环氧乙烷基共聚物
1.本公开涉及可降解环氧乙烷基共聚物,以及形成这些可降解环氧乙烷基共聚物的方法。
背景技术:
2.聚亚烷基二醇类作为用作合成洗涤剂或聚氨酯的原料的特种聚合物有着悠久的历史。这 种聚合物是水溶性或者油性液体,它们最终会进入环境系统或者废水系统。因为它们不能被 回收或焚烧,所以对于进入这些系统的物质来说,可降解性是必需的。例如,聚亚烷基二醇 类包括聚环氧乙烷(peo)和聚环氧丙烷(ppo,1,2-环氧丙烷的聚合物)
3.聚环氧乙烷(peo),通常被称为聚乙二醇,由于其具有独特的性能(例如化学稳定性、 其亲水性、其生物相容性、尤其是其难以被免疫系统识别的隐身效应等)已成为美国fda批 准用于临床的聚合物。官能团在链端的存在使得生物活性分子与peo能够结合(即聚乙二醇 化)。因此,可以将所谓的“聚乙二醇化货物”在不被免疫系统识别的情况下运输至靶点。 为了延长循环时间并改善其空间屏蔽效应,防止其被肾脏清除,经聚乙二醇化的结合物的流 体力学尺寸应超过6-8nm,该尺寸是肾小球滤过作用的阈值。然而,由于peo的非降解性, 以及其潜在的体内生物蓄积性,所使用peo的摩尔质量不应超过40kg/mol。
4.为了克服这个问题,已通过将可降解键引入peo主链内而给peo分子链赋予可降解性, 在这方面已经付出了很多努力。最常用的策略是通过遥爪型peo的缩聚,包括将酯类、二硫 化物、乙缩醛、肟、亚胺或碳酸酯键引入peo缩聚物中。然而,后面这种peo衍生物显示 出非常宽的多分散性和不确定的结构。
5.经典的策略包括将环氧乙烷与其它单体发生阴离子共聚合反应,然后将可降解键引入 peo主链内。例如,可以将共聚合的环氧乙烷(eo)与环氧氯丙烷经过高效的消除反应,从 而在peo主链内生成可降解的亚甲基环氧乙烷(meo)重复单元。类似地,可以通过阴离子 开环聚合反应(arop)使环氧乙烷与3,4-环氧-1-丁烯(epb)发生共聚合,然后可以使epb 的烯丙基部分异构化为ph可裂解的乙烯基醚类。另外一种策略包括对所制备的peo或市售 的peo进行后氧化处理,以生成沿其主链分布的可水解键。例如,经过芬顿反应(fentonreaction),在中性ph条件下,半缩醛可由过氧化氢和氯化铁无规地引入peo主链。另外还 有报道显示,聚乙二醇类聚合物(pegs)在钌的催化作用下,发生聚合后加氧官能化反应, 生成酸可裂解的聚乙二醇-羟基乙酸共聚物。尽管后面这两种方法赋予可降解聚乙二醇以确定 的结构以及较低的多分散性,但也存在以下弊端:这种聚合物的合成所需的步骤较多,而且 在聚合后步骤中,与官能团的相容性问题应加以解决。
6.聚左旋丙交酯(plla)是另一种重要的聚合物,由于其生物相容性和可降解性以及其来 自生物资源的可获得性因而广泛地应用于生物医学领域。然而,由于其高结晶性、疏水性和 可降解性,已发现plla具有不同于peo的生物医学应用。左旋丙交酯(lla)与其它单体 的共聚合代表了调整其物理性能以适应各种生物医学应用的一般策略。例如,已通过多种单 体与lla的序列聚合反应(sequential polymerization)获得了二嵌段共聚物或三嵌
段共聚物。 关于环氧化物单体(即环氧乙烷),在文献中仅报道了有限数量的描述它们与lla的共聚合 的研究。有一项报道,除了设法由peo大分子引发剂进行plla嵌段增长外,还提到了使用 多种铝(al)系催化剂和锡-铝(sn-al)双金属催化剂来制备具有宽分子量分布的lla-eo 多嵌段共聚物。另一项报道是采用经典的范登堡(vandenberg)催化剂以获得高摩尔质量的 lla-eo无规共聚物。在这些报道的每个报道中,主要的焦点是使用各种配位催化剂来研究 lla与环氧化物的“可共聚性”并且对最终获得的共聚物类型进行表征:即在第一种情况下 获得的多嵌段共聚物和在第二种情况下的无规共聚物。
7.将co2掺入聚合物中不仅可以充分利用该温室气体以解决全球变暖问题而且给聚合物赋 予可降解性能,这对于面临白色污染的全世界来说是非常可取的。因此,自从1960年代由inoue 进行的开创性工作到现在,环氧化物与co2的共聚合已成为热点课题。虽然人们已对大多数 环氧化物的这种共聚合进行了充分研究,对于环氧乙烷(eo)来说,虽然通过乙烯氧化的年 生产规模约为3000万吨,目前甚至以乙醇为原料通过生物法生产,但在这方面的研究还不够。 作为最简单的环氧化物,eo与其它环氧化物单体相比,在(共)聚合过程中有其独特性。首 先,由于它的摩尔质量最小,对co2的利用率也最高。如果eo与co2的共聚合是交替的, 可以掺入50质量%的co2,所产生的聚碳酸乙烯酯(pec)是完全疏水的、生物相容的和生 物可降解的,其可以用作离子导电电解质、弹性体,特别是生物材料。具有高碳酸乙烯酯(ec) 含量的peceo的合成最初是使用znet2/h2o催化体系而实现,这可以生产具有在81%-94% 范围内的碳酸酯含量的peceo。然而,该聚合不能很好地控制并且所获得的peceo显示较 宽的摩尔质量分布。使用钴基金属有机催化剂,获得具有良好交替结构的聚碳酸乙烯酯 (pec)。尽管取得了这些成就,但存在与金属催化剂相关的缺点,比如终产物的着色、金 属残留物的毒性、以及催化合成工艺步骤较多。
8.第二个特性是,由eo均聚反应得到的无任何co2掺入的peceo(即peo)是亲水性的, 这与由其它环氧化物经过开环聚合(rop)所产生的聚醚类完全不同。也有人尝试利用不稳 定碳酸酯作为可降解键来赋予可降解的性能。在碱存在下和苛刻反应条件下(160-200℃), 碳酸乙烯酯(ec)的开环聚合(rop)可以给peo提供在10-25mol%范围内的碳酸酯含量, 然而不能很好地控制聚合,赋予聚合物的亲水性不知何故丢失,有一篇报道中有仅有的例外, 其中获得了“类peg(peg-like)”的聚醚碳酸酯(摩尔质量高达10000g/mol,pdi《1.6), 其碳酸酯含量较低(《10mol%)。
9.自从在2016年发现了无金属co2/环氧化物共聚合以来,三乙基硼烷(teb)介导(催化) 的(共)聚合越来越普及用于聚碳酸酯、聚醚、聚酯的合成以及最近的聚氨酯的合成。尤其 是,通过调整单体与引发剂之间的投料比以及co2的聚合压力,可以容易地调整所合成聚碳 酸酯的摩尔质量(从百万到千g/mol)和碳酸酯含量(50-95%)。
10.因此,亟需具有改善可降解性的类peo聚合物和聚醚共聚物,以用于各种工业、医学和 化妆品用途。
技术实现要素:
11.总体而言,本公开的实施方案描述了可降解的聚醚型共聚物,包括无规共聚物、递变共 聚物和嵌段共聚物。例如,本公开描述了合成下列物质的材料和方法:可降解的疏水性eo 基共聚物(例如,聚碳酸乙烯酯)、可降解的两亲性eo基嵌段共聚物(例如,聚醚碳酸
有选自以下的化学式:{y
+
,ro-}、{y
+
,rcoo-}、{x
+
,n
3-}和{x
+
,cl-};其中y
+
是选自k
+
、 t-bup
4+
和t-bup
2+
;其中x
+
是选自nbu
4+
、pbu
4+
、noct
4+
和ppn
+
;其中ro-是选自是选自ch3o(ch2)2o(ch2)2o-和h2c=chch2o-,其中rcoo-是脂肪族或 芳香族羧酸酯。在某些情况下,引发剂是四丁基琥珀酸铵、四丁基氯化铵、四辛基氯化铵或 双(三苯基膦)氯化亚胺。环氧乙烷单体与引发剂的摩尔比可以在约1000:1至约50:1的范围内。 活化剂与引发剂的摩尔比可以在约5:1至约1:1范围内。该方法还可以包括以在约1至约30 巴范围内的恒定压力通入二氧化碳。在一个或多个实施方案中,环酯是存在的并且可以是丙 交酯或者是选自由以下组成的组:l-丙交酯(左旋丙交酯)、d-丙交酯(右旋丙交酯)、内 消旋丙交酯以及它们的混合物。在一个或多个实施方案中,环酐是存在的并且是选自由芳香 族和脂肪族酸酐组成的组。在某些情况下,环酐是邻苯二甲酸酐、琥珀酸酐、二甘醇酸酐或 马来酸酐。
21.在另一个方面,本公开的实施方案包括一种形成可降解嵌段共聚物的方法,其包括:在 溶剂、烷基硼烷活化剂和鎓盐引发剂的存在下,使第一环氧乙烷单体与二氧化碳、环酯或环 酐接触,以形成具有聚醚键和碳酸酯或酯键的第一嵌段,其中碳酸酯或酯在共聚物主链中的 含量不超过共聚物的50重量%;以及使第一嵌段与第二环氧乙烷单体接触,以形成与第一嵌 段连接的第二嵌段。该可降解嵌段共聚物可以在无金属的条件下形成。第一环氧乙烷单体可 以选自由以下组成的组:
[0022][0023]
其中r3和r4各自独立地选自由烷基组成的组,这些烷基包括饱和和不饱和的、芳香族 和环状烷基、含叠氮化物的烷基以及含杂原子的烷基,其中杂原子是卤素、n、o、p、si、 se或s。在一个或多个实施方案中,n、p、s和se原子已被氧化。n杂原子可以被季铵化。 在某些情况下,环氧乙烷单体是环氧乙烷。第二环氧乙烷单体可以是环氧乙烷。可降解嵌段 共聚物可以在同一锅中通过序列开环聚合反应而形成。该方法还可以包括在使第一嵌段与第 二环氧乙烷单体接触之前释放二氧化碳。
[0024]
本公开的另一个方面描述了一种通过以下工艺而制备的可降解嵌段共聚物,该工艺包括: 在溶剂、烷基硼烷活化剂和鎓盐引发剂的存在下,使第一环氧乙烷单体与二氧化碳、环酯或 环酐接触,以形成具有聚醚键和酯键或碳酸酯键的第一嵌段,其中碳酸酯在共聚物主链中的 含量最高为共聚物的50重量%;以及使第一嵌段与第二环氧乙烷单体接触,以形成与第一嵌 段连接的第二嵌段。该嵌段共聚物可以是二嵌段共聚物或三嵌段共聚物。该嵌段共聚物可以 是aba型三嵌段共聚物,其包括亲水性的a嵌段(含有第一嵌段)和疏水
性的b嵌段(含 有第二嵌段)。a嵌段在aba型嵌段共聚物中的含量可以在20重量%至80重量%的范围内。 在某些情况下,a嵌段在aba型嵌段共聚物中的含量可以在40重量%至70%重量的范围内。 b嵌段可以是环氧乙烷与co2的共聚物,其中碳酸酯的含量最高为b嵌段的50重量%。该嵌 段共聚物可以具有选自由以下组成的组中的至少一种性能:该嵌段共聚物具有至少0.05g/l 的临界胶束浓度;该嵌段共聚物形成流体力学半径至少为的的胶束;以及,该嵌段共聚 物具有在2-9或11-18范围内的亲水亲油平衡值(hlb)。
[0025]
在下面的描述中陈述了一个或多个实施例。基于描述和权利要求,本公开的其它技术特 征、目的和优点将是显而易见的。
附图说明
[0026]
本书面公开内容描述了非限制性和非详尽的示例性实施方案。另外,在未必按比例绘制 的附图中,在所有的若干视图中相同的数字基本上描述相似的部分。具有不同字母后缀的相 同数字代表基本上相似部分的不同情况。这些附图一般是通过举例而非限制的方式来说明本 文件中所论述的各种实施方案。
[0027]
参考在附图中所描绘的示例性实施方案,在附图中:
[0028]
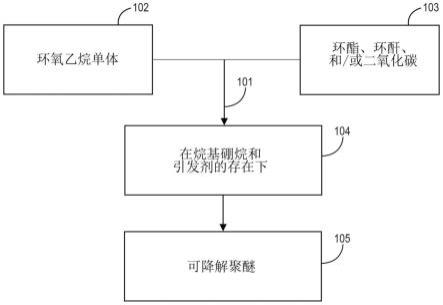
图1是根据本公开一个或多个实施方案的、形成可降解共聚物的方法的流程图。
[0029]
图2是根据本公开一个或多个实施方案的、形成可降解共聚物的反应图式的示意图。r1 和r2独立地选自脂肪族基和芳香族基,并且可以是饱和的或不饱和的。
[0030]
图3是根据本公开一个或多个实施方案的、形成可降解星形聚醚的方法的流程图。
[0031]
图4是根据本公开一个或多个实施方案的、p(eo-co-lla)无规共聚物(表1的第7项) 的代表性核磁共振氢谱图(1h nmr spectrum)。
[0032]
图5是根据本公开一个或多个实施方案的、目标聚合度(dp)为100至500的各种共聚 物样品(表1)的凝胶渗透色谱(gpc)图形视图。
[0033]
图6是根据本公开一个或多个实施方案的、共聚物(表1第21项)的红外(ir)光谱, 图中示出了叠氮化物掺入。
[0034]
图7是根据本公开一个或多个实施方案的、溶解于甲苯的p4/pmba(表2第1、2、3项) 的竞聚率图。
[0035]
图8是根据本公开一个或多个实施方案的、溶解于甲苯的tbacl(表2第4、5、6项) 的竞聚率图。
[0036]
图9是根据本公开一个或多个实施方案的、溶解于甲苯的ppncl(表2第7、8、9项) 的竞聚率图。
[0037]
图10是根据本公开一个或多个实施方案的、具有不同酯组成的共聚物p(eo-co-lla)的 差示扫描量热法(dsc)谱图。
[0038]
图11是根据本公开一个或多个实施方案的、在降解前后共聚物p(eo-co-lla)(表1第 12项)的gpc叠加谱图。
[0039]
图12示出了根据本公开一个或多个实施方案的均聚星形peo(peo homostars) (pvdox-eo)的合成的反应图式。
[0040]
图13示出了根据本公开一个或多个实施方案的表4第21项的1h nmr表征。
[0041]
图14示出了根据本公开一个或多个实施方案的表4第21项的gpc图。
[0042]
图15示出了根据本公开一个或多个实施方案的表4第22项的1h nmr表征。
[0043]
图16示出了根据本公开一个或多个实施方案的表4第22项的gpc图。
[0044]
图17示出了根据本公开一个或多个实施方案的表4第23项的1h nmr表征。
[0045]
图18示出了根据本公开一个或多个实施方案的表4第23项的gpc图。
[0046]
图19示出了根据本公开一个或多个实施方案的表4第24项的1h nmr表征。
[0047]
图20示出了根据本公开一个或多个实施方案的表4第24项的gpc图。
[0048]
图21示出了根据本公开一个或多个实施方案的表4第25项的1h nmr表征。
[0049]
图22示出了根据本公开一个或多个实施方案的表4第25项的gpc图。
[0050]
图23示出了根据本公开一个或多个实施方案的表4第26项的1h nmr表征。
[0051]
图24示出了根据本公开一个或多个实施方案的表4第26项的gpc图。
[0052]
图25示出了根据本公开一个或多个实施方案的表5第27项的gpc图。
[0053]
图26示出了根据本公开一个或多个实施方案的表5第28项的gpc图。
[0054]
图27示出了根据本公开一个或多个实施方案的表5第30项的gpc图。
[0055]
图28示出了根据本公开一个或多个实施方案的表5第33项的gpc图。
[0056]
图29示出了根据本公开一个或多个实施方案的表5第33项的1h nmr表征。
[0057]
图30a-图30c示出了(a)p(eo-ec-eo)三嵌段共聚物的1h nmr谱图(cdcl3作为溶 剂);(b)p(eo-ec-eo)三嵌段共聚物及它们的相应的前体中间嵌段的尺寸排阻色谱(sec) 图;以及(c)以i1/i3值(利用荧光光谱法得到)对浓度(g/l)对数的拟合图,测定临界胶 束浓度(cmc)值,其中插图是利用动态光散射(dls)光谱法所获得的粒径分布图。
[0058]
图31a-图31f示出了(a)具有不同碳酸酯含量的eo与co2的共聚物的1h nmr谱图 (溶剂为cdcl3);(b)升温速率为10k/min的热重分析(tga)曲线;(c)升温速率为 10k/min的dsc曲线;(d)显示了在peceo表面上的水滴随时间的变化;(e)显示在 ph为8.5下peceo的降解的1h nmr谱图(溶剂为cdcl3);以及(f)age、eo和co2的三元共聚物的1h nmr谱图(溶剂为cdcl3)。
[0059]
图32示出了根据本公开一个或多个实施方案的、通过eo与co2的共聚合可以合成的各 种产物,包括:疏水性的聚碳酸乙烯酯、两亲性的三嵌段聚环氧乙烷-b-碳酸乙烯酯-环氧乙烷 共聚物、以及亲水性的可降解聚环氧乙烷。
[0060]
图33示出了根据本公开一个或多个实施方案的、eo与co2共聚合的粗产物的代表性1h nmr(溶剂是cdcl3)谱图。
[0061]
图34示出了纯peceo的代表性1h nmr谱图(cdcl3为溶剂)。
[0062]
图35示出了用于降解试验的peceo(第23项)的sec图(洗脱液为thf)。
[0063]
图36示出了在ph值为13的情况下,peceo(第23项)的降解产物的1h nmr谱图(溶 剂是cdcl3)。
[0064]
图37示出了在ph值为13的情况下,peceo(第23项)的降解产物的sec图(洗脱 液为thf)。
[0065]
图38示出了在ph值为8.5的情况下,在不同的降解时间,peceo(第23项)的降解产 物的sec图(洗脱液为thf)。
[0066]
图39示出了在ph值为7.4的情况下,peceo(第23项)的降解产物的1h nmr谱图 (溶剂为cdcl3)。
