一种氨基乙酸的合成方法与流程
1.本发明涉及有机合成的技术领域,具体涉及一种氨基乙酸的合成方法。
背景技术:
2.氨基乙酸又名甘氨酸,是结构最简单的氨基酸类化合物,其作为一种重要的精细化工中间体和化工原料,广泛用于医药、农药、食品、化肥、饲料等各个领域。随着人们生活水平的不断提高,氨基乙酸在食品、医药行业的需求不断扩大。
3.目前,氨基乙酸的合成方法主要有氯乙酸氨解法、天然蛋白质水解法和strecker法。国外多数采用以氰化钠为主原料的strecker法生产甘氨酸,其优点是产品易精制、生产成本低;缺点是氰化钠是剧毒物、条件苛刻、工艺路线较长。国内普遍采用氯乙酸氨解法生产甘氨酸,如专利cn1058957、cn1136035、cn1803763、cn1896049b等均报道了采用氯乙酸氨解法制备氨基乙酸的技术方案,这些技术方案均以乌洛托品为催化剂。传统氯乙酸氨解法得到的氨基乙酸
‑
氯化铵混晶中氨基乙酸含量低,仅为30%~50%左右。氨基乙酸
‑
氯化铵混晶通过醇析法精制后,得到的氨基乙酸收率较低,仅为80%左右。虽然通过提升反应温度和加大乌洛托品的量可以提高氨基乙酸的收率,但是随着反应温度的升高,副产物亚氨基二乙酸和氨基三乙酸的含量增加。并且,由于乌洛托品热稳定性差,随着反应温度升高,乌洛托品分解生成甲醛和氨,导致催化剂损失,反应液和产品颜色加深。
4.为解决传统催化剂乌洛托品在制备氨基乙酸中的技术问题,现有专利提出了相应的技术方案。专利cn111196768a采用两步法合成氨基乙酸。该技术方案使用吡啶碱类化合物替代乌洛托品催化剂,首先在低温下合成氯乙酸铵,进而以吡啶碱催化氯乙酸铵与氨反应生产氨基乙酸。该技术方案生成的氨基乙酸纯度较高,并且实现了混合溶剂和催化剂的循环使用,但是仍存在以下问题:反应步骤繁琐,收率低;使用毒性大的甲醇作溶剂;吡啶碱类化合物价格昂贵,增加生产成本,不利于工业化生产。
5.专利cn108558687a以氯乙酸、氨气为原料,在取代吡啶类催化剂存在下,氯乙酸在醇相中进行氨化反应,得到的反应液经多次过滤、洗涤等操作后分别得到氨基乙酸和氯化铵。该技术方案同样解决了传统催化剂乌洛托品热稳定性差、易损失等弊端,合成的氨基乙酸纯度较高,且催化剂可以循环使用。但仍存在以下缺点:催化效率仍不理想,催化剂用量为3mol%时,反应总收率仅为78.5%;反应溶剂为甲醇、乙醇等且用量大,增加回收工段的成本;取代吡啶类催化剂价格昂贵,增加生产成本,不利于工业化生产。
6.专利cn111187173a将传统的氯乙酸氨解法拆分成“氨溶解”、“成盐反应”、“氨化反应”三个工序。(1)配制氨水;(2)将氯乙酸溶液与配制的氨水混合发生成盐反应,得到溶液a;(3)将含有乌洛托品和氨水的混合溶液分多个部分加入溶液a中进行氨化反应后,经冷却、结晶、分离得到甘氨酸
‑
氯化铵混晶和母液。上述母液可用于配置步骤(1)中的氨水或步骤(2)中的氯乙酸溶液,继续进行上述反应实现连续化的甘氨酸合成。该技术方案有效避免了传统工艺方案的缺陷,而且实现了乌洛托品的循环利用。实验结果显示,催化剂用量为10mol%时,混晶中氨基乙酸含量为50%,甘氨酸收率为96%。该技术方案存在的问题是催
化剂用量较大,混晶中副产物多、氨基乙酸的含量偏低,操作步骤繁琐、成本高等问题。
7.使用廉价、高效、稳定的催化剂合成氨基乙酸的方法未见报道。彭春雪等报道了以三乙胺作为缚酸剂合成氨基乙酸的方法(彭春雪,詹志平,刘三六,等.以三乙胺为缚酸剂合成甘氨酸的实验研究[j].山东化工,2017,46(7):44
‑
47.)。该方法以三乙胺为缚酸剂,以多聚甲醛为催化剂,以甲醇为溶剂,通过氯乙酸的氨化反应合成氨基乙酸。该反应的收率最高可达92%,甘氨酸主含量可达98.5%。但是该方法中的催化剂仍是乌洛托品,三乙胺仅作为缚酸剂,用量高达115mol%。
技术实现要素:
[0008]
