为所得双氯芬酸钠的高分辨质谱
‑
液相色谱图,其m/z为[m+h]
+
=296.0277(双氯芬酸钠在质谱条件下被质子化,m/z=296.0277对应双氯芬酸的质谱峰位,即能够证明得到了双氯芬酸钠);图2为所得双氯芬酸钠的高效液相色谱图,根据色谱测试结果可知,纯度为99.761%。
[0084]
实施例4
[0085]
将0.5mol实施例1所制得的邻氨基苯乙酸甲酯和45ml二甲苯加入到反应容器中,在冰浴条件下,搅拌均匀,缓慢滴加0.55mol的氯乙酰氯,使反应容器内部温度低于50℃,滴加完成后,缓慢升温至110℃,反应5h,反应过程中用吸收瓶吸收产生的hcl。
[0086]
反应结束后,冷却至65℃,加入0.6mol的2,6
‑
二氯苯酚、0.6mol碳酸钠、8.5g聚乙二醇
‑
600,搅拌条件下,升温至110℃,反应24h后,加入0.6mol的氢氧化钠,继续保温反应10h,反应结束后,再加入0.6mol的氢氧化钠,保温在80℃下反应10h,加入饱和氯化钠溶液10ml,搅拌冷却至室温,静置分层,对有机相用无水硫酸镁干燥、过滤,减压蒸馏去除溶剂,得到双氯芬酸钠粗产品。
[0087]
将所述粗产品中加入水150ml,活性炭脱色,冷却结晶得到141.73g双氯芬酸钠,为类白色固体,收率约为89.1%,其hplc纯度为99.802%。
[0088]
以上结合具体实施方式和/或范例性实例以及附图对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。本发明的保护范围以所附权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1