[0067]
图40示出了在ph值为7.4的情况下,peceo(第23项)的降解产物的sec图(洗脱 液为thf)。
[0068]
图41示出了在ph值为6.5
±
0.1的情况下,peceo(第23项)的降解产物的1h nmr 谱图(溶剂为cdcl3)。
[0069]
图42示出了在ph值为6.5的情况下,peceo(第23项)的降解产物的sec图(洗脱 液为thf)。
[0070]
图43示出了在eo/age比例为20,在1巴的co2压力下获得的共聚物的sec图(洗脱 液为thf)。
[0071]
图44示出了在eo/age比例为10,在1巴的co2压力下获得的共聚物的sec图(洗脱 液为thf)。
[0072]
图45示出了在eo/age比例为20,在1巴的co2压力下获得的共聚物的1h nmr(溶 剂为cdcl3)谱图。
[0073]
图46示出了反应图式a-c:通过teb介导的环氧乙烷与co2的共聚合而形成(a)可 降解疏水性共聚物、(b)两亲性共聚物以及(c)官能化peg类似物的合成路线。
具体实施方式
[0074]
本发明涉及用于形成可降解聚醚类等的方法。这些可降解聚醚可以包含具有含量可控且 可调的可降解酯键(例如,酯单元)或可降解碳酸酯键(例如,碳酸酯单元),这些可降解 键被掺入星形聚醚的聚醚主链或多官能核中。例如,本发明的实施方案包括制备成无规共聚 物的可降解聚醚类,这些聚醚类包含来自环酯(例如,l-丙交酯)、环酐的酯单元以及/或者 来自二氧化碳的碳酸酯单元,这些单元无规地掺入聚醚主链中。本发明的实施方案包括制备 成线型二嵌段共聚物或三嵌段共聚物的可降解聚醚类,其包含连接到单官能或二官能(大分 子)寡聚疏水物的聚醚的臂,这些疏水物包含碳酸酯单元或酯单元。本发明的实施方案还包 括制备成星形聚合物的可降解聚醚,这种星形聚合物包含连接到包含碳酸酯单元或酯单元的 多官能核的聚醚的臂。本文中所公开的方法提供对掺入可降解聚醚中的酯单元和碳酸酯单元 的量和/或长度的控制。例如,在一个实施方案中,可降解聚醚可以包含约5%的掺入聚醚主 链中的酯单元,每个酯单元的平均长度为大约两个相邻酯基或更短。通过以这种方式将可降 解键并入聚合物主链中,而给在不改变这些聚合物固有特性的情况下给该聚醚赋予可降解的 特征。
[0075]
本公开的可降解聚醚可以由环氧乙烷单体与环酯、环酐或二氧化碳通过阴离子开环共聚 合反应而直接地制备。阴离子共聚合反应可以在活化剂(即,烷基硼烷)和引发剂的存在下 进行。活化剂的存在可以选择性地提高环氧乙烷单体的反应性,并且抑制酯交换反应和/或环 碳酸酯的形成。例如,在化学计量条件下,活化剂和引发剂可以发生反应以形成酸根型配合 物。该酸根型配合物可以用于引发阴离子共聚合。在一些实施方案中,该酸根型配合物的增 加不具有足够的亲核性以激活环氧乙烷单体,在这种情况下可以以相对于引发剂化学计量过 量来提供活化剂,以确保环氧乙烷单体的活化。通过以这种方式进行,可以精确地控制可降 解键的含量,从而给明确的可降解醚赋予可控的摩尔质量和较窄的多分散性。另外,这种方 法是通用的,并且可以用于合成官能化的线型和/或支化的聚环氧乙烷、以及可降解的聚环氧 乙烷星形聚合物等。
[0076]
在一些实施方案中,这些可降解聚醚可以进一步制备成双官能或异聚双官能聚醚类,用 于修饰生物分子。例如,可降解聚醚可以形成为使得可降解聚醚类的末端具有官能团,这些 官能团允许生物活性分子与聚环氧乙烷经过通常称为聚乙二醇化的方式而相结合。因此,本 公开的实施方案进一步描述了经修饰的生物分子,这些经修饰的生物分子包含与本公开的可 降解聚醚相结合的生物活性分子。这样,诸如肽类、蛋白类和酶类等的生物活性分子可以经 由与可降解聚醚共价结合的方式进行修饰。
[0077]
定义
[0078]
下文中所用的术语具有如下定义。本文中的所有其它术语和短语应根据本领域的技术人 员所理解的通常含义进行解释。
[0079]
本文中所用的术语“可降解聚醚”是指包含可降解键的任何聚醚。例如,这些可降解键 可以提供在聚合物主链中,或者在介于聚合物主链与聚合物的一个或多个末端官能团之间的 基团中,或者在星形聚合物的多官能核中,或者其它位置。在星形聚合物的的语境中,可降 解星形聚醚可以包括具有多官能核的均聚星形(homostars)聚醚或异聚星形(heterostars)聚 醚,这些核包含可降解键。
[0080]
本文中所用的术语“可降解键”是指任何能够被降解的聚合物的单元或链段。术语“可 降解键”包括酯单元和碳酸酯单元。因此,“酯单元”、“可降解酯键”、以及“碳酸酯单 元”和“可降解碳酸酯键”等在本文中可互换地使用。这些化学键降解的机制可以取决于其 目标应用。例如,这些可降解键可以是水解键、酶解键、ph可降解键、酸可降解键等。
[0081]
本文中所用的术语“环酯”包括单酯类、环二酯类、环三酯类等。环酯的一个非限制性 示例是丙交酯。本文中所用的术语“丙交酯”可以指代丙交酯的三种立体异构型中的一种或 多种。丙交酯的三种立体异构型包括左旋丙交酯(l-lactide)、右旋丙交酯(d-lactide)和内 消旋丙交酯(meso-lactide)。
[0082]
本文中所用的术语“环酐”包括脂肪族酸酐和芳香族酸酐,环酯的非限制性示例是邻苯 二甲酸酐、琥珀酸酐、二甘醇酸酐或戊二酸酐。
[0083]
本文中所用的术语“酯单元”是指任何包含至少一个酯基的聚合物的链段。该聚合物可 以包含多个酯单元。每个酯单元可以包含一个或多个相邻的酯基。通常用化学式(-rc(=o)or
’ꢀ‑
)a来表示酯基,其中a的值至少为1,r和r’是通用的,且不作具体限定,而是可以取决 于该酯基来自于哪种单体。例如,每个酯单元可以包含一个或多个相邻的丙交酯。聚合物中 的酯单元可以用平均长度来描述,其中酯单元的平均长度可以指存在于聚合物中的相邻酯基 的平均数。
[0084]
本文中所用的术语“碳酸酯单元”是指任何包含至少一个碳酸酯基聚合物的链段。一种 聚合物可以包含多个碳酸酯单元。每个碳酸酯单元可以包含一个或多个相邻的碳酸酯基。通 常用化学式(-roc(=o)or
’‑
)a来表示碳酸酯基,其中a至少为1,其中r和r’是通用的, 不作具体限制,并且可以取决于碳酸酯基来自于哪种单体。例如,酯基可以包含一个或多个 相邻的碳酸单乙酯。聚合物的酯单元可以用平均长度来描述,其中酯单元的平均长度可以指 存在于聚合物中的相邻碳酸酯基的平均数。
[0085]
本文中所用的术语“脂肪族”或“脂肪族基”是指烃部分,其中该烃部分可以是直链的 (例如,不分枝或线型的)、支链的或环状的,和/或可以是完全饱和的,或者含有一个或多 个不饱和单元,只要不是芳香族的。术语“不饱和”是指具有一个或多个双键和/或三键
的部 分。因此,术语“脂肪族”包括烷基、环烷基、烯基、环烯基、炔基或环炔基、以及它们的 组合。脂肪族基可以包含30个碳原子或更少,或者包含在1至30范围内的任意数目的碳原 子,或者在1至30个碳原子范围内的任何递增数目。脂肪族基的非限制性示例包括线型或支 链的烷基、烯基和炔基、以及它们的混合物,如(环烷基)烷基、(环烯基)烷基和(环烷基)烯 基等。
[0086]
本文中所用的术语“烷基”是指饱和的、直链或支链的烃基,其中已有氢原子从脂肪族 部分中去除。烷基可以任选择地包含具有1至20个碳原子的直链或支链。烷基的非限制性示 例包括:甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、仲戊基、异戊 基、正戊基、新戊基、正己基、仲己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、 正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷 基、正十九烷基、正二十烷基、1,1-二甲基丙基、1,2-二甲基丙基、2,2-二甲基丙基、1-乙基丙 基、正己基、1-乙基-2-甲基丙基、1,1,2-三甲基丙基、1-乙基丁基、1-甲基丁基、2-甲基丁基、 1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、2-乙 基丁基、2-甲基戊基、3-甲基戊基等。
[0087]
本文中所用的术语“烯基”是指衍生自从具有至少一个碳-碳双键的直链或支链的脂肪族 部分中去除氢原子的基团。本文中所用的术语“炔基”是指衍生自从具有至少一个碳-碳三键 的直链或支链的脂肪族部分中去除氢原子的基团。烯基的非限制性示例包括:乙烯基、丙烯 基、丁烯基、1-甲基-2-丁烯-1-基、烯丙基、1,3-丁二烯基和丙二烯基。炔基的非限制性示例 包括:乙炔基、2-丙炔基和1-丙炔基。当“烯烃”指代h-r化合物或h-r部分时,其中r是 烯基。
[0088]
本文中所用的术语“脂环族的”、“碳环”或“碳环形的”是指具有3至20个碳原子的 饱和或部分不饱和的环状脂肪族单环或多环(包括稠合、桥连和螺-稠合)的环系统。脂环基 团可任选地具有3-15个碳原子、任选地3-12个、任选地3-10个、任选地3-8个、和/或任选 地3-6个碳原子。术语“脂环族的”、“碳环”或“碳环形的”还包含与一个或多个芳香族 环或非芳香族环稠合的脂肪族环,例如四氢萘环,其中接合点位于脂肪族环上。碳环基可以 是多环的,例如双环或三环。应当理解的是,脂环基可以包含带有一个或多个连接性或非连 接性烷基取代基的脂环族环,例如-ch
2-环己基。碳环的非限制性示例包括:环丙烷、环丁烷、 环戊烷、环己烷、双环[2,2,1]庚烷、降冰片烯、苯环、环己烯、萘、螺[4,5]癸烷、环庚烷、金 刚烷和环辛烷。
[0089]
本文中所用的术语“杂脂肪族基”(包括杂烷基、杂烯基和杂炔基)是指如上文所定义 的脂肪族基,其额外地含有一个或多个杂原子。杂脂肪族基可以任选地含有2-21个原子、任 选地2-16个、任选地2-13个、任选地2-11个、任选地2-9个,和/或任选地2-7个原子,其 中至少一个原子是碳原子。杂原子的非限制性示例包括o、s、n、p和si。在杂脂肪族基具 有两个或更多杂原子的情况下,这些杂原子可以是相同的或不同的。杂脂肪族基可以是经取 代的或未经取代的、支链的或不分枝的、环状的或无环的,并且包含饱和、不饱和、或部分 不饱和的基团。
[0090]
本文中所用的术语“脂环基”是指具有3-20个碳原子的饱和或部分不饱和的环状脂肪族 单环或多环的(包括稠合、桥连和螺-稠合)环系统。脂环基可以任选地具有3-15个碳原子, 任选地3-12个、任选地3-10个、任选地3-8个、和/或任选地3-6个碳原子。术语“脂
环族的
”ꢀ
包括环烷基、环烯基和环炔基。应当理解的是,脂环基团可以包含带有一个或多个连接性或 非连接性烷基取代基(例如-ch
2-环己基)的脂环族环。具体地,c
3-20
环烷基的示例包括:环 丙基、环丁基、环戊基、环己基、环庚基、金刚烷基和环辛基。
[0091]
本文中所用的术语“杂脂环基团”是指如上文所定义的脂环基团,其除了碳原子之外, 还具有一个或多个环杂原子,这些杂原子任选地选自o、s、n、p和si。杂脂环基团可以任 选地包含1-4个杂原子,这些杂原子可以的相同的或不同的。杂脂环基团可以任选地含有5 至20个原子、任选地5至14个、和/或任选地5至12个原子。
[0092]
本文中所用的术语“芳基”、“芳香基团”或“芳基环”是指具有5至20个碳原子的单 环或多环的环系统,其中该系统中的至少一个环是芳香族环,并且其中各环含有3至12个环 成员。术语“芳基”可以单独使用,或者用作如芳烷基、芳烷氧基或芳氧基烷基等更大基团 的一部分。芳基的非限制性示例包括:苯基、甲基苯基、(二甲基)苯基、乙基苯基、联苯基、 茚基、蒽基、萘基或薁基等。术语“芳香基团”包括例如茚满、苯并呋喃、邻苯二甲酰亚胺、 菲啶和四氢萘等稠环类。当“芳烃”指代化合物h-r时,其中r是芳基。
[0093]
本文中所用的术语“杂芳基”,在单独使用或用作另一个术语(如杂芳烷基或杂芳烷氧 基)的一部分时,是指具有5至14个环原子的单环或多环基,除了碳原子之外还具有1至5 个杂原子。术语“杂原子”是指氮、氧或硫,并且包括任何氧化形式的氮或硫、以及任何季 铵化形式的氮。术语“杂芳基”还包括其中的杂芳基环与一个或多个芳基环、脂环族环或杂 环基环相稠合的基团,其中接合的基团或接合点位于杂芳香族环上。杂芳基的非限制性示例 包括:吲哚基、异吲哚基、苯并噻吩基、苯并呋喃基、二苯并呋喃基、吲唑基、苯并咪唑基、 苯并噻唑基、喹啉基、异喹啉基、噌啉基、酞嗪基、喹唑啉基、喹喔啉基、4氢-喹嗪基、咔 唑基、吖啶基、吩嗪基、吩噻嗪基、吩噁嗪基、四氢喹啉基、四氢异喹啉基、呋喃基、咪唑 基、吲哚基、吲唑基、甲基吡啶基、噁唑基、吡啶基、吡咯基、嘧啶基、吡嗪基、喹啉基、 喹唑啉基、喹喔啉基、噻吩基、三嗪基和吡啶并[2,3-b]-1,4-噁嗪-3(4h)-酮。
[0094]
本文中所用的术语“芳烷基”是指如前文所定义的烷基,其中一个氢原子被芳基和/或杂 芳基取代,从而形成杂芳烷基,其中任选地烷基、芳基和/或杂芳基部分独立地被取代。当引 用于杂环的环原子时,术语“氮”包括经取代的氮。芳烷基的非限制性示例是苯甲基(benzyl、 bn)和2-苯基-乙基。
[0095]
脂环基团、杂脂环基团、芳基和杂芳基的非限制性示例包括但不限于:环己基、苯基、 吖啶、苯并咪唑、苯并呋喃、苯并噻吩、苯并噁唑、苯并噻唑、咔唑、噌啉、二噁英、二氧 六环、二氧戊环、二噻烷、二噻嗪、二噻唑、二噻茂烷、呋喃、咪唑、咪唑啉、咪唑啶、吲 哚、吲哚啉、吲哚嗪、吲唑、异吲哚、异喹啉、异噁唑、异噻唑、吗啉、萘啶、噁唑、噁二 唑、噁噻唑、噁噻唑烷、噁嗪、噁二嗪、吩嗪、吩噻嗪、吩噁嗪、酞嗪、哌嗪、哌啶、蝶啶、 嘌呤、吡喃、吡嗪、吡唑、吡唑啉、吡唑烷、哒嗪、吡啶、嘧啶、吡咯、四氢吡咯、吡咯啉、 喹啉、喹喔啉、喹唑啉、喹嗪、四氢呋喃、四嗪、四唑、噻吩、噻二嗪、噻二唑、噻三唑、 噻嗪、噻唑、硫代吗啉、硫杂萘、噻喃、三嗪、三唑和三噻烷。
[0096]
本文中所用的术语“卤化物”、“卤代”和“卤素”可以互换地使用,并且表示氟原子、 氯原子、溴原子、碘原子等,任选地表示氟原子、溴原子或氯原子,任选地表示氟原子。术 语“卤代烷基”包括氟化基或氯化基,包括全氟化合物。卤代烷基的非限制性示例包括:氟 甲基、二氟甲基、三氟甲基、氟乙基、二氟乙基、三氟乙基、氯甲基、溴甲基、碘甲基等。
[0097]
本文中所用的术语“烷芳基”是指如前面所定义的芳基和/或杂芳基,其中一个或多个氢 原子被如前面定义的烷基和/或杂烷基所取代。
[0098]
本文中所用的术语“烷氧基”是指-or基团,其中r是如本文所定义的烷基和/或杂烷基。 烷氧基的非限制性示例包括:-och3、-och2ch3、-och2ch2ch3、-och(ch3)2、-och(ch2)2、
ꢀ‑
oc3h6、-oc4h8、-oc5h
10
、-oc6h
12
、-och2c3h6、-och2c4h8、-och2c5h
10
、-och2c6h
12
等。烷氧基的非限制性示例还包括:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异 丁氧基、仲丁氧基、叔丁氧基、正戊氧基、异戊氧基、仲戊氧基、正己氧基、异己氧基、正 庚氧基、正辛氧基、正壬氧基、正癸氧基、正十一烷氧基、正十二烷氧基、正十三烷氧基、 正十四烷氧基、正十五烷氧基、正十六烷氧基、正十七烷氧基、正十八烷氧基、正十九烷氧 基、正二十烷氧基、1,1-二甲基丙氧基、1,2-二甲基丙氧基、2,2-二甲基丙氧基、2-甲基丁氧基、 1-乙基-2-甲基丙氧基、1,1,2-三甲基丙氧基、1,1-二甲基丁氧基、1,2-二甲基丁氧基、2,2-二甲 基丁氧基、2,3-二甲基丁氧基、1,3-二甲基丁氧基、2-乙基丁氧基、2-甲基戊氧基和3-甲基戊 氧基等。
[0099]
本文中所用的术语“烯氧基”、“炔氧基”、“芳氧基”、“芳烷氧基”、“杂芳氧基
”ꢀ
和“酰氧基”是指定义为-or的基团,其中r分别是烯基、炔基、芳基、芳烷基、杂芳基和 酰基。示例包括但不限于:芳氧基(如-o-ph)和芳烷氧基(如-och
2-ph(-obn)和-och2ch
2-ph)。
[0100]