针对现有技术中由氯乙酸和成氨基乙酸的催化剂多为乌洛托品以及产品中氯化铵残留高、收率低等问题,本发明提供一种氨基乙酸的合成方法,以解决上述技术问题。本发明以氯乙酸为起始原料,采用脂肪胺类作为催化剂,在氨气或氨水作用下,制备生成氨基乙酸。本发明使用的催化剂相比于乌洛托品,具有良好的热稳定性,反应温度较高时不会分解,保证了较高的催化剂有效量;并且通过催化剂的空间位阻调控,可以有效抑制二级胺和三级胺副产物的生成,从而选择性的生成氨基乙酸。本发明可在较宽的温度范围内获得优异的反应收率,并且粗品中副产物少、氨基乙酸含量高。
[0009]
本发明的技术方案如下:
[0010]
一种氨基乙酸的合成方法,以氯乙酸或氯乙酸铵为原料,氨气或氨水为胺化剂,在脂肪胺催化下,合成氨基乙酸;反应方程式如下:
[0011][0012]
其中,r1,r2,r3各自独立的为h或c1~c4的烷基。
[0013]
优选的,所述脂肪胺选自正丁胺、二正丁胺、二正丙胺、二乙基甲胺、二异丙基乙胺、三甲胺、三乙胺、三正丁胺或二甲基乙胺;优选三乙胺、正丁胺、二正丁胺或三正丁胺。
[0014]
具体合成步骤如下:
[0015]
(1)将脂肪胺催化剂加入反应容器中,控制温度预热;
[0016]
(2)将氯乙酸配置成一定浓度的水溶液,移至氯乙酸水溶液滴加装置中;
[0017]
(3)取适量氨水,移至氨水滴加装置中;
[0018]
(4)控制氯乙酸溶液及氨水的滴加速度,滴加温度控制在25~45℃之间;
[0019]
(5)滴加完毕后控制反应温度为40~85℃之间,反应时间为1~4小时;
[0020]
(6)反应结束后,将反应液进行减压蒸馏、过滤,制得氨基乙酸
‑
氯化铵混晶湿品,湿品在65℃下真空干燥至恒重得到粗品;滤液可直接用于或者补加一定量催化剂后用于下一批次的氨基乙酸合成;
[0021]
(7)50℃下,使用98%的乙醇水混合液对粗品进行重结晶,经过滤、醇洗、干燥后,得到氨基乙酸。将滤液冷却结晶、过滤得到氯化铵,滤液重复使用;
[0022]
优选的,所述步骤(1)中,氯乙酸与催化剂的投料摩尔比为1:0.005~0.05,优选为1:0.02~0.05。
[0023]
优选的,所述步骤(1)中,预热温度为20℃~40℃。
[0024]
优选的,所述步骤(2)中,氯乙酸水溶液浓度为60%~65%,优选为62%~63%。
[0025]
优选的,所述步骤(3)中,氨水浓度为25%~28%。
[0026]
优选的,所述步骤(4)中,氨水的滴加速度为8.0g/min~15.0g/min,优选为10.0g/min~13.0g/min,氯乙酸溶液的滴加速度为13g/min~22.0g/min,优选为15.0g/min~20.0g/min。控制氯乙酸和氨水的滴加速度目的在于:一是滴加过快会导致反应液局部ph过大,产生过多的副反应产物;二是滴加过程是一个放热的过程,控制滴加速度有利于控制反应体系的温度,避免因局部过热导致的杂质含量升高。
[0027]
优选的,所述步骤(4)中,氨水及氯乙酸溶液滴加时间控制在1~2小时,优选为1.5小时。
[0028]
优选的,所述步骤(5)中,反应温度控制在50~80℃之间。
[0029]
优选的,所述步骤(6)中,减压蒸馏压力为
‑
0.1mpa,温度控制在60~70℃。
[0030]
优选的,所述步骤(6)中,减压蒸馏得到的纯水可用于配制氯乙酸或者氨水溶液。
[0031]
本发明的有益效果为:
[0032]
(1)本发明首次使用了脂肪胺类化合物作为合成氨基乙酸的催化剂,氨基乙酸的总收率最高可达99.0%,混晶中氯化铵的含量在41.6%~42.9%之间,接近理论值(理论含量为41.6%)。
[0033]
(2)本发明使用的脂肪胺催化剂热稳定性好,在40℃~85℃之间均可高效的催化氯乙酸的氨化反应,而传统催化剂乌洛托品的使用温度一般在60℃~75℃。相比之下,本发明使用的催化剂比乌洛托品有着更好的反应适应性。
[0034]