本文中所用的术语“任选地取代”表示在任选地经取代的基团中一个或多个氢原子被合 适的取代基所取代。除非另有说明,“任选地取代”的基团可以在该基团每一个可取代的位 置具有合适的取代基,而且对于任何给定的结构,如果其中多个可被取代的位置被多个选自 特定组的取代基取代时,每个位置上的取代基可以相同,也可以不同。本公开所构想的取代 基的组合,任选地为可以形成稳定化合物的那些取代基。适用于本发明的取代基的非限制性 示例包括:卤素、羟基、硝基、羧酸酯、碳酸酯、烷氧基、芳氧基、烷硫基、芳硫基、杂芳 氧基、烷基芳基、氨基、酰氨基、亚胺、腈、甲硅烷基、甲硅烷基醚、酯、亚砜、磺酰基、 乙炔化物、亚膦酸酯、磺酸酯,或任选取代的脂肪族基、杂脂肪族基、脂环基团、杂脂环基 团、芳基或杂芳基(例如,任选地被卤素、羟基、硝基、碳酸酯、烷氧基、芳氧基、烷硫基、 芳硫基、氨基、亚胺、腈、甲硅烷基、亚砜、磺酰基、亚膦酸酯、磺酸酯或乙炔化物取代) 等。
[0101]
a.可降解聚醚
[0102]
本公开的实施方案描述了可降解聚醚,其含有掺入其中的可降解键,而且这些可降解键 的含量可调可控。在一些实施方案中,可降解聚醚可制备为无规共聚物,其中酯单元或碳酸 酯单元无规则地掺入聚醚主链。例如,该可降解聚醚可以包含衍生自环酯(例如丙交酯)、 环酐(邻苯二甲酸酐)的酯单元或者衍生自二氧化碳的碳酸酯单元,并且这些单元掺入聚环 氧乙烷主链。这种可降解聚醚的非限制性示例包括聚环氧乙烷-丙交酯共聚物(poly(ethyleneoxide-co-lactide))和聚环氧乙烷-碳酸乙酯共聚物(poly(ethylene oxide-co-ethyl carbonate))等。 可降解聚醚也可以制备为二官能或异聚二官能共聚物,其中该可降解聚醚的末端可以具有多 个适于生物结合和应用的官能团。在其它实施方案中,可降解聚醚可制备为线型二嵌段、线 型三嵌段共聚物或星形聚合物,其中碳酸酯单元或酯单元掺入其线型单官能臂、二官能臂或 多官能核,并且所述核具有与其连接的聚醚臂。这种可降解聚醚的非限制性示例包括均聚星 形聚环氧乙烷,其连接到可降解聚碳酸酯核。
[0103]
在一些实施方案中,聚合物的主链包括聚环氧乙烷。例如,聚合物主链可以是聚环
氧乙 烷主链,该主链其可以是线型或支链的、经取代或未经取代的、以及官能化或非官能化的。 在一个实施方案中,聚环氧乙烷主链通常可以用以下化学式来表示:
[0104]
(-cr
2-cr
2-o-)n[0105]
其中,各r独立地选自氢、烷基、杂烷基、环烷基、烯基、杂烯基、环烯基、炔基、杂 炔基、环炔基、烷氧基、芳基、杂芳基、芳烷基、亚烷基、烷芳基、亚烷芳基(aralkylene)、 卤素、或者它们的组合,其中每个基团可以是经取代或未经取代的、官能化或非官能化的; 其中n至少为1。在一个实施方案中,聚环氧乙烷主链是官能化的线型聚环氧乙烷。在一个 实施方案中,聚环氧乙烷主链是官能化的支链聚环氧乙烷。
[0106]
衍生自环酯、环酐的酯单元或者衍生自二氧化碳的碳酸酯单元可以掺入(例如,无规掺 入)聚环氧乙烷主链中或者掺入可降解聚醚(例如,均聚星形聚环氧乙烷)的多官能核中。 本文中所用的术语“酯单元”是指包含至少一个酯基(例如,-rc(=o)or
’‑
)的共聚物的任 何链段。例如,在一个实施方案中,酯单元可以包含一个或多个相邻的丙交酯单元(例如, 左旋丙交酯单元),其中丙交酯单元由以下化学结构表示:
[0107][0108]
术语“碳酸酯单元”是指包含至少一个碳酸酯基(例如,-roc(=o)or
’‑
)的共聚物的 任何链段。例如,在一个实施方案中,碳酸酯单元可以包含一个或多个相邻的碳酸单乙酯单 元,其中碳酸单乙酯单元由以下化学结构表示:
[0109][0110]
在一个实施方案中,可降解聚醚可以由以下化学结构表示:
[0111][0112]
其中,m《n或m《《n;其中x选自cl、br、n3、oh、o-、ch2=chch2o-、或它们的 组合。
[0113]
在一个实施方案中,可降解聚醚可以由以下化学结构表示:
[0114][0115]
其中m《n或m《《n;其中x选自cl、br、n3、oh、o-、ch2=chch2o-、或它们的组 合。
[0116]
在一个实施方案中,可降解聚醚的核可以由以下化学结构表示:
组合。
[0122]
在一个实施方案中,可降解聚醚可以是线型亲水性共聚物(例如,peoec),任选地由 以下化学结构表示的官能化peg类似物:
[0123][0124]
其中m《n或m《《n;x和y可以为0或正整数,x+y《m;其中*是-ch
2-o-ch2ch=ch2或者可以是通过硫醇-烯烃点击反应(thiol-ene click reaction)或通过缩醛形成而衍生的任何基 团。
[0125]
可降解聚醚可以包含带官能团的侧链,从而允许生物活性分子与聚环氧乙烷的接合。例 如,本公开的方法包括制备官能化可降解聚醚共聚物的方法,其包括:在溶剂、烷基硼烷活 化剂和鎓盐引发剂的存在下,使环氧乙烷和官能化环氧化物与二氧化碳接触,以形成聚合物 主链中掺入有可降解碳酸酯键以及具有侧基官能团的聚环氧乙烷,其中聚合物中碳酸酯的含 量约为20重量%或更少。例如,为了维持peo的性能,碳酸酯的含量应该低于10mol%,例 如约5mol%。这种烷基硼烷活化剂可以选自三乙基硼烷、三苯基硼烷、三丁基硼烷、三甲基 硼烷、三异丁基硼烷、以及它们的组合。鎓盐引发剂可以选自上述的组。可降解聚醚可以在 无金属的条件下形成。官能化的可降解聚合物可以是三元共聚物或嵌段共聚物。该方法可以 包括以至少10:1(例如约20:1或更大)范围内的摩尔比投入环氧乙烷和官能化环氧化物。聚 合可以包括环氧乙烷与环酯、环酐或二氧化碳的共聚合;或者首先进行环氧乙烷与环酯、环 酐或二氧化碳的序列聚合(sequential polymerization),然后在第二步骤中进行环氧乙烷的均 聚反应。反应物可以溶解于配位溶剂,如四氢呋喃。环氧乙烷与溶剂可以以介于3:1至约1:1 的比例(v:v)存在。引发剂可以具有选自以下的化学式:{y
+
,ro-}、{y
+
,rcoo-}、{x
+
,n
3-} 和{x
+
,cl-};其中y
+
选自k
+
、t-bup
4+
和t-bup
2+
;其中x
+
选自nbu
4+
、pbu
4+
、noct
4+
和ppn
+
; 其中ro-选自:ch3o(ch2)2o(ch2)2o-和 h2c=chch2o-,其中rcoo-是脂肪族或芳香族羧酸酯,任选地鎓盐引发剂是四丁基琥珀酸铵、 四丁基氯化铵、四辛基氯化铵或双(三苯基膦)氯化亚胺。环氧乙烷与引发剂可以以介于约 5000:1至约1000:1的摩尔比存在。活化剂与引发剂可以以介于2:1至1:1的摩尔比存在。可 以将二氧化碳以小于4巴(如2巴或更少、或约1巴)的恒定压力通入反应器中。共聚物可 以具有至少80kg/mol的摩尔质量(数)。该共聚物的分散度可以小于1.2,比如小于1.15或 约为1.1。在某些情况下,官能化环氧化物选自由以下组成的组:叠氮烷基缩水甘油醚、丙烯 基缩水甘油醚、异丙亚基甘油缩水甘油醚、乙氧基乙基缩水甘油醚、乙氧基乙烯基缩水甘油 醚、n,n-二苄基氨基缩水甘油、甲基丙烯酸缩水甘油酯、1,2-环氧基-5-己烯、1,2-环氧-7-辛烯、 1,2-环氧-9-葵烯、或者它们的组合。可以执行聚合后修饰,即硫醇-烯烃官能化。这些官能化 共聚物可以通过不同方式进行衍生,例如通过与侧链上的巯基反应而生成聚硫醚,例如与醇 反应而生成侧链聚缩醛。
[0126]
这些可降解聚醚是可以清晰确定的,并且具有介于大于约0kg/mol至约50kg/mol,甚至 高达约850kg/mol的摩尔质量。在一个实施方案中,该可降解聚醚的摩尔质量约为24kg/mol 或更小。在一些实施方案中,这些可降解聚醚的摩尔质量可以约为50kg/mol、约为35kg/mol 或更小、约为30kg/mol或更小、约为25kg/mol或更小、约为24kg/mol或更小、约为23kg/mol 或更小、约为22kg/mol或更小、约为21kg/mol或更小、约为20kg/mol或更小、约为19kg/mol 或更小、约为18kg/mol或更小、约为17kg/mol或更小、约为16kg/mol或更小、约为15kg/mol 或更小、约为14kg/mol或更小、约为13kg/mol或更小、约为12kg/mol或更小、约为11kg/mol 或更小、约为10kg/mol或更小、约为9kg/mol或更小、约为8kg/mol或更小、约为7kg/mol 或更小、约为6kg/mol或更小、约为5kg/mol或更小、约为4kg/mol或更小、约为3kg/mol 或更小、约为2kg/mol或更小、或者约为1kg/mol或更小。在其它实施方案中,可降解星形 聚醚的摩尔质量约为850kg/mol或更小,或者是在0kg/mol至850kg/mol之间的任意值或区 间。
[0127]
这些可降解聚醚也可以具有窄的多分散性。在一个实施方案中,可降解聚醚的多分散性 指数可以在约1至约1.6的范围内。例如,这些可降解聚醚的多分散性指数可以为约1.6、约 1.5、约1.4、约1.30、约1.29、约1.28、约1.27、约1.26、约1.25、约1.24、约1.23、约1.22、 约1.21、约1.20、约1.19、约1.18、约1.17、约1.16、约1.15、约1.14、约1.13、约1.12、 约1.11、约1.10、约1.09、约1.08、约1.07、约1.06、约1.05、约1.04、约1.03、约1.02、 约1.01、或者约1.00。
[0128]
图1是根据本公开一个或多个实施方案的、利用阴离子开环共聚合而形成可降解聚醚的 方法的流程图。如图1所示,方法100可以通过以下步骤进行:在烷基硼烷和引发剂104的 存在下,使环氧乙烷单体102与环酯、环酐和/或二氧化碳103接触101,以形成聚合物主链 中掺入有可降解碳酸酯键或可降解酯键的聚醚105。图2示出了形成可降解聚醚的反应图式 的示意图。
[0129]
接触通常是通过使环氧乙烷单体、环酯、环酐、二氧化碳、烷基硼烷和/或引发剂进行物 理接触、直接接触或紧密接触而进行。各种组分或化学物质的接触,可以按任意顺序,同时 或相继地进行,而不作具体限定。每种化学物质可以都在溶剂中接触,例如非极性溶剂或弱 极性溶剂。例如,在一个实施方案中,这种溶剂可以选自甲苯和四氢呋喃,以及其它这类溶 剂。可以在介于约0℃至约100℃范围内或者其中的任何值或区间的温度下进行接触。优选地, 在室温附近的温度下进行接触,例如介于约20℃至约30℃之间的温度。接触的持续时间应该 使其足以进行共聚合反应。例如,接触的持续时间可以在介于约1分钟至约1000分钟的范围 内,或者在某些情况下更长。
[0130]
在包括诸如丙交酯等环酯的实施方案中,活化剂和引发剂可以任选地分别与环氧乙烷单 体和环酯接触。例如,在一个实施方案中,活化剂和引发剂可以先在溶剂内接触以形成第一 溶液,然后环氧乙烷单体和环酯可以分别在溶剂内接触以形成第二溶液。然后第一溶液和第 二溶液再进行接触,任选地在搅拌条件下接触,从而使反应进行。在引发剂含有两种组分的 实施方案中,引发剂可以任选地在与活化剂接触之前形成。例如,在一个实施方案中,引发 剂前体物质可以在溶剂内接触,以生成引发剂,然后引发剂可再与活化剂在溶剂内相接触, 以形成第一溶液。在涉及二氧化碳的实施方案中,引发剂和二氧化碳可以任选地接触,再溶 于溶剂中以形成第一溶液,然后活化剂可以与溶剂接触以形成第二溶
液。第一溶液和第二溶 液可以再进行接触,然后可加入环氧乙烷单体,从而使反应进行(例如,在1巴的二氧化碳 压力下反应)。
[0131]
在选定链长或者有所需链长的情况下,环氧乙烷单体与环酯、环酯或二氧化碳的摩尔比 可以进行选择和调整,以获得具有不同酯单元或碳酸酯单元含量(和可调含量)的可降解聚 醚。一般来讲,环氧乙烷单体的加入量相对于环酯、环酐或二氧化碳是化学计量过量的。例 如,环氧乙烷单体与环酯的摩尔比可以介于约1.01:1至约10:1之间。在一个实施方案中,摩 尔比可以为约2:1、约3:1、约4:1、约5:1、约6:1、约7:1、约8:1、约9:1、约10:1,或者是 这些比例之间的任何递增值。
[0132]
合适的环氧乙烷单体包含下式表示的单体:
[0133][0134]
其中r1和r2可以各自独立地不存在,或者各自独立地选自氢、烷基、杂烷基、环烷基、 烯基、杂烯基、环烯基、炔基、杂炔基、环炔基、烷氧基、芳基、杂芳基、芳烷基、亚烷基、 烷芳基、亚烷芳基、卤素、或它们的组合,其中每个基团可以是经取代或未取代的。在一些 实施方案中,r1和r2连接起来并形成稠环,例如环结构中含有5个或更多的碳原子,其中任 何一个碳原子可以任选地被杂原子取代。在一些实施方案中,r1和/或r2包含一个或多个附 加的环氧乙烷单体。例如,在一些实施方案中,环氧乙烷单体可以表征为双环氧化合物单体、 三环氧化合物单体等。合适的环氧乙烷单体的非限制性示例包括:
[0135][0136]
其中r3和r4各自独立地是一种或多种烷基,包括饱和和不饱和的、芳香族的、环状烷 基、含杂原子(例如,卤化物、n3、o、s等)烷基。这些示例都应该是非限制性的,同理, 可以用于本公开的其它环氧乙烷单体,而不脱离本公开的保护范围。例如,r3和r4可以各自 独立地选自由以下烷基组成的组:包括饱和和不饱和的、芳香族烷基和环状烷基、含叠氮化 物的烷基,以及含杂原子的烷基,其中的杂原子为卤素、n、o、p、si、se或s,其中n、p、 s、se原子任选地被氧化,n原子任选地被季铵化,任选地,环氧乙烷单体是环氧乙烷。
[0137]
环酯可以选自任何环状化合物(例如,环烷烃和环烯烃等),并且其具有一个或多个被 化学式为-c(o)o-的酯单元/基团取代的碳原子。合适的环酯包括但不限于:环状单酯、环状 二酯、环状三酯等。合适的环酯的非限制性示例包括丙交酯、三亚甲基碳酸酯、乙交酯、ββ 丁内酯、δ内戊内酯、γ内丁内酯、γ内戊内酯、4-甲基二氢-2(3h)-呋喃酮、α喃甲基-基酮丁 内酯、ε内己内酯、1,3-二氧戊环-2-酮、碳酸丙烯酯、4-甲基-1,3-二氧六环-2-酮、1,3-二氧杂 环庚烷-2-酮、5-c
1-4
烷氧基-1,3-二氧六环-2-酮、以及它们的混合物或衍
生物;以上这些中的 任一种都可以是未取代或经取代的。在一些优选实施方案中,环酯包括丙交酯单体。丙交酯 单体可以选自l-丙交酯、d-丙交酯、内消旋丙交酯、以及它们的组合。进一步地,丙交酯单 体可以是经取代或未经取代的。例如,丙交酯的甲基可被一个或多个选自氢、烷基、杂烷基、 环烷基、烯基、杂烯基、环烯基、炔基、杂炔基、环炔基、烷氧基、芳基、杂芳基、芳烷基、 亚烷基、烷芳基、亚烷芳基、卤素、或它们的组合的取代基所取代,以上这些中的任一种都 可以是经取代或未经取代的。上述取代基不应该是限定性的,本领域技术人员已知的任何取 代基都可用于被发明。
[0138]
环酐可以是饱和的环酐和不饱和的环酐、或者是其混合物。“饱和的”酸酐包括那些不 具有活性烯基不饱和键的酸酐类,但可具有芳香族环。示例性的饱和环酐包括但不限于:琥 珀酸酐、邻苯二甲酸酐、四氢邻苯二甲酸酐、烷基和芳基取代的琥珀酸酐、饱和卤代环酐(例 如,四溴邻苯二甲酸酐)以及它们的混合物。在某些情况下,环酐选自不饱和环酐(即,具 有烯基不饱和键的环酐)或选自不饱和环酐和饱和环酐的混合物。不饱和环酐的例子包括但 不限于:马来酸酐、柠康酸酐、衣康酸酐、不饱和卤代环酐以及它们的混合物。在一个或多 个实施方案中,环酐可以是一种或多种芳香族酐和脂肪族酐,例如邻苯二甲酸酐、琥珀酸酐、 二甘醇酸酐、戊二酸酐、马来酸酐以及它们的混合物。
[0139]
可以对活化剂进行选择,以实现以下的一个或多个:选择性地活化环氧乙烷单体,与引 发剂形成酸根型配合物,抑制酯交换反应,以及抑制环碳酸酯的形成。烷基硼烷通常是以相 对于引发剂化学计量过量来提供。在一个实施方案中,烷基硼烷和引发剂的比例可以为约5:1。 在一些实施方案中,烷基硼烷与引发剂的比例是在约1:1至约5:1的范围内。在其它实施方案 中,烷基硼烷与引发剂的比例可以为约2:1、约3:1、约4:1、约5:1、约6:1、约7:1、约8:1、 约9:1、约10:1、或者更大。本文中所述方法中所使用的活化剂可以是烷基硼烷,如三烷基硼 烷。合适活化剂的非限制性示例包括:三乙基硼烷、三苯基硼烷、三丁基硼烷、三甲基硼烷、 三异丁基硼烷、以及它们的组合。在某些实施方案中,烷基硼烷是三乙基硼烷。
[0140]
与活化剂形成酸根型配合物的引发剂可以包括盐类或有机碱。盐类和有机碱可以包括与 阴离子相关或混合的有机阳离子或碱金属。例如,在一个实施方案中,引发剂包括与具有有 机取代基的醇盐相关或混合的有机阳离子。在一个实施方案中,引发剂包括与具有有机取代 基的醇盐相关或混合的碱金属。在一个实施方案中,引发剂包括与叠氮化物相关或混合的有 机阳离子。在一个实施方案中,引发剂包括与卤素相关或混合的有机阳离子。
[0141]
有机阳离子可以基于磷腈(phosphazenium)、铵(ammonium)和磷(phosphonium)中 的一种或多种。例如,在一个实施方案中,有机阳离子可以基于磷腈碱,例如t-bu-py,其中 y为2或4;或者铵盐或磷盐,其中氮或磷与四个烷基相连,每个烷基可以是相同或不同的。 碱金属可以包括任何碱金属。例如,在一个实施方案中,碱金属可以选自锂、钾、钠、以及 它们的组合。阴离子可以包括任何带负电荷的物质。例如,在一个实施方案中,阴离子可以 选自羟基、酯类、酸类、醇盐(alkoxide)、叠氮化物和卤素。醇盐可由任何具有至少一个羟 基的醇而制备。可以使用任何卤素。例如,在一个实施方案中,卤素可以选自cl-和br-。
[0142]
在一个实施方案中,引发剂可以选自以下化学式:
[0143]
{y
+
,ro-}、{y
+
,rcoo-}、{x
+
,n
3-}和{x
+
,cl-};
[0144]
其中y
+
选自k
+
、t-bup
4+
、和t-bup
2+
;x
+
是选自nbu
4+
、pbu
4+
、noct
4+
和ppn
+
;ro-选自ch3o(ch2)2o(ch2)2o-、h2c=chch2o-、例 如,引发剂可以选自和/或制备自对甲基苄醇(pmba)与磷腈碱(t-bup4)、二乙二醇单甲 醚(dgme)与t-bup4、双酚a(bpa)与t-bup4、pmba与磷腈碱(t-bup2)、四丁基氯化 铵(tbac或bu4ncl)、双(三苯基膦)氯化亚胺(ppncl)、四辛基氯化铵(toacl或oct4cl)、 四丁基氯化膦(tbpcl)、叠氮化四丁基铵(tbaa)、以及烯丙醇与t-bup4。
[0145]
引发剂可以是单官能引发剂,例如四丁基氯化铵、四辛基氯化铵和双(三苯基膦)氯化亚 胺,也可以是双官能引发剂,例如四丁基琥珀酸铵(tbas)。引发剂可以根据文献进行合成 和纯化。
[0146]
在一个实施方案中,形成可降解聚醚的方法可以如以下所示的反应图式进行:
[0147][0148]
r4n
+
(铵),r4p
+
(磷),p4h
+
(磷腈)
[0149]
ci,br,n3,-o,ch2=chch2o-,rco2[0150]
本公开的实施方案进一步描述了经修饰的生物分子,这些经修饰的生物分子包含与可降 解聚醚结合的生物活性分子,该可降解聚醚具有掺入聚环氧乙烷主链中的酯单元或碳酸酯单 元。通常,该生物活性分子是通过与可降解聚醚的共价结合而进行修饰。该生物活性分子可 以选自蛋白类、肽类、酶类、医药化学品或有机基团、以及它们的组合。该可降解聚醚可以 包含本公开的共聚物中的任一种。
[0151]
b.可降解星形聚醚
[0152]
图3是根据本公开一个或多个实施方案的、形成可降解星形聚醚的方法的流程图。如图 3所示,该方法300可以通过以下步骤进行:在引发剂和第一量的烷基硼烷存在下,使双环 氧化物单体与二氧化碳、环酯和/或环酐接触301。在此步骤中,双环氧化物单体可以与二氧 化碳、环酯和/或环酐发生共聚合(例如,通过阴离子开环共聚合),以产生具有碳酸酯单元 和/或酯单元的多官能核。例如,碳酸酯单元可以衍生自二氧化碳,以产生可降解的碳酸酯键。 酯单元可以衍生自环酯或环酐,以产生可降解酯键。碳酸酯单元和/或酯单元的
存在可以给所 产生的多官能核赋予可降解性。这种多官能核的示例包括但不限于:聚碳酸酯核、聚醚核、 聚酯核等。
[0153]
接触301可以通过按任意顺序相继或同时地将引发剂、溶剂、烷基硼烷、双环氧化物单 体、二氧化碳、和/或环酯、和/或环酐加入反应容器,任选地可以在机械搅拌下加入。例如, 在一些实施方案中,合适的制备顺序包括在机械搅拌或无机械搅拌的条件下,相继地将引发 剂加入反应容器,接着相继地加入溶剂、烷基硼烷和双环氧化物单体。在加入前述组分中的 一种或多种时,可以将二氧化碳、环酯和/或环酐加入反应容器中,并且可以进行共聚合反应。 在共聚合反应期间或者在共聚合反应过程中,在引发剂和烷基硼烷的存在下,双环氧化物单 体的环氧环可以开环,并且各自可以与二氧化碳、环酯和/或环酐进行共聚合。这样,双环氧 化物单体可以作为交联剂,将至少两个聚合物链连接起来,通过共聚合形成各聚合物链。
[0154]
合适的引发剂、溶剂和/或烷基硼烷已在上文有描述,因此这里不再赘述。双环氧化物单 体可以选自包含至少两个环氧化物的任何单体。合适的双环氧化物单体的示例包括二氧化乙 烯基环己烯及其衍生物。其它合适的双环氧化物单体包括但不限于:二氧化丁二烯;1,2,3,4
‑ꢀ
二环氧丁烷;1,2,7,8-二环氧辛烷;1,2,5,6-二环氧环辛烷;二环戊二烯双环氧化物;聚(乙二醇 二缩水甘油);甘油二缩水甘油等二缩水甘油醚类,以及如1,3-丙二醇、1,4-丁二醇、1,6-己二 醇、环己烷-1,4-二醇、环己烷-1,1-二甲醇、环己烷-1,2-二甲醇、环己烷-1,3-二甲醇、环己烷-1,4
‑ꢀ
二甲醇、二甘醇、对苯二酚、间苯二酚、4,4-异亚丙基双酚、萘二酚等的二缩水甘油醚类等; 或者它们的衍生物。虽然对这些双环氧化物单体进行了描述,但本公开中也可以使用其它多 官能环氧化物,包括例如三环氧化物等。
[0155]
交联的范围或程度会影响所形成的多官能核的可降解性。例如,虽然交联可以基于试剂 的选择和反应条件等,但高度的交联可能不会产生可降解多官能核而形成凝胶。因此,在进 行共聚合时,可取的是将双环氧化物单体的交联的范围或程度保持或维持在低水平至中等水 平。例如,这可通过使用低到中等量的双环氧化物单体来实现。例如,在一些实施方案中, 将双环氧化物单体与引发剂的摩尔比保持在低于约10,但不大于约20。例如,双环氧化物单 体与引发剂的摩尔比可以为约20或更少、约19或更少、约18或更少、约17或更少、约16 或更少、约15或更少、约14或更少、约13或更少、约12或更少、约11或更少,优选地为 约10或更少、约9或更少、约8或更少、约7或更少、约6或更少,或者更优选地为约5或 更少、约4或更少、约3或更少、约2或更少,或者其中的任意值或区间。
[0156]
双环氧化物单体与溶剂的体积比可以在约1:1至约1:10的范围内。例如,在一些实施方 案中,双环氧化物单体与溶剂的体积比为约1:1、约1:1.5、约1:2、约1:2.5、约1:3、约1:3.5、 约1:4、约1:4.5、约1:5、约1:5.5、约1:6、约1:6.5、约1:7、约1:7.5、约1:8、约1:8.5、约 1:9、约1:9.5、或约1:10,或者在它们之间的任何区间或者其中的任何值。
[0157]
可以在介于约0.01巴至约25巴之间的压力下,将二氧化碳通入反应容器。例如,在一 些实施方案中,可以在介于约5巴至约15巴之间的压力下通入二氧化碳,优选地约10巴。 在其它实施方案中,在以下压力下通入二氧化碳:约1巴、约2巴、约3巴、约4巴、约5 巴、约6巴、约7巴、约8巴、约9巴、约10巴、约11巴、约12巴、约13巴、约14巴、 约15巴、约16巴、约17巴、约18巴、约19巴、约20巴、约21巴、约22巴、约23巴、 约24巴、或约25巴,或者是其中的任何值或区间。
[0158]
执行步骤301的温度可以在约0℃至约100℃的范围内。在一些实施方案中,进行接触的 温度是在约50℃至约80℃的范围内。例如,进行接触的温度可以在约50℃、约51℃、约52℃、 约53℃、约54℃、约55℃、约56℃、约57℃、约58℃、约59℃、约60℃、约61℃、约62℃、 约63℃、约64℃、约65℃、约66℃、约67℃、约68℃、约69℃、约70℃、约71℃、约72℃、 约73℃、约74℃、约75℃、约76℃、约77℃、约78℃、约79℃、或约80℃,或者在其间 的任意值或其中的任何区间。另外,进行接触的持续时间可以约为1周或更少,优选地少于 约24小时,或者更优选地少于约17小时,例如约15小时。
[0159]
当在步骤301中形成多官能核以及任选地使反应容器冷却时,可降解星形聚醚的臂可以 聚合。因此,在步骤302中,在第二量的烷基硼烷存在下,将来自步骤301的可降解多官能 核与环氧乙烷单体进行接触。在随后的反应中,环氧乙烷单体发生聚合,从而产生连接到可 降解多官能核的聚醚的臂,由此形成可降解星形聚醚。在一些实施方案中,星形聚醚的臂在 化学上是相同的,从而提供均聚星形物。在一些实施方案中,可以使两种或更多种环氧乙烷 单体发生反应,或者可以使除环氧乙烷单体以外的单体发生反应,以提供具有不同臂的异聚 星形物,或者带有包含共聚物的臂的星形聚合物(例如,嵌段共聚物),或者其它类型的聚 合物。
[0160]
为了形成星形聚醚的臂,可以将环氧乙烷单体加入反应容器中。合适的环氧乙烷单体已 在上文中进行了描述,因此这里不再赘述。在一些实施方案中,在将未反应的二氧化碳清除 或释放之后,将包含环氧乙烷单体的溶液、溶剂和第二量的烷基硼烷注入反应容器中。在涉 及环酯的实施方案中,可以使步骤301中的反应进行,直到环酯完全或全部消耗,或者可以 将未反应的环酯从反应容器中分离和/或去除。在加入环氧乙烷单体、溶剂和第二量的烷基硼 烷时,可以任选地在机械搅拌下,使聚合反应进行,以形成星形聚合物的聚醚臂。
[0161]
环氧乙烷单体与溶剂的体积比可以在约1:1至约1:20的范围内。例如,在一些实施方案 中,环氧乙烷单体与溶剂的体积比为约1:1、约1:2、约1:3、约1:4、约1:5、约1:6、约1:7、 约1:8、约1:9、约1:10、约1:11、约1:12、约1:13、约1:14、约1:15、约1:16、约1:17、约 1:18、约1:19、或者约1:20,优选地为约1:5至约1:15,或者更优选地为约1:10。
[0162]
尽管不是必须的,但在一些实施方案中,在本发明方法的步骤301中所加入的烷基硼烷 是以相对于引发剂为化学计量而加入反应容器,它们各自反应而生成酸根型配合物,酸根型 配合物可以用于活化步骤301中的共聚合。在某些情况下,可将步骤302中第二量的烷基硼 烷加入反应容器,使得烷基硼烷以化学计量过量而存在,以活化环氧乙烷和开环聚合。在一 些实施方案中,过量的烷基硼烷可用于活化聚醚臂聚合中的环氧乙烷单体。在一些实施方案 中,烷基硼烷的第一量与第二量是相同的。在一些实施方案中,烷基硼烷的第一量与第二量 是不同的。例如,在一些实施方案中,烷基硼烷的第一量少于烷基硼烷的第二量。在一些实 施方案中,烷基硼烷的第一量大于烷基硼烷的第二量。
[0163]
在一些实施方案中,在接触302时或者在整个环氧乙烷单体的聚合中,或者在这两种情 况下,可将多官能核与烷基硼烷的摩尔比选定或维持在约1:1至约1:10的范围内。例如,在 一些实施方案中,将多官能核与烷基硼烷的摩尔比选定或维持在约1:1、约1:1.5、约1:2、约 1:2.5、约1:3、约1:3.5、约1:4、约1:4.5、约1:5、约1:5.5、约1:6、约1:6.5、约1:7、约1:7.5、 约1:8、约1:8.5、约1:9、约1:9.5、或约1:10,或者其中的任何值或区间。优选地,双
环氧化 物单体与烷基硼烷的摩尔比是在约1:3至约1:5的范围内,或者其中的任何值,更优选地约 1:3。
[0164]
执行步骤302的温度可以在约0℃至约100℃的范围内。在一些实施方案中,在约30℃ 至约50℃范围内的温度下进行接触。例如,接触可以在约30℃、约31℃、约32℃、约33℃、 约34℃、约35℃、约36℃、约37℃、约38℃、约39℃、约40℃、约41℃、约42℃、约43℃、 约44℃、约45℃、约46℃、约47℃、约48℃、约49℃、或约50℃、或者在它们之间的任 意值或其中的任何区间的温度下进行。优选地,聚合反应是在约40℃的温度下进行。另外, 进行接触的持续时间可以是约1周或更少,优选地约24小时或更少。
[0165]
在进一步的步骤303(未示出)中,可以用溶解于醇(例如甲醇)的酸(例如hcl)使 来自步骤302的反应混合物淬灭。为了获得细微的产物,可以将粗产物溶解和/或沉淀于二乙 醚中,然后进行离心和干燥。
[0166]
c.疏水性共聚物
[0167]
本公开还涉及环氧乙烷单体与二氧化碳、环酯或环酐反应生成的疏水性共聚物。用于形 成疏水性共聚物的合成路线可在无金属的条件下进行。在这些实施方案中,引发体系只需包 括有机阳离子和硼烷作为活化剂,用于在无金属的条件下进行共聚合。
[0168]
该方法可以包括第一个步骤:在活化剂(包括三烷基硼烷)、溶剂和引发剂的存在下, 使环氧乙烷单体(例如,eo)与二氧化碳、环酯或环酐接触。该方法可以包括在通入二氧化 碳或加入环酯或环酐之前将活化剂从环氧乙烷单体中加以分离,以防止发生均聚合。环氧乙 烷单体可包括以下中的一种或多种:环氧乙烷、环氧丙烷、1-环氧丁烷、1-环氧己烷、1-环氧 辛烷、环氧苯乙烷、环氧环己烷、烯丙基缩水甘油醚、和丁基缩水甘油醚。
[0169]
接触和/或加入可指代使两种或更多的组分接近,比如在物理和/或化学上接近。在许多实 施方案中,接触可包括在反应容器中加入和/或混合入两种或更多的组分,和/或将足以使至少 两种组分在物理和/或化学上接近的气态组分通入反应室(包括反应容器)。在某些情况下, 接触包括确保混合物是均匀的。在许多实施方案中,接触和加入通常是在相同的活化剂和引 发剂的存在下进行。在其它实施方案中,接触和加入可在不同的活化剂和/或不同的引发剂的 存在下进行。
[0170]
接触可以在约0℃至约100℃范围内或者其中的任何值或区间的温度下进行。优选地,接 触是在室温附近进行,例如在约20℃至约30℃的范围内的温度。接触的持续时间应当足以进 行共聚合反应。例如,接触的持续时间可以在约1分钟至约1000分钟的范围内(例如,约 15小时),或者在某些情况下更长(例如,长达约50小时)。
[0171]
可以使用上述的任何活化剂和引发剂。在许多实施方案中,活化剂包括以下中的一种或 多种:三乙基硼烷(teb)、三甲基硼烷、三异丁基硼烷和三苯基硼烷。在其它实施方案中, 活化剂可包括烷基硼烷和/或烷基铝。引发剂可包括有机阳离子,例如磷腈、铵和磷。引发剂 可以是单官能引发剂或双官能引发剂。
[0172]
在一些实施方案中,以相对于引发剂的化学计量将活化剂加入反应容器中,它们各自反 应而形成酸根型配合物,酸根型配合物可以用于活化共聚合。可以对活化剂进行选择以实现 以下中的一个或多个:选择性地活化环氧乙烷单体,与引发剂形成酸根型配合物,抑制酯交 换反应,以及抑制环碳酸酯的形成。烷基硼烷活化剂通常是以相对于引发剂化学计量过量来 加入。在一个实施方案中,烷基硼烷和引发剂的比例可以在约1:1至约5:1
的范围内。在一些 实施方案中,烷基硼烷和引发剂的比例可以为约1.2:1、约1.4:1、约1.6:1、约1.8:1、约2:1、 约2.2:1、或更大。第一个步骤中所使用的活化剂可以是烷基硼烷。合适的活化剂的非限制性 示例包括:三乙基硼烷、三苯基硼烷、三丁基硼烷、三甲基硼烷、三异丁基硼烷,以及它们 的组合。在某些实施方案中,烷基硼烷是三乙基硼烷。第一个步骤中所使用的引发剂可以是 如上所述的单官能或双官能引发剂,比如tbacl、toacl、ppncl、或tbas。例如,为了 合成具有高碳酸酯含量的聚碳酸酯第一嵌段,活化剂可以是teb,引发剂可以是tbas,并 且活化剂与引发剂的比例约为1:1-1.6:1。
[0173]
例如,根据本发明的实施方案,可以选择性地对合成路线进行修改,以将碳酸酯含量或 酯含量调整为约50%至约100%、约80%至约99%、以及约90%至约95%。通过增加碳酸酯 含量或酯含量,可以降低共聚物的润湿性。例如,选择性修改可以包括增加所形成共聚物中 的碳酸酯或酯的量。在某些情况下,疏水性共聚物具有至少约50%的碳酸酯含量或酯含量, 比如约80%、约85%、约90%、约91%、约92%、约93%、或约95%。通过改变单体与引发 剂的投料比以及二氧化碳的聚合压力,可以调整碳酸酯含量。
[0174]
例如,可以在约0.01巴至约30巴范围内的压力下将二氧化碳通入反应容器。在一些实 施方案中,可以在约1巴至约30巴之间范围内的压力下通入二氧化碳。在其它实施方案中, 在约1巴、约2巴、约3巴、约4巴、约5巴、约6巴、约7巴、约8巴、约9巴、约10巴、 约11巴、约12巴、约13巴、约14巴、约15巴、约16巴、约17巴、约18巴、约19巴、 约20巴、约21巴、约22巴、约23巴、约24巴、约25巴、约26巴、约27巴、约28巴、 约29巴、或约30巴,或者是其中的任何值或区间的压力下通入二氧化碳。在某些情况下, 在30巴的压力下将二氧化碳通入反应容器。