(3)本发明使用的脂肪胺催化剂的催化效率高,催化剂用量在0.5mol%~5.0mol%之间,即可高效地催化氯乙酸的氨化反应。而传统催化剂乌洛托品的用量一般为3.0mol%~10.0mol%之间,甚至更高。
[0035]
(4)本发明通过催化剂空间位阻的调控,可有效抑制亚氨基二乙酸等副产物的生成,提高产品质量。
[0036]
(5)本发明的催化剂可循环使用,操作条件较现有技术更加温和、简便,总反应氨消耗接近理论当量,氨消耗不超过理论当量5%。
附图说明
[0037]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,对于本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0038]
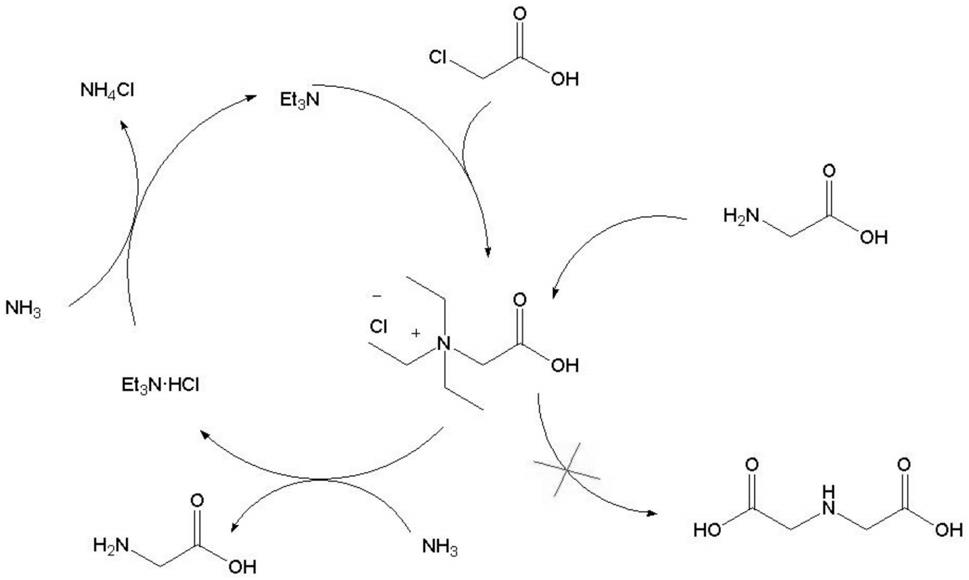
图1是本发明实施例1催化反应机理图。
具体实施方式
[0039]
为了使本技术领域的人员更好地理解本发明中的技术方案,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都应当属于本发明保护
的范围。
[0040]
实施例1
[0041]
(1)将三乙胺39.7g(0.39mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制30℃预热;
[0042]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0043]
(3)取1600ml 25%的氨水,转移至氨水滴加装置;
[0044]
(4)控制氯乙酸溶液及氨水滴加速度分别为16.0g/min、12.0g/min,温度控制在30℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2小时滴加完毕;
[0045]
(5)滴加完毕后升温至60℃,保温反应2小时;
[0046]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1326.5g,其中副产物氯化铵含量为41.8%;
[0047]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸741.2g,收率为93.3%,滴定法检测含量为98.9%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0048]
以实施例1为例阐述一下本发明的催化机理:首先三乙胺与氯乙酸进行加成反应生成季铵盐中间体,该中间体存在较大的空间位阻;进而氨与季铵盐中间体发生亲核反应,生成氨基乙酸和三乙胺盐酸盐。由于氨基乙酸的空间位阻较大,不易与季铵盐中间体发生亲核反应,因而可以有效的抑制亚氨基二乙酸等副产物的生成。
[0049]
实施例2
[0050]
(1)将三正丁胺72.7g(0.39mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制40℃预热;
[0051]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0052]
(3)取1600ml 25%的氨水,转移至氨水滴加装置;
[0053]
(4)控制氯乙酸溶液及氨水滴加速度分别为16.0g/min、12.0g/min,温度控制在40℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2小时滴加完毕;
[0054]
(5)滴加完毕后升温至60℃,保温反应2小时;
[0055]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1345.6g,其中副产物氯化铵含量为41.7%;