[0175]
根据本发明的实施方案,可以选择性地修改合成路线,以获得特定的目标聚合度(dp)。
[0176]
环氧乙烷单体与溶剂的体积比可以在约0.1:1至约1:5的范围内变化。例如,在一些实施 方案中,环氧乙烷单体与溶剂的体积比为约0.5:1、约1:1、及约1:2。溶剂可以选自配位溶剂 和非极性溶剂。例如,溶剂可以是thf、甲苯、或己烷。在某些情况下,溶剂是thf。
[0177]
在一些实施方案中,第一环氧乙烷单体是以相对于引发剂为化学计量而加入反应容器中。 例如,第一环氧乙烷单体与引发剂的比例可以是约100至约4000,例如约100、约200、约 500、约1000、约2000、和约4000。在某些情况下,第一环氧乙烷单体与引发剂的比例为100。
[0178]
在一个实施方案中,可降解聚醚可以是由以下化学结构表示的疏水性共聚物:
[0179][0180]
(例如,peceo),其中n《m或n《《m;其中x选自cl、br、n3、oh、o-、ch2=chch2o-、 或者它们的组合。因此,疏水性共聚物的特征在于,具有与eo含量相比相对较高的碳酸酯 (ec)含量。碳酸酯含量可以在大于50%至约99%的范围内变化。
[0181]
d.可降解嵌段共聚物
[0182]
本公开还涉及可降解嵌段共聚物,包括二嵌段共聚物和三嵌段共聚物,如ab型和/
或aba 型嵌段共聚物,其中嵌段a不同于嵌段b。这种可降解嵌段共聚物可以是使用具有不同润湿 性的嵌段的两亲性共聚物。这些嵌段共聚物可包含由上述任何聚醚共聚物所组成的嵌段,例 如根据方法100的共聚物和上述疏水性聚醚,嵌段可以以任意方式排列。例如,在某些情况 下,嵌段a是亲水性嵌段,例如可以是peo基嵌段(例如,纯聚环氧乙烷或含有酯键或碳酸 酯键的聚环氧乙烷);而嵌段b是疏水性嵌段,例如co2基聚碳酸酯嵌段,其中含有的碳酸 酯含量高于嵌段a(例如,聚碳酸乙烯酯-环氧乙烷共聚物(poly(ethylene carbonate-co-ethyleneoxide),peceo)),或者环酯基或环酐基聚乙烯嵌段,其中含有的酯含量高于嵌段a。在 某些情况下,这种嵌段共聚物的亲水亲油平衡值(hlb)介于约2-9之间或约11-18之间。 嵌段b可以包括环氧乙烷与co2的疏水性共聚物,按该嵌段的重量计,其中的碳酸酯含量至 少为60%,例如,至少为约70%、至少为约75%、至少为约80%、至少为约85%、至少为约 90%、约90-98%、或者约92-95%。在某些情况下,按该共聚物的重量计,嵌段b中的碳酸 酯含量或酯含量可以不超过60%(例如,不高于共聚物的50%)。嵌段a可以包含环氧乙烷 与co2的亲水性共聚物,按重量计,该共聚物具有不大于50%,比如不大于25%、不大于20%、 不大于15%、不大于12%、不大于10%、不大于7%、不大于5%、或不大于3%的碳酸酯含 量。因此,嵌段a和嵌段b可以包含可降解的碳酸酯键。
[0183]
本公开的实施方案包括aba型嵌段共聚物,该共聚物包含亲水性嵌段a(其含有聚乙烯 主链)和疏水性嵌段b(其含有基于co2的聚碳酸酯主链)。在某些情况下,按aba型嵌 段共聚物的重量计,嵌段a的含量为20%至80%,例如约30%至约70%、约35%至约75%、 约40%至约60%。aba型嵌段共聚物可以用临界胶束浓度(cmc)进行表征。例如,在25℃ 下,本公开的aba型嵌段共聚物的cmc可以至少为0.003g/l,例如至少约0.005g/l、或至 少约0.08g/l。aba型嵌段共聚物可以形成流体力学半径至少为约的胶束。例如,由本 公开的aba型嵌段共聚物所形成胶束的流体力学半径可以至少为约例如至少约或至少约胶束直径可在室温下(例如25℃)测量。
[0184]
本公开中的方法提供了多种简易、可扩展、可修改的合成路线。用于形成可降解嵌段共 聚物的合成路线可以在无金属的条件下进行。在这些实施方案中,引发体系只需包括鎓盐引 发剂和三烷基硼烷作为活化剂,用于在无金属的条件下进行共聚合。
[0185]
这种方法可以包括第一个步骤,在三烷基硼烷活化剂、溶剂和引发剂的存在下,使第一 环氧乙烷单体(例如,eo)和二氧化碳接触,以形成第一嵌段(例如,聚碳酸酯嵌段),然 后加入第二环氧乙烷单体,以形成与第一嵌段连接的第二嵌段(例如,聚环氧烷嵌段)。例 如,这种嵌段共聚物可以通过一锅法,以环氧乙烷的序列开环聚合反应来制备。该方法可以 包括排出二氧化碳,然后加入第二环氧乙烷单体。然而,在某些情况下,二氧化碳的存在可 以提供包含聚醚共聚物的第二嵌段,并且这种聚醚共聚物带有可降解碳酸酯键(例如,无规 共聚物或梯度共聚物)。
[0186]
第一环氧乙烷单体可以包括以下中的一种或多种:环氧乙烷、环氧丙烷、1-环氧丁烷、 1-环氧己烷、1-环氧辛烷、环氧苯乙烷、环氧环己烷、烯丙基缩水甘油醚和丁基缩水甘油醚。 第二环氧乙烷单体可以与第一环氧乙烷单体相同,或者第一环氧乙烷单体与第二环氧乙烷单 体可以是不同的。在某些情况下,第一环氧乙烷单体和第二环氧乙烷单体是环氧乙烷。
[0187]
接触和/或加入可指代使两种或更多的组分接近,例如在物理和/或化学上接近。
在许多实 施方案中,接触可包括在反应容器中加入和/或混合两种或更多的组分,和/或用足以使至少两 种组分在物理和/或化学上接近的气态组分通入反应室(包括反应容器)。在许多实施方案中, 接触和加入通常是在相同的活化剂和引发剂的存在下进行。在其它实施方案中,接触和加入 可在不同的活化剂和/或不同的引发剂的存在下进行。
[0188]
接触可以在约0℃至约100℃范围内或者其中的任何值或区间的温度下进行。优选地,接 触是在室温附近的温度下进行,例如在约20℃至约30℃范围内的温度下进行。接触的持续时 间应当足以进行共聚合反应。例如,第一个接触步骤的接触持续时间可以在约1分钟至约1000 分钟的范围内(例如,约15小时),或者在某些情况下更长(例如,长达约50小时)。第 二个接触步骤的持续时间可以在约1分钟至约300分钟的范围内(例如,约4小时),或者 在某些情况下更长(例如,长达约50小时)。
[0189]
在一些实施方案中,以相对于引发剂的化学计量将活化剂加入反应容器中。可以对活化 剂进行选择以实现以下中的一个或多个:选择性地活化环氧乙烷单体,与引发剂形成酸根型 配合物,抑制酯交换反应,以及抑制环碳酸酯的形成。烷基硼烷活化剂通常以相对于引发剂 化学计量过量来提供。在一个实施方案中,烷基硼烷与引发剂的比例可以在约1:1至约5:1 之间的范围内。在一些实施方案中,烷基硼烷和引发剂的比例可以为约1.2:1、约1.4:1、约 1.6:1、约1.8:1、约2:1、约2.2:1、或者甚至更高。第一个步骤中所使用的活化剂可以是烷基 硼烷。合适的活化剂的非限制性示例包括:三乙基硼烷、三苯基硼烷、三丁基硼烷、三甲基 硼烷、三异丁基硼烷、以及它们的组合。在某些实施方案中,烷基硼烷是三乙基硼烷。第一 步骤中所使用的引发剂可以是如上所述的单官能或双官能引发剂,比如tbacl、toacl、 ppncl、或者tbas。例如,为了合成具有高碳酸酯含量的聚碳酸酯第一嵌段,活化剂可以是 teb,引发剂可以是tbas,并且活化剂和引发剂的比率为约1:1-1.6:1。
[0190]
可以根据本发明的实施方案选择性地对用于形成可降解嵌段共聚物的合成路线进行修 改,以将第一嵌段的碳酸酯含量调整为约50%至约100%、约60%至约99%、约80%至约98%、 或者约90%至约95%。在某些情况下,第一嵌段具有至少约50%的碳酸酯含量,例如约80%、 约85%、约90%、约91%、约92%、约93%、或者约95%。通过增加碳酸酯含量可以降低第 一嵌段的润湿性并提高疏水性。该选择性修改可以包括增加所形成共聚物中碳酸酯的量。
[0191]
通过改变单体与引发剂的投料比以及二氧化碳的压力,可以调整第一嵌段的碳酸酯含量 (50-95%)。例如,在第一个步骤中,可以在约0.01巴至约30巴范围内的压力下,将二氧 化碳通入反应容器。在一些实施方案中,可以在约1巴至约30巴范围内的压力下,通入二氧 化碳。在其它实施方案中,在以下的压力下通入二氧化碳:约1巴、约2巴、约3巴、约4 巴、约5巴、约6巴、约7巴、约8巴、约9巴、约10巴、约11巴、约12巴、约13巴、 约14巴、约15巴、约16巴、约17巴、约18巴、约19巴、约20巴、约21巴、约22巴、 约23巴、约24巴、约25巴、约26巴、约27巴、约28巴、约29巴、或约30巴,或者其 中的任何值或区间。在某些情况下,在30巴的压力下将二氧化碳通入反应容器。
[0192]
可以选择性地对用于形成可降解嵌段共聚物的合成路线进行修改,以获得嵌段或嵌段共 聚物的特定目标聚合度(dp)。例如,可以改变活化剂和环氧乙烷单体的比例,以提供聚合 度为200-4,000(比如约500)的第一嵌段。
[0193]
环氧乙烷单体与溶剂的体积比可以在约0.1:1至约1:5的范围内变化。例如,在一
些实施 方案中,环氧乙烷单体与溶剂的体积比为约0.5:1、约1:1、和约1:2。溶剂可以选自配位溶剂 和非极性溶剂。例如,溶剂可以是thf、甲苯、或己烷。在某些情况下,溶剂是thf。
[0194]
在一些实施方案中,以相对于引发剂的化学计量将第一环氧乙烷单体加入反应容器中。 例如,第一环氧乙烷单体与引发剂的比例可以介于约100至约1000之间,例如约100、约200、 约500、和约1000。在某些情况下,第一环氧乙烷单体与引发剂的比例介于约100至约500 的范围内。
[0195]
在第一步骤中生成的第一嵌段用作第二步骤的大分子引发剂。可以基于目标dp改变大 分子引发剂与第二环氧乙烷单体的比例。例如,可以使用相对于大分子引发剂化学计量过量 (至少50、至少100、至少125、或至少150)的第二环氧乙烷单体,而获得目标聚合度(dp) 至少为25(例如约35-40、或约75-80、或更大)的可降解嵌段成分。
[0196]
在某些情况下,方法包括释放二氧化碳并加入第二环氧乙烷单体,以形成均聚嵌段(比 如peo)。例如,本公开的实施方案包括由化学结构(vii)表示的两亲性嵌段共聚物(例如, p(eo-eceo-eo)):
[0197][0198]
其中m《n或m《《n。peo的重量分数可以变化,从而给共聚物赋予不同的表面活化性能。 通过控制peo含量,可以对可降解嵌段共聚物的润湿性进行调节。嵌段共聚物的peo含量 可以为至少40重量%、至少重量50%、至少60重量%或更高。本公开的两亲性嵌段共聚物 可以具有亲水性(即,hlb》10)或疏水性(亲脂性,即hlb《10)。具有不同hlb值的两 亲性嵌段共聚物的混合物可以用于增强分散性或者水体系中的去污和/或除油性能。临界胶束 浓度(cmc)也可用来表征本公开的两亲性共聚物的表面活性效率。
[0199]
以下的实施例是用来说明上述的发明而不应被理解成缩小其范围。本领域技术人员将容 易地认识到,实施本发明的许多其它方式也是建议的,并且可以作出多种变型和修改而仍属 于本公开的范围内。
[0200]
实施例1
[0201]
通过环氧乙烷与l-丙交酯的阴离子共聚合而制备可降解聚环氧乙烷
[0202]
以下的实施例描述了一种用于制备可降解聚环氧乙烷(peo)的简便方法。在三乙基硼 烷的存在下,通过环氧乙烷与l-丙交酯(lla)的阴离子共聚合,而将极低含量的lla无规 地掺入peo的主链中。借助于三乙基硼烷的路易斯酸,减弱lla的反应性,并且抑制酯交 换反应。eo与lla的共聚合形成p(eo-co-lla)样品,其具有低含量至中等含量的酯单元、 受控的摩尔质量、和窄的多分散性。利用kelen-t
ü
dos法和meyer-lowry末端模型法测定了 竞聚率。利用差示扫描量热法(dsc)对所形成的共聚物进行了进一步研究;进行了水解实 验以显示这些peo样品的可降解性。
[0203]
本实施例中所提供工作的目的是:通过eo与lla的阴离子共聚合,将低至极低百分比 的lla单元掺入peo链内,从而在不改变它们的亲水性、结晶性等固有特性的情况下给这 些peo链赋予可降解性。具体地研究了三乙基硼烷在eo与lla的阴离子共聚合中的作用。 不
同于给lla-eo共聚物提供宽摩尔质量分布且通常摩尔质量分布不确定的配位催化途径, 由硼活化的eo与lla的阴离子共聚合反应产生明确的p(eo-co-lla)共聚物样品,这些样品 显示出窄的多分散性,并且eo和lla单元的含量可调(参见下面的反应图式)。下面的反 应图式示出了使用三乙基硼烷作为活化剂的环氧乙烷与l-丙交酯的阴离子开环聚合的反应图 式。
[0204][0205]y+
:k
+
,t-bup
4+
,t-bup
2+
[0206]
x
+
:nbu
4+
,pbu
4+
,noct
4+
,ppn
+
[双(三苯基膦)亚胺]
[0207]
实验部分
[0208]
通用方法
[0209]
所有反应均在braun labmaster手套箱内,在干燥无氧的氩气气氛中进行。环氧乙烷(eo)、 l-丙交酯(lla)、二乙二醇单甲醚(dgme)、对甲基苄醇(pmba)、双酚a(bpa)、 t-bup4、t-bup2、四丁基氯化铵(tbacl)、双(三苯基膦)氯化亚胺(ppncl)、四辛基氯化 铵(toacl)、四丁基氯化膦(tbpcl)、叠氮化四丁基铵(tbaa)、丙烯醇(allyl a)购 自aldrich公司。使用前,将四氢呋喃(thf)和甲苯(tol)在钠/二苯甲酮混合物上进行蒸 馏。在搅拌两天后,将1,4-二氧六环在cah2上进行蒸馏。通过将环氧乙烷在cah2上搅拌一 天而对环氧乙烷进行纯化,接着蒸馏至含有正丁基锂(n-buli)的烧瓶中。然后将其搅拌几 小时,接着将其进一步蒸馏。通过将lla从乙酸乙酯中重结晶两次,接着从干燥二氧六环中 进行冷冻干燥,而对lla进行纯化。通过将二乙二醇单甲醚从甲苯中进行共沸蒸馏,而对二 乙二醇单甲醚进行纯化。将pmba和bpa从二氧六环中进行冷冻干燥。溶于cdcl3中,并 将所有1h和
13
c nmr谱记录于核磁共振分析仪(bruker avance iii-400mhz)中。用配备 有styragel hr2色谱柱的凝胶渗透色谱仪(viscotek ve2001)使用四氢呋喃(thf)作为 洗脱剂(流速为1ml/min),对gpc图形进行记录。使用窄分子量分布(mw)的聚苯乙烯 标准品来校准仪器。用梅特勒托利多差示扫描量热仪(mettler toledo dsc1/tc100)在空气 气氛中进行dsc测量。将样品以10℃/min的加热/冷却速率,首先从室温加热至200℃以消 除热历史,然后冷却至-100℃,最后再次加热至200℃。重复此循环,直到记录到恒定的熔融 温度和冷却温度(tm和tc)。
[0210]
使用四丁基氯化铵(tbacl)作为引发剂的合成p(eo-co-lla)共聚物的代表性过程:使 用经预干燥的30ml玻璃希丁克管(schlenk tube,80mm
×
28mm)进行该反应,该管带有 旋塞阀并且配备有磁力搅拌棒。在氩气气氛中,首先将约86μl的三乙基硼烷(约0.086mmol) 加入到在玻璃schlenk管中的tbacl(约4.8mg,约0.017mmol)甲苯(约0.5ml)溶液中。 然后将lla(约100mg,约0.69mmol)和环氧乙烷(约150mg,约3.47mmol)溶于甲苯 (约1ml)的预混溶液加入到引发剂-硼烷体系中。搅拌下在室温(约25℃)下进行约4小 时的聚合反应。然后用数滴5%hcl的甲醇溶液使反应淬灭,再使聚合物在冷二乙醚中沉淀。 在过
滤后将所获得的聚合物在真空干燥箱中干燥,并利用gpc和nmr进行表征。
[0211]
使用t-bup4引发剂的合成p(eo-co-lla)的代表性过程:使用经预干燥的30ml玻璃 schlenk管(80mm
×
28mm)进行该反应,该管是带有旋塞阀并且配备有磁力搅拌棒。在氩 气气氛中,将pmba(约4.3mg,约0.035mmol)的甲苯(约0.5ml)溶液、t-bup4溶液(约 35μl,约0.035mmol)加入反应瓶中,在大约室温下搅拌数分钟。然后,将三乙基硼烷(约 176μl,约0.176mmol)和lla(约75mg,约0.520mmol)与环氧乙烷(约152mg,约3.47 mmol)的预混单体的甲苯(约1ml)溶液相继地加入到引发剂-硼烷体系中,搅拌下在大约 室温下进行约1小时的聚合反应。用数滴5%hcl的甲醇溶液使反应淬灭,并在冷二乙醚中 沉淀。将过滤后所获得的聚合物在真空干燥箱中干燥,利用gpc和nmr进行表征。
[0212]
结果和讨论
[0213]
环氧乙烷与l-丙交酯的共聚合
[0214]
因为eo和lla表现出非常不同的反应性,并且因为与lla相对应的单体单元在阴离 子条件下容易发生酯交换反应,所以在甚至使用弱碱作为引发剂的情况下,eo与lla也不 能发生共聚合。例如,在醇和弱碱(如t-bup2)的存在下,需要约3天时间完成eo的共聚 合,而在相同条件下仅需约1分钟便实现lla的完全转化。这已是利用配位化学使这两种单 体发生共聚合的主要原因。
[0215]
意味着必须采用单体的配位步骤的催化过程具有优点,例如能够形成长链,但它们也有 缺点,例如链的结构未必明确且链的摩尔质量分布较宽。在本实施例中提出了一种用于eo 与lla共聚合的新方法。该新方法不是基于纯粹的阴离子物质,而是基于涉及路易斯酸(即 三乙基硼烷)和碱(其通常是醇盐)的酸根型配合物。酸根型配合物用于在不形成环碳酸酯 的情况下使环氧化物与co2的成功共聚合,这通常是要使用纯净的离子物质来获得的。同样 地,本发明实施例还发现硼基酸根型配合物能够非常有效地引发叠氮缩水甘油醚(一种之前 从未能聚合的环氧化物单体)的共聚合,并且这种共聚合反应是受控的。在上述两个实施例 的每个实施例中,除了硼基酸根型配合物以外,还必须加入游离的三烷基硼以使单体活化以 便产生聚合物,因为增长的酸根型配合物通常不具有足够的亲核性。
[0216][0217]
起初尝试使用pmba/t-bup4作为引发剂体系并使用teb作为路易斯酸,以形成导致共 聚合的酸根型配合物,从而使eo和lla发生均聚合。在5当量过量的teb(用以使单体活 化)存在下进行两种均聚合。在eo的情况下,如预计的,在四氢呋喃或者甲苯中均能提供 具有预计分子量的样品:显然游离teb的存在对于触发共聚合是必不可少的(参见表1的第 1和第2项)。相反,在lla的情况(参见上方的反应图式)下,尽管存在5当量过量的teb, 但几乎未观察到任何均聚合(转化率低于1%),并且进一步加入过量的teb也无助于提高 lla的转化率(参见表1的第3和第4项)。
[0218]
令人关注的是,像lla的单体,当其接触到纯净的离子物质时会很快地发生均聚
合,在 这种情况下保持“原状(put)”并且在硼基酸根型配合物存在下未发生开环。然后,对在5 当量teb存在下lla与eo的共聚合进行了研究。将约15-20mol%的lla投入到反应介质 中,以观察低含量的酯是否可以掺入peo主链中。在所有的以下实验中,将作为非极性溶剂 的甲苯使用于eo与lla的共聚合。在聚合后,将反应混合物倒入冷冻的醚中以收集所有的 聚合物产物,并利用gpc和nmr进行表征。图4示出了代表性的1h nmr谱。如图4所示, 在5.2ppm和1.5ppm(峰a、b)处以及在3.5ppm(峰c)处,清楚地检测到lla和eo单 元的特征峰,从而说明了酯单元的掺入。在4.3ppm和4.10ppm处的峰g和峰h分别对应于 在eo与lla单元-ch2ch2oocch(ch3)oocch(ch3)o-之间连接的eo的亚甲基质子和lla 的次甲基质子、以及在lla与eo单元-occh(ch3)oocch(ch3)och2ch2o-之间连接的lla 单元的次甲基质子。两种聚合物peo和plla单元的特征峰和连接峰的存在,说明了无规共 聚物的形成。峰g与峰h的积分比接近3,说明lla的酯交换反应可忽略不计。基于nmr 数据,然后可以计算出lla单元的含量,并且使用引发剂对甲基苯醇的峰(在4.5ppm、7.1 ppm、2.3ppm处的峰d、e、f)作为参考可以估算所获得共聚物的摩尔质量(请参见表1中 所列出的相关数据)。利用方程式llla=(si
5.21 ppm
+2si
4.10 ppm
)/2si
4.10 ppm
确定plla的平均 链段长度等于约1.57,其中si是各个峰的积分强度。这就意味着,平均地,沿聚合物主链发 现少于两个lla单元是相邻的,从而证实lla的竞聚率(r
lla
)的值非常低。按照相同的过 程,在teb的存在下,利用pmba/t-bup4体系引发共聚合,并且将不同的摩尔质量作为目 标(表1的第6-12项)。由nmr获得的各种样品的摩尔质量值接近理论值;实现了高达24 kg/mol的摩尔质量,并且在所有情况下酯含量保持在5%左右。当改变lla与eo的投料比 时,所获得共聚物中的酯含量发生变化(表1第11项)。利用gpc对所获得共聚物样品的 分析显示了摩尔质量分布较窄的单峰图形(图5);后者接近于理论值及通过nmr计算得到 的值。因此证明,在这些条件下,eo与lla以一种“活性(living)”的方式发生共聚合, 并且酯交换反应被完全抑制。
[0219]
可以认为,不仅在“活性”条件下首次成功进行了使像eo和lla这样不同的单体之间 发生共聚合的尝试,而且证明了可以将极低含量的酯键掺入聚醚链中,这是先前从未实现的 成就。当在thf(其是一种弱极性的溶剂,表1第5项)中进行反应时,观察到失去对共聚 物样品摩尔质量的控制,表明发生了酯交换反应,因此很有可能发生了“反咬(back-biting)
”ꢀ
反应。
[0220]
在teb的存在下,除了利用pmba/tbup4体系所引发的聚合反应之外,也将其它有机引 发剂的铵和磷卤化物(如tbacl和tbpcl)用于共聚合。得到了类似的结果,但在分离的共 聚物内部的酯含量往往稍高于从醇盐/tbup4体系中所产生的共聚物(表1的第15、19和20 项)。这可能是由于在与醇盐相关的各种阳离子的存在下(参见下文),eo与lla之间的 反应性存在差异。也使用各种引发剂制备了二官能和异聚二官能共聚物。例如,当选用双酚 a作为引发剂时,得到包含约3%酯含量的两种羟端基peo(表1第13项);如果以烯丙醇 和四丁基叠氮化物作为引发剂(表1的第21和22项)开始反应,则生成了带具有乙烯基和 叠氮化物端基的共聚物样品(图6),这是对于生物结合和应用而言意义重大且强有力的官 能团。
[0221]
表1使用不同引发体系的eo与lla的无规共聚结果
[0222][0223]
除非另有说明,将对甲基苄醇(pmba)与p4和p2联用,p4=t-bup4,p2=t-bup2,a二乙二醇单 甲醚用作醇,b双酚a用作醇。
c m
n(theo)
=(m
p
/n1),m
p
=聚合物的总回收重量,ni=引发剂的摩尔数。d酯 含量和m
n(nmr)
的计算基于1hnmr。e以thf作为洗脱剂,用聚苯乙烯标准品作校准,测定gpc。
[0224]
为了鉴定所形成的共聚物的性质,使用pmba/tbup4、tbacl和ppncl作为引发剂,在 相同的初始进料比下,收集了动力学数据并测定了单体转化率。将相关的聚合数据列于表2 中。随着聚合时间的增加,酯含量逐渐地增加,尽管速率远低于醚含量的速率,这表明eo 的消耗快得多。通过以不同的时间间隔终止共聚合并且对相应的共聚物的组成进行分析,从 而计算了竞聚率值r
eo
和r
lla
并且测定了自增长或其它单体掺入的趋势。可使用各种方法来 计算竞聚率,包括mayo-lewis法、fineman-ross法和kelen-t
ü
dos法等。在本实施例中,利 用kelen-t
ü
dos法计算竞聚率。在tbup4/醇盐体系的情况下,eo的竞聚率r
eo
为6.27,lla 的竞聚率r
lla
为0.08。而在使用tbacl作为引发剂的情况下,发现r
eo
为1.67,r
lla
为0.15 (参见图7-图9)。在相同的进料比下,随着与醇盐相关的阳离子尺寸的减小,eo的反应性 下降,因此掺入了更多的酯单元。然而,在这两种情况下,eo的反应性仍远高于lla的反 应性,这导致在所获得共聚物p(eo-co-lla)中lla单元含量较低且酯链段非常短。
[0225]
表2具有不同转化率的eo与lla的共聚合结果
[0226][0227]
除非另有说明,将pmba与p4联用,p4=t-bup4,am
n(theo)
=(m
p
/ni),m
p
=聚合物的总回收重量,ni=
[0228]
引发剂的摩尔数。b酯含量和m
n(nmr)
的计算基于1h nmr。c以thf作为洗脱剂,用聚苯乙烯标准品 作校准,测定gpc。
[0229]
因为kelen-t
ü
dos法对于“瞬时”组成和相当低的转化率而言效果最好,所以使用非末 端链共聚模型(bsl)来测定这两种单体的竞聚率r
eo
和r
lla
。该模型假定增长物质的反应性 仅取决于进入的单体的反应性,并且忽略以活性物质为特征的最后单体的性质;直至完全转 化它都是适用的。基于表3中所示数据计算了竞聚率,并且得到大概结果:以p
4+
作为抗衡阳 离子时,r
lla
=0.17
±
0.04、r
eo
=5.37
±
0.4;以tba
+
作为抗衡阳离子时,r
lla
=0.49
±
0.08、r
eo
=2.07
±
0.25;以ppn
+
作为抗衡阳离子时,r
lla
=0.14
±
0.01,r
eo
=6.61
±
0.67。在用三种不同 的阳离子(p4
+
、tba
+
、ppn
+
)进行研究的三种情况下,竞聚率的乘积r
eo
×rlla
非常接近于 1,利用1h nmr证实了该特征,该特征表明梯度共聚物的形成。还尝试使用ml的末端模型 来测定竞聚率r
eo
和r
lla
。假定所形成的共聚物具有梯度性质,并且没有嵌段或交替的倾向。 我们从lowry和meyer的共聚物转化率依赖型通用方程式开始,将竞聚率的简单关系推导成 转化率的函数。因此,ml的末端模型提供了在teb存在下eo与lla的共聚合,以及以下 的竞聚率值(表3):对于p
4+
,r
lla
=0.19
±
0.02、r
eo
=5.15
±
0.556;对于tba
+
,r
lla
=0.50
ꢀ±
0.07、r
eo
=2.03
±
0.27;对于ppn
+
,r
lla
=0.15
±
0.01、r
eo
=6.49
±
0.46。
[0230][0231]
作为本研究的一个目的是尽可能精确地控制酯单元在peo链中的掺入,并且如有可能将 其限定在一个百分比(~5%),利用dsc对所获得共聚物样品的热性能进行了检查。对peo 的熔融转变都进行了测定,并与纯peo的熔融转变进行了比较,随着更多的酯单元掺入peo 主链中,所获得共聚物的熔融温度(tm)逐渐降低。当掺入约3%的酯时,tm从58.9℃降低 至约52.3℃。当酯键进一步递增至约7%时,tm进一步降低至约38.5℃;当掺入约14%的酯 单元时,tm则降低至约28.6℃(图10)。特别是在后一种情况下,由于更多酯单元的掺入, 因而测得在约-17.6℃下明显的冷结晶转变。然而,未检测出plla的熔融转变温度,即使样 品中含有约14%的酯,说明沿peo主链掺入了非常短的plla链段。作为对比,在含有约 17%酯单元的多嵌段p(eo-co-lla)共聚物的情况下,明确地测定出plla的熔融温度(tm)。
[0232]
最后,对p(eo-co-lla)进行降解,以检查peo链段的平均长度。将共聚物溶解于约0.5 m naoh的40:60甲醇:水溶液中,搅拌约两天以使酯键水解。利用1h nmr对在这种处理后 所回收的聚合物进行表征,结果表明酯键完全降解并消失。利用gpc对降解后的peo的摩 尔质量进行了分析。如图11所示,共聚物样品的摩尔质量从最初的约24kg/mol下降至约3 kg/mol。
[0233]
总之,通过eo与lla的阴离子开环共聚合反应,以可控的方式直接地制备了具有低多 分散性和明确结构的可降解聚环氧乙烷。teb的存在选择性地增加eo的反应性,并抑制酯 交换反应。该方法是通用的,不仅可用于合成官能化线型peo类,还可以合成具有高摩尔质 量的支链peo类并且不用担心可降解性问题。此外,无金属合成使该方法更有望用于生物医 学用途。
[0234]
实施例2
[0235]
通过环氧乙烷与丙交酯或二氧化碳的阴离子共聚合而制备可降解聚环氧乙烷
[0236]
以下所示的反应图式中示出了在三烷基硼烷存在下,通过eo与丙交酯或二氧化碳的阴 离子共聚合反应,以直接的方式形成可降解peg。如本实施例中所描述,非常低含量(约5%) 的丙交酯和二氧化碳的无规掺入导致在peg主链内形成酯键或碳酸酯键,这给所获得peg 赋予可降解性;另外,所获得共聚物依然保留其亲水性和明确的结构。这样可以制备异聚二 官能封端可降解peg,或者经衍生化以结合生物应用的分子。
[0237][0238]
r4n
+
(铵),r4p
+
(磷),p4h
+
(磷腈)
[0239]
gi.br,n3,-o,ch2=chgh2o-,rco2[0240]
方法
[0241]
使用四丁基氯化铵(tbacl)作为引发剂的合成聚环氧乙烷-l-丙交酯共聚物的代表性过 程:使用经预干燥的30ml玻璃schlenk管(80mm
×
28mm)进行该反应,该管带有旋塞阀 并且配备有磁力搅拌棒。在氩气气氛中,首先将86μl的三乙基硼烷(0.086mmol)加入到 在玻璃schlenk管中的tbacl(4.8mg,17μmol)甲苯(0.5ml)溶液中。然后将l-丙交酯 (100mg,0.69mmol)和环氧乙烷(150mg,3.47mmol)的1ml甲苯预混溶液加入到引发 剂-硼烷体系中。搅拌下,在室温(约25℃)下进行4小时聚合反应。然后,用数滴5%hcl 的甲醇溶液使反应淬灭,再使聚合物溶液在冷二乙醚中沉淀。在过滤后,将所获得聚合物在 真空干燥箱中干燥,并利用gpc和nmr进行表征。
[0242]
使用p4为引发剂合成聚环氧乙烷-l-丙交酯共聚物的代表性过程:使用经预干燥的30ml 玻璃schlenk管(80mm
×
28mm)进行该反应,该管带有旋塞阀并且配备有磁力搅拌棒。在 氩气气氛中,将对甲基苄醇(pmba)(4.3mg,35μmol)的甲苯(0.5ml)溶液、t-bup4溶液(44μl,0.044mmol)加入反应瓶中,在室温下搅拌数分钟。然后,将三乙基硼烷(176 μl,0.176mmol)以及l-丙交酯(75mg,0.520mmol)与环氧乙烷(152mg,3.47mmol) 的预混单体的甲苯(1ml)溶液相继地加入到引发剂-硼烷体系中,搅拌下在室温下进行1小 时共聚合反应。用数滴5%hcl的甲醇溶液使反应淬灭,并在冷二乙醚中沉淀。将过滤后所获 得的聚合物在真空干燥箱中干燥,利用gpc和nmr进行表征。
[0243]
使用四丁基氯化铵(tbacl)作为引发剂合成聚环氧乙烷-碳酸乙烯酯共聚物的代表性过 程:使用经预干燥的30ml玻璃schlenk管(80mm
×
28mm)进行该反应,该管带有旋塞阀 和隔膜并且配备有磁力搅拌棒。首先在经干燥的二氧化碳气氛中,加入27.8mg的tbacl(100 μmol),接着溶解于2ml的thf。将溶于thf的三乙基硼烷(150μl,0.15mmol)和eo (1ml,20mmol)相继地注入管中。在1巴的二氧化碳压力和室温下,进行12小时共聚合 反应。用数滴5%hcl的甲醇溶液使反应淬灭,并在冷二乙醚中沉淀。将过滤后所获得的聚 合物在真空干燥箱中干燥,并利用gpc和nmr进行表征。
[0244]
图13-图24提供了下表4中所给出某些项的1h nmr谱和gpc谱图。
[0245]
表4:eo分别与lla和co2进行无规共聚合的结果
[0246][0247]
除非另有说明,对甲基苄醇(pmba)与p4和p2联用,p4=t-bup4,p2=t-bup2,lla=左旋丙交酯, eo=环氧乙烷,thf=四氢呋喃,tol=甲苯,tbacl=四丁基氯化铵,ppncl=双(三苯基膦)氯化亚胺, toacl=四辛基氯化铵,tbpcl=四丁基氯化膦,tbaa=叠氮化四丁基铵。以thf作为洗脱剂,用聚 苯乙烯标准品作校准进行gpc测定。a双酚a用作醇,b二乙二醇单甲醚用作醇。c聚合反应在1巴 的co2氛围下进行。d聚合反应在2巴的co2氛围下进行。e聚合反应在4巴的co2氛围下进行。
[0248]
实施例3
[0249]
含有可降解聚碳酸酯核的星形peo的合成
[0250]
利用先核后臂法(core first approach)制备可降解星形peo,其中核具有碳酸酯键,以赋 予其可降解性,如图12中所示。这里,将二环氧化物,二氧化乙烯基环己烯(vdox)用作 交联剂,通过与co2的共聚合而形成可降解聚碳酸酯核。为了获得具有低交联度的可溶性聚 碳酸酯核,使用非常低量的二环氧化物,并且vdox与鎓盐引发剂的比例保持在低于约10。 一旦核形成,将co2逐渐释放后,将环氧乙烷连同thf溶剂注入相同的parr反应器中,随 后在约40℃和搅拌下进行环氧乙烷与所生成聚碳酸酯核的聚合反应。聚合条件和结果如表5 所示。
[0251]
利用先核后臂法由聚碳酸酯核(pvdox-eo)合成星形聚环氧乙烷的代表性过程:在100 ml parr反应器中进行反应,该反应器具有内置加料孔,将其在120℃下干燥过夜,然后在手 套箱中抽真空约3小时。为了说明该合成过程,将被命名为pvdox1-eo1的星形peo样品 (表5中的第27项)作为代表。首先将ppncl(0.114g,0.2mmol)加入反应器中,接着加 入thf(约2.5ml)和teb(0.2ml,约1当量)。将二氧化乙烯基环己烯(约0.14g,约 1mmol)加入反应混合物中,然后将反应器关闭并从手套箱中取出,以便在约10巴的co2压力下进行加料。在约80℃的温度下进行约15小时聚合反应。将反应器冷却后,将co2缓 慢释放至最低水平,将eo(约2.6ml,约60mmol)、teb(约0.6ml)和thf(约20ml) 经过加料孔加入到反应器中,在约40℃下进行约15小时的聚合反应。最后,用盐酸的甲醇 溶液(约1mol/l)淬灭反应混合物。通过在二乙醚中沉淀以纯化所得到的粗产物,离心并在 真空干燥箱中约40℃下干燥约15小时,以获得最终产物(产率=约90%)。
[0252]
图25-图29提供了下表5中所给出某些项的1h nmr谱和gpc谱图。
[0253]
表5利用先核后臂法合成的pvdox-eo星形聚合物的概述
[0254][0255]a除非另有说明,用ppncl作引发剂,在50-80℃、10巴co2压力下,制备pvdox,其中vdox/thf 的体积比为1:2.5,接着在40℃下将所得的内核与eo聚合24h,并保持pvdox与teb的比例为1:3, eo/thf的体积比为1:10。b以聚苯乙烯标准品为参照,以thf作洗脱剂进行测定。c通过凝胶渗透 色谱-多角度激光散射(gpc-malls)联用技术进行测定。
d n
am
=m
w,ls
×
armwt%/m
n,peo
。
e vdox/thf 的比为1:2。f将单官能cho加入交联剂内,使vdox/cho的比为1:1,vdox/thf的比为1:2.5。g用nbu4cl引发聚合,在80℃下反应3h。
[0256]
实施例4
[0257]
环氧乙烷与co2通过无金属共聚合制备疏水性聚碳酸乙烯酯、 两亲性聚醚碳酸酯和亲水性可降解聚环氧乙烷
[0258]
本实施例描述了在三乙基硼烷(teb)的存在下,用鎓盐(os)作引发剂,在无金属条 件下,通过环氧乙烷(eo)与co2的(均聚)共聚合制备三种类型的eo基共聚物。首先, 在co2聚合压力介于10-30巴下,teb与os的摩尔比在1-1.2当量的范围内并溶于thf或 己烷,制备碳酸酯含量高于90%的疏水性聚碳酸乙烯酯-环氧乙烷共聚物(peceo);接着, 用以上制得的peceo(碳酸酯含量》91%)作为大分子引发剂,在一锅法中通过eo的序列开 环聚合反应,制
备两亲性peo-b-peceo-b-peos;最后,在低压co2下(1-2巴)、teb与 os的摩尔比为1.2-2.0当量,进行eo的共聚合,从而使亲水性peoec的碳酸酯含量低于10%; 在相同的条件下,使烯丙基缩水甘油醚(age)与eo和co2发生三元共聚反应,以在peo 主链内引入官能度。两亲性peo-b-peceo-b-peo样品作为非离子表面活性剂,对其临界胶 束浓度(cmc)和胶束尺寸进行了测定。亲水性peoec的类peo性能,通过热重分析法 (tga)、差示扫描量热法(dsc)和润湿性试验进行表征,并在不同的条件下对它们的降 解性能进行了进一步研究。
[0259]
利用teb介导的聚合体系的多样性和可调性与eo的特性相结合,本实例描述了eo与 co2的共聚合以及eo的开环聚合(rop),从而评价了适用于不同用途的一系列eo基聚合 物,其涵盖了疏水性peceo、两亲性peo-peceo-peo和亲水性peoec(参见图46的反应 图式)。首先,利用鎓盐引发eo与co2的共聚合反应,可获得高碳酸酯含量(》90%)的 peceo(参见图46a的反应图式)。基于上述过程,在eo与co2的共聚合反应之后,采用 一锅法、通过eo的序列rop,提供了两亲性peo-peceo-peo三嵌段共聚物(参见图46b 的反应图式),这将会是优良的非离子表面活性剂。实际上,与市售的同类产品(poloxamers 或pluronics的peo-ppo-peo)相比,其使用廉价和丰富的co2作为原料,并给 peo-peceo-peo赋予可降解性,这将产生深远的经济和环境效益。最后,通过调节co2的 聚合压力,就可以形成碳酸酯含量非常低(约5mol%)的、可降解的类peo型peoec,参 见图46c的反应图式。此外,其它官能化环氧化物,例如烯丙基缩水甘油醚(age)可以与 eo和co2发生三元共聚,旨在引入官能团从而作为药物递送载体用于后续的结合。应该注 意的是,在所有情况下,所得的eo基聚合物不含有任何金属残留物,在用作生物材料和表 面活性剂时不用担心个人健康问题。
[0260]
结果和讨论
[0261]
表6使用teb1合成具有不同碳酸酯含量的peceo的共聚合数据
[0262][0263]1在室温下进行15小时共聚合反应。2基于粗产物的1h nmr谱图而确定。3基于纯产物 的1h nmr谱图而计算。4利用sec法测定,用dmf作洗脱液(第1-15项),用thf作洗 脱液(第16-23项),并用线型聚环氧化乙烷标准品作为参照。5利用此公式计算产率:产率 =所得聚
合物的重量/所加入eo的重量+碳酸酯含量
×
所加入eo的重量。6反应持续时间为 45个小时。7将age用作共聚单体,并且age/eo的比例为20:1。8利用此公式计算产率: 产率=所得聚合物的重量/(所加入eo的重量+碳酸酯含量
×
所加入eo的重量+114.14
×
age 掺入含量
×
所加入eo的摩尔数)。9将age用作共聚单体,并且age/eo的比例为10:1。缩 写:tbacl=四丁基氯化铵;ppncl=双(三苯基膦)氯化亚胺;toacl=四辛基氯化铵;tbas =四丁基琥珀酸铵。
[0264]
a.通过eo/co2的共聚合反应合成peceo
[0265]
自从首次报道了teb催化的co2/po(cho)共聚合反应之后,最近也对这种用于co2与 其它环氧化物共聚合的反应体系的多样性进行了研究,其共聚合的结果表明,环氧化物与co2的共聚活性在很大程度上依赖于环氧化物环上的侧基,侧基取代基的位阻和官能度可以降低 环氧化物的活性,尽管相对于引发剂,teb需要加入的量更多,以此来促进共聚反应,并抑 制环碳酸酯的形成。在所有的环氧化物中,eo是最简单的,而且没有任何取代基,因此在 rop的过程中也最活跃。实际上,在类似的条件下(teb相对于tbacl为2当量),与po 相比,eo和co2的共聚合赋予peceo以相对于环状更高的线型选择性,但是碳酸酯含量较 低(参见表6第1项)。从粗产物的1h nmr谱图可以看出(图33),线型聚碳酸乙烯酯(pec) 的质子信号出现在4.31-4.23ppm处,而环状碳酸乙烯酯(cec)的质子信号出现在4.46ppm 处,从而得出其线型相对于环状的聚合选择性为96%;根据纯品的1h nmr谱图(图34), 酯键的峰出现在3.66-3.58ppm处,线型碳酸酯键的峰出现在4.31-4.23ppm处,根据这些峰 的积分强度可以得出所得共聚物中碳酸酯的含量相当低(15mol%)。很明显,与po相比, eo的反应性较高,再加上teb的活化,都有助于形成醚的均聚合反应,而不是共聚合反应。 为了避免eo的过度活化,以及适当抑制eo的均聚合反应,尝试加入少量的硼烷(teb相 对于引发剂为1当量),在1巴的co2压力和室温下在thf中进行共聚合反应(表6第2项)。 相反,所获得peceo中碳酸酯的含量显著升高至81%,而线型对环状的聚合选择性降为82%。 当用非极性溶剂己烷代替配位溶剂thf之后,所获得peceo中的碳酸酯含量升高至91%, 产率较低为47%,而选择性(81%)几乎保持不变(参见表6的第3项)。由于己烷的非解 离效应,使醇盐/teb酸根型配合物的结合更加紧密,也都抑制了均聚合反应,但也表现出较 低的活性,造成了聚合物的产率降低。使用ppncl(参见表6的第4项)和toacl(参见表 6的第5项)等引发剂可以提高选择性(分别为87%和98%)和增加产率(分别为58%和70%), 然而,与tbacl相比,碳酸酯含量降低(分别为85%和81%),这与含有大体积鎓离子的酸 根型配合物的活性较高有关。稍微增加teb的量,使teb相对于tbacl为1.2当量(表6 第6项)导致选择性提高至95%,而碳酸酯含量降低(64%)。为了掺入更多的co2(》90%) 而不会失去其线型对环状的选择性,在保持这种共聚合反应条件(teb相对于tbacl为1.2 当量,己烷作溶剂)的基础上,将co2的压力从10巴升高至20巴,结果发现co2压力对聚 碳酸酯含量产生了的重大影响,所获得的peceo中,碳酸酯的含量从64%提高到92%,选 择性适中(85%),产率为46%(参见表6第7项)。如果进一步减少共聚合体系中非极性 溶剂己烷的体积,即eo/己烷的体积比=1:1(第8项),结果产生的peceo,其线型对环状 的选择性增加至95%,碳酸酯含量为90%,产率为69%。按照这种思路,通过提高co2压力 来增加所获得peceo中的碳酸酯含量,采用配位溶剂thf、在30巴的co2压力下,也获得 了类似的良好共聚合结果(92%的碳酸酯含量,参见表6第9项)。在teb相对于双官能引 发剂tbas为1当量、30巴的co2压力下进行共聚合反
应(表6第10项),得到了peceo 二醇,其含有95%的碳酸酯键、产率为87%、cec可以忽略不计,其可用作制备如ppc多元 醇等聚氨酯的前体。
[0266]
在目标聚合度为500的前提下,对用于合成具有高ec含量和高选择性的peceo的优化 条件进行了进一步研究。有意义的是,在10巴的低co2压力下,使用相对于四丁基铵为1 当量的teb,以thf为溶剂,可得到高碳酸酯含量(》92%)的peceo,其在选用tbacl 的情况下,选择性为89%(表6第11项),在选用tbas的情况下,cec可以忽略不计(表 6第12项)。相反,使用ppncl和toacl等大体积氯化鎓,可以得到碳酸酯含量》90%的 peceo,其线型对环状的选择性较高(》93%),而且产率较高(表6的第13-15项)。很明 显,在高目标聚合度体系中,thf溶剂中的高活性鎓盐酸根型配合物以某种方式被其低浓度 抵消了,正是这种抵消作用,才获得了以上良好的共聚合结果,并且无需将eo与非极性溶 剂混合,也无需升高co2的聚合压力。用dmf作洗脱液、使用聚环氧乙烷校准,通过sec 的分析,发现所得peceo的摩尔质量明显低于预期值,但无论是在哪种情况下,都表现出 单峰性和窄分子量分布。用toacl作引发剂,通过将反应时间从15小时延长至45小时,产 率达到57%、m
n(gpc)
高达21kg/mol,而且并没有降低选择性,也没有减少co2的掺入(表6 的第16项)。
[0267]
b.两亲性peo-b-peceo-b-peo三嵌段共聚物的合成
[0268]
成功制备了高碳酸酯含量的peceo之后,尝试了以前一步骤中的产物作为大分子引发 剂,通过eo的序列rop来合成两亲性三嵌段共聚物。由于teb介导的体系对于eo/co2的共聚合以及eo的rop均有效果,因此可以用图46中反应图式b所示的一锅法合成在结 构上与peo-ppo-peo表面活性剂类似的三嵌段共聚物:首先,使用双官能引发剂tbas,在 表6第10项所述的反应条件下进行eo与co2的共聚合,排出co2后加入另外的eo单体, 进行eo的rop。通常,在大分子链转移剂存在下,通过co2/环氧化物的“永生(immortal)
”ꢀ
共聚合来制备co2基两亲性共聚物,在这里,只给出了peo-pc和pc-peo-pc这两种两亲性 共聚物的结构。就发明人所知,含有peo的非离子表面活性剂,在其两端具有亲水性嵌段, 在其中部具有co2基疏水性聚碳酸酯,这种非离子表面活性剂之前还未见报道。以这种方式, 制备了具有不同摩尔质量和组成的三种两亲性共聚物,在其中间的peceo嵌段中含有大致 相同含量的碳酸酯(91mol%)。从它们的1h nmr谱图中,可观察到pec带来的峰,以及 经过eo的rop后peo带来的更高强度的峰(图30a)由琥珀酸酯的亚甲基质子引起的峰位 于2.66ppm处,可用它来计算三嵌段共聚物的摩尔质量。三嵌段共聚物的sec图形分析表明, 经过第二个步骤中eo的rop后,其前体出现了明显的位移,证明三嵌段共聚物已成功形成 (图30b)。这三种共聚物样品的相关表征数据列于表7中。p(eo-eceo-eo)1和 p(eo-eceo-eo)2具有相同的pec中间嵌段,但peo的重量分数不同,p(eo-eceo-eo)1中 peo的重量分数是45%,而p(eo-eceo-eo)2的是65%。p(eo-eceo-eo)3和p(eo-eceo-eo)2具有相同的peo重量分数(65%),但p(eo-eceo-eo)3的摩尔质量高于p(eo-eceo-eo)2。
[0269]
表7由eo和co2制备的两亲性三嵌段共聚物的表征数据
[0270][0271]1用dmf作洗脱液,peo为标准品,在60℃下测定sec;2根据1h nmr谱图,使用以 下公式计算:m
nnmr
=(i
4.37
×
88+i
3.63
×
44)/i
2.66
;3根据1h nmr谱图,使用以下公式计算:peowt%=100
×i3.63
×
44/(i
4.37
×
88+i
3.63
×
44);4使用griffin法计算hlb(亲水亲油平衡值), hlb=20
×
亲水嵌段的重量分数;5在25℃下利用自荧光光谱法,用芘作荧光探针获得;以及 6
在25℃下利用动态光散射法进行测定。
[0272]
通过荧光光谱法用芘作荧光探针,测定这些共聚物的cmc值。分别测定在373nm和384 nm处的荧光强度,再以其比值(i1/i3)相对于聚合物浓度作图,以此作为计算浓度的函数, 见图30c。以两条最佳拟合线的交叉点的浓度值作为cmc值。p(eo-eceo-eo)1在25℃下的 cmc值为0.0037g/l或0.0074mm。该值显著地低于具有相似hlb值的peo-ppo-peo表面 活性剂聚合物。例如,普朗尼克(pluronic)共聚物p104(hlb=8)在25℃下具有0.508mm 的cmc值。由此可以得出结论,peceo的疏水性高于ppc,而且在一定程度上掺入eo(9 mol%)对peceo的疏水性能影响不大。此外,在ppc-peo-ppc具有相似的hlb值(9.9) 的情况下,这种三嵌段共聚物是不溶于水的,说明结构对胶束效应的重要性。将亲水性peo 的重量分数从45%增加至65%,造成其cmc值升高至0.081g/l。p(eo-eceo-eo)3的cmc 值(0.055g/l)低于p(eo-eceo-eo)2。利用动态光散射法,测定了两亲性三嵌段共聚物形成 的胶束尺寸(图30c插图)。p(eo-eceo-eo)1、p(eo-eceo-eo)2和p(eo-eceo-eo)3的流 体力学半径(rh)分别为70、83和可以观察到,这些三嵌段共聚物形成的胶束的尺 寸分布较窄,介于0.04-0.1之间。与p(eo-eceo-eo)2相比,p(eo-eceo-eo)3具有相同的 hlb值、较低的cmc值和较大的rh值,这就意味着其更容易形成胶束,而且需要更多的分 子来屏蔽较长的疏水性嵌段,这种趋势与前面的报道一致。作为比较,peo-b-ppo-b-peo三 嵌段共聚物(pluronics p-85),其宣称的摩尔质量为4.5kg/mol,含有49wt%的peo链段, 在25℃下它形成的胶束的rh值为
[0273]
c.类peo型peoec和官能peoec的合成
[0274]
为了保持peo的性能并赋予其可降解性,掺入的co2不应高于10mol%,最佳含量约为 5mol%。基于之前的报道,可以有两种策略:在co2/环氧化物共聚合过程中,可以通过调整 teb与引发剂的比例或者改变co2的压力,以调节碳酸酯的含量。如表6所示(第17-24项), 通过增加teb相对于引发剂的投料量,或者是降低co2的压力,或者同时采用这两种方法, 以获得低碳酸酯含量的聚环氧乙烷-碳酸乙烯酯共聚物(peoec)。例如,使用相对于引发剂 tbacl为2当量的teb,在4巴的压力下,进行eo与co2的共聚合反应,可使peoec中 的碳酸酯含量显著降低(11%,表6第17项);进一步降低co2的压力到2巴和1巴,结果 相应的碳酸酯含量分别降至4.6%和1.7%(表6第18和19项)。使用少于2当量的teb, 例如1.4和1.2
当量,将co2的聚合压力保持在1巴以下,进行同样的共聚合反应,结果所制 得的peoec中的碳酸酯含量从1.7%分别上升至2.9%和6.9%(表6的第20和21项)。通 过比较第19项、第22项、第21项和第23项的所得结果,表明在相同条件下的共聚合反应, 只是目标聚合度从200提高到了500,所制得的peoec中的碳酸酯含量增加了。另一方面, 在第22和23项的情况下,相对降低teb和eo的比例,而降低eo的活化程度,结果造成 了更多的co2掺入。目标聚合度为4000,使用相对于tbas为1.6当量的teb,可以在没有 任何困难的情况下合成高摩尔质量(208kg/mol)、碳酸酯含量为4.4%的peoec(表6第24 项)。值得一提的是,在这种共聚合期间,没有检测到环碳酸乙烯酯,并且所有所获得的peoec 样品都是水溶性的,通过sec进行表征(使用thf作为洗脱剂,peo作为标准品),发现 其具有窄分散性(在1.03-1.16的范围内)。
[0275]
为了克服在可降解peoec亲水物中缺少官能团的不足,可以引入第三官能单体(比如 age),其中叠氮缩水甘油醚可以通过其与eo和co2的三元共聚合而掺入,以满足多种生 物应用的目的。作为一个概念证明,选择age用于三元共聚合。在恒定的1巴co2压力下, 使用相对于tbas为1.6当量的teb,其中eo/age投料比为20/1,可以获得碳酸酯含量为 5.8%、age含量为3.0%的p(eo-ec-age)(表6第25项)。图45提供了共聚物的1h nmr 谱图,图43提供了共聚物的sec(thf)图形谱图。在eo/age投料比为10/1时,碳酸酯 含量为8.9%、age含量为3.2%(表6第26项)。图31f提供了后一个共聚物的1h nmr 谱图,图44提供了共聚物的sec(thf)图形的谱图。在1h nmr谱图中,烯丙基的特征峰 位于5.88ppm和约5.23ppm处,证实了age的并入。如果仔细检查就会发现,其中一些age 单体是随着co2掺入(5.03ppm和4.96ppm),并与醚键和前一个碳酸酯键连在一起,这种 掺入类型在掺入总量(3.2%)中占75%。在4.96ppm处观察到的信号,对应的是与peo键 相邻的pagec中的甲烷质子,再结合age的反应活性远低于eo(源于其空间位阻效应和 双键与thb的相互作用),表明该共聚物的递变性。这些共聚物中的双键可以通过硫醇-烯 烃click化学反应而进一步衍生,与此同时给peo赋予可降解性和官能化。
[0276]
对可降解peoec的物理性质进行表征,以peceo作为参照。它们的不同ec含量已清 楚地示于图31a中的1h nmr谱图中。随着碳酸酯含量的增加,热稳定性降低。如图31b所 示,通过热重分析(tga)的表征可以看出,高碳酸酯含量的peceo样品表现出清晰的“两 段式”降解行为,其中第一段开始于约160℃,对应于碳酸酯键的断裂,第二段位于约350℃, 这是由于醚键的断裂。相反地,与高碳酸酯含量的peceo样品相比,低碳酸酯含量的peoec 表现出更高的稳定性。不同碳酸酯含量的peoec对应的t
5%
值如下:11%,335℃(表6第 17项);4.6%,358℃(表6第18项);1.7%,361℃(表6第19项)。
[0277]
peoec的类peo性能,也通过差示扫描量热法(dsc)和润湿性试验得以见证。对于 这三种低碳酸酯含量的peoec样品,分别检测到了熔融尖峰(图31c),11%的碳酸酯含量, 33℃(表6第17项);4.6%的碳酸酯含量,50℃(表6第18项);1.7%的碳酸酯含量,56℃ (表6第19项),其中后者与纯peo(58.9℃)非常接近。在peoec内微量的掺入co2, 并没有改变peos的结晶行为。对于高碳酸酯含量的peceo样品,当碳酸酯含量从81%、92% 升高到95%,只发现了玻璃化转变温度(tg)分别为3、7和11℃。
[0278]
采用溶剂浇铸共聚物表面水滴的接触角(ca)来评价peoec的润湿性或亲水性。图31d 显示了随着时间变化,ca的演变过程。碳酸酯含量为95%的peceo样品(表6第10项) 的
ca为91
°
,并且随时间保持基本恒定,显示了其疏水性。水滴在碳酸酯含量为11%的peoec 样品(表6的第17项)表面上形成的ca最初是64
°
,然后迅速在表面上扩散,以致其ca 变为14
°
。碳酸酯含量为1.7%的peoec样品(表6第19项),随着观察的时间推移,其ca 从31
°
变为15
°
。两种peoec样品的低ca证明了它们的亲水性质。
[0279]
了解到制备的peoecs具有类peo性能,对它们的降解行为进行了研究。为了方便地追 踪降解过程,将高摩尔质量的peoec样品(表6第24项,图35)用于此实验,其m
n(gpc)
=208 kg/mol,碳酸酯含量为4.4%。第一次降解在苛刻的条件下进行,以确保其完全降解,这是在 ph=13的naoh水溶液中完成的,温度为25℃,用时两天。将浓缩产物用1h nmr和gpc 进行表征。与原始的聚合物的1h nmr谱图比较(图31e),在4.34ppm处对应于碳酸酯键 的峰消失了(图36),说明完全降解。同时,利用gpc分析摩尔质量,发现降解后降至1.1 kg/mol,而且具有非常宽的多分散性(图37)。接着,在温和的条件下进行降解:(1)ph=8.5, 25℃(常用于表面修饰、细胞培养、酶测定和电泳实验);(2)ph=7.4,37℃(模拟体液环 境);(3)ph=6.5,25℃(模拟肿瘤的微酸性微环境)。如图31e中所示,缓冲液(ph=8.5, 25℃)中的碳酸酯含量从4.4%降至1.2%,同时其摩尔质量降至1.6kg/mol(图38)。在生理 条件下(ph=7.4,37℃),降解较慢,一个月后,碳酸酯含量降至3.0%,而摩尔质量降至2.9 kg/mol(图39和图40)。在微酸性环境下,降解最慢,碳酸酯含量降至4.1%,而摩尔质量 仅降至6.2kg/mol(图41和图42)。在任何情况下,降解后peoec的摩尔质量显著地降至 40kg/mol的肾阈值以下,确保服用后能从身体完全排出。
[0280]
结论
[0281]
利用teb介导聚合体系的多样性以及eo单体的特性,通过调整co2的掺入程度,可以 合成从疏水性、两亲性到亲水性的多种eo基共聚物。由于eo相对于其同系物的高反应性, 在相对于引发剂的少量teb的条件下,结合高co2压力或者使用非极性聚合介质,eo与co2的共聚合反应可以制备碳酸酯含量超过90%的peceo。eo的序列rop之后,采用一锅法, 获得一种新型的两亲性共聚物peo-b-peceo-b-peo,这种以疏水性嵌段为中心的co2基aba 型表面活性剂,之前从未用其它方法合成。同时,碳酸酯含量低于10%的peoec可以通过 在低co2压力下获得,尽管所得聚合物具有可降解性,但其依然保持类peo性能。在这项 工作中,为了提高co2的价值化,在使用普通eo同时使用了co2,co2不仅可以作为廉价 的c1碳源,而且用作可降解部分,这会在学术界和工业界引起高度关注。
[0282]
实验部分
[0283]
材料
[0284]
除非另有说明,所有的试剂均购自sigma-aldrich公司。在室温下,将环氧乙烷(eo) 在钠屑上搅拌过夜,然后进行真空蒸馏,以进行eo的纯化。将经纯化的eo保存于schlenk 瓶中,并置于手套箱中的冷冻室内。在60℃下,将四氢呋喃(thf)在cah2上干燥过夜, 然后进行真空蒸馏。在真空状态下,将thf从正丁基锂溶液中再次蒸馏出来,以获得极度干 燥的thf。通过重结晶或沉淀法对单官能引发剂四丁基氯化铵(bu4ncl)、四辛基氯化铵 (oct4cl)和双(三苯基膦)氯化亚胺(ppncl)进行纯化,然后在五氧化二磷(p2o5)的存在 下进行真空干燥两天。根据文献的方法合成双官能引发剂四丁基琥珀酸铵(tbas),再用与 单官能引发剂同样的方法进行纯化。二氧化碳(co2)购自abdullah hashim industrial&gasco.,并在临用前使其通过纯化柱(vici metronics)。
[0285]
仪器及方法
[0286]
以cdcl3为溶剂,在核磁共振分析仪(bruker avance iii-400mhz)中记录1h nmr 谱。高ec含量的peceo的sec图形分析,通过agilent 1260infinity系统进行,其配有两 根polargel-m色谱柱。将n,n-二甲基甲酰胺(dmf)用作洗脱液,并将流速设置为1ml/min。 使该系统在45℃下平衡,用线型聚环氧乙烷标准品对分子量进行校准。在配备有styragel hr2 和styragel hr4色谱柱的viscotek ve2001系统上,获得具有低ec含量的peceo的sec图 形。将该系统在35℃下用thf以1.00ml/min的流速进行平衡。该系统用线型聚环氧乙烷标 准品校准。热重分析(tga)在mettler toledo tga/dsc分析仪上进行。在n2气氛中,将样 品从25℃加热至850℃,升温速率为10℃/min。差示扫描量热法(dsc)的测定使用mettlertoledo dsc1/tc100系统,在氮气氛围下,升温速率为10℃/min。第二次扫描的曲线用来测 定玻璃化转变温度(tg)。使用easydrop标准接触角测量仪来测定接触角。将水滴 (3μl)滴在浇铸在玻璃板上的peceo样品表面上,记录不同时间的图像。临界胶束浓度 (cmc)的测定采用荧光光谱法,并以芘(1
×
10-6
mol/l)作为探针。在thermo lumina荧 光分光计中,对不同浓度的一系列表面活性剂聚合物的水溶液的荧光光谱进行记录,温度为 25℃,激发波长为340nm。将373nm和384nm的荧光强度比值(i1/i3)相对于浓度作图, 以此作为计算浓度的函数,再以两条最佳拟合线的交叉点的浓度值作为cmc值。通过动态 光散射(dls)法对流体力学直径(dh)和胶束的尺寸分布进行测定,所用仪器为配备有30 mw氦氖激光器的malvern zetasizer nano zs装置,该激光器的工作波长为632.8nm。用于 dls测量的聚合物浓度为1mg/ml。
[0287]
聚合物合成
[0288]
具有高ec含量的peceo的合成。经典的过程如下:在手套箱内,在预干燥的50ml parr 反应器中,加入tbacl(0.057g,0.2mmol)、thf(1ml)、teb(1m,240ul)和eo (1ml,20mmol)。将teb单独地放入2ml的玻璃小瓶内,以防止在通入co2前eo发 生均聚合。在组装反应器之后,将其从手套箱内取出,再通入co2,使其压力达到20巴。 剧烈晃动反应器,以确保反应物均匀混合,然后在室温下,将反应体系搅拌15小时。再将反 应器排气,用2ml hcl的甲醇溶液(1m)淬灭聚合反应。将二氯甲烷加入反应混合物,便 于溶解聚合物。取出一份所得混合物用于1h nmr分析,以得到聚合的选择性数据。用甲醇 进行沉淀并再溶解于二氯甲烷,重复循环多次以纯化反应混合物。将聚合物在室温下真空干 燥,以达到恒重,以此去除残留的溶剂。
[0289]
低ec含量的peceo的合成。经典的过程如下:在手套箱内,在预干燥的50mlparr反 应器中,加入tbacl(0.057g,0.2mmol)、thf(1ml)、teb(1m,480ul)和eo(1ml, 20mmol)。将et3b单独地置于2ml玻璃小瓶内,以防止在通入co2之前eo发生均聚合。 将配备有气压计(精度为0.2巴)的调节器连接在co2充气管路之间。在整个聚合反应过程 中,保持反应器与充气管路之间的连接。以1巴的恒定压力将co2通入反应器。剧烈摇动反 应器,以确保反应物均匀混合,然后在室温下,将反应体系搅拌15小时。再将反应器排气, 用2ml hcl的甲醇溶液(1m)使聚合反应淬灭。取出一份所得混合物用于1h nmr分析, 以得到聚合的选择性数据。然后通过用二乙醚进行沉淀再溶解于thf,重复循环多次以纯化 反应混合物。通过将聚合物在室温下进行真空干燥以达到恒重,以将残留的溶剂去除。
[0290]
两亲性嵌段共聚物的合成。三嵌段共聚物的合成以两步法进行。第一步是合成高
ec含 量的peceo,作为中间嵌段。使用tbas作为引发剂,加入1当量的teb,在30巴的co2压力下合成上述peceo。对于合成p(eo-ec-eo)1和p(eo-ec-eo)2,目标聚合度为38;对 于合成p(eo-ec-eo)3,目标聚合度为78。在室温下将反应介质搅拌15小时,将反应器进行 排气,再转移入手套箱中。取出一份上述步骤得到的混合物,用于1h nmr和sec分析。对 于合成p(eo-ec-eo)1,将58当量的eo加入反应器内,用于增长亲水性嵌段;对于合成 p(eo-ec-eo)2和p(eo-ec-eo)3,加入117当量的eo。摇动后留下所形成的反应介质再搅拌 4小时。用二乙醚进行沉淀再溶解于thf,重复循环多次以纯化嵌段共聚物。通过将聚合物 在室温下真空干燥至恒重以将残留的溶剂去除。
[0291]
降解试验
[0292]
在强碱条件下进行完全降解试验,采用naoh 10mm与水性聚合物(表6第10项)的 水溶液(2mg/ml)。在25℃下100rpm搅拌上述溶液,持续48小时。当通过冷冻干燥去除 水之后,取出一份残留物溶于cdcl3,用于1h nmr分析,再取出另外一份溶于thf中,用 于sec表征。还在25℃、ph为8.5(tris-hcl,100mm)的条件下,对随时间推移的降解行 为进行了分析。每周取样品进行分析。在37℃下使用磷酸盐缓冲液,在模拟的生理条件下, 对聚合物的降解进行了研究。使用ph为7.4
±
0.1的磷酸盐缓冲液来模拟体液,并且使用ph 为6.5
±
0.1的磷酸盐缓冲液来模拟在肿瘤组织周围的微酸微环境。一个月后,对样品进行分 析。
[0293]
本公开的其它实施方案是可能的。尽管以上的描述含有很多特定性,但不应被理解成限 制本公开的范围,而仅提供对本公开的部分优选实施方案的说明。也可设想到的是,可做出 各实施方案的具体特征和方面的各种组合或亚组合,并且仍然落在本公开的保护范围内。应 当理解的是,所公开各实施方案的各种特征和方面可以互相组合或替换,从而形成各种实施 方案。因此,至少部分的本公开的范围不旨在受到上述具体公开实施方案的限制。
[0294]
因此,本公开的保护范围应当由所附权利要求及它们的法律等同物所确定。因此,应当 理解的是,本公开的保护范围完全包括对于本领域技术人员而言为显而易见的其它实施方案, 因此本公开的保护范围仅由所附权利要求所限定,其中除非明确说明,以单数形式对要素的 指代并不旨在表示“一个和仅一个”,而是表示“一个或多个”。对于本领域技术人员为已 知的上述优选实施方案中的要素的所有结构的、化学的、功能的等同物,均明确地以引用的 方式并入本公文中,并且旨在为权利要求所覆盖。此外,装置或方法未必解决本公开要解决 的各个问题或每个问题,因为它是由本公开的权利要求所包含。此外,无论在权利要求中是 否明确地陈述了要素、组分或方法步骤,本公开的要素、组分或方法步骤并非意图专用于共 众。
[0295]
为了说明和描述的目的,上面已给出了对本公开各种优选实施方案的描述。其并非旨在 是详尽地公开或者将本公开局限于具体实施方案,显而易见地根据以上教导许多种修改或变 型是可行的。选择并描述如上所述的示例性实施方案,是为了最佳地解释本公开的原理及其 实际应用,从而使本领域技术人员在各种实施方案中更好地利用本公开,并且各种修改是为 了适应所设想的具体用途。本公开的保护范围旨在由所附权利要求所限定。
[0296]
已描述了各种实施方案。这些和其它实施方案落入所附权利要求的范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1