[0056]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸761.1g,收率为95.8%,滴定法检测含量为99.0%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0057]
实施例3
[0058]
(1)将二正丁胺68.5g(0.53mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制40℃预热;
[0059]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0060]
(3)取1600ml 28%的氨水,转移至氨水滴加装置;
[0061]
(4)控制氯乙酸溶液及氨水滴加速度分别为15.0g/min、10.0g/min,温度控制在40
℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2.5小时滴加完毕;
[0062]
(5)滴加完毕后升温至50℃,保温反应4小时;
[0063]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1269.4g,其中副产物氯化铵含量为42.9%;
[0064]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸737.3g,收率为92.8%,滴定法检测含量为98.1%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0065]
实施例4
[0066]
(1)将正丁胺38.8g(0.53mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制35℃预热;
[0067]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0068]
(3)取1600ml 28%氨水,转移至氨水滴加装置;
[0069]
(4)控制氯乙酸溶液及氨水滴加速度分别为15.0g/min、10.0g/min,温度控制在40℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2.5小时滴加完毕;
[0070]
(5)滴加完毕后升温至50℃,保温反应4小时;
[0071]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1201.4g,其中副产物氯化铵含量为42.4%;
[0072]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸684.0g,收率为86.1%,滴定法检测含量为97.8%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0073]
实施例5
[0074]
(1)将二异丙基乙基胺6.85g(0.053mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制30℃预热;
[0075]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0076]
(3)取1600ml 28%氨水,转移至氨水滴加装置;
[0077]
(4)控制氯乙酸溶液及氨水滴加速度分别为15.0g/min、10.0g/min,温度控制在30℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2.5小时滴加完毕;
[0078]
(5)滴加完毕后升温至80℃,保温反应2小时;
[0079]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1023.1g,其中副产物氯化铵含量为42.6%;
[0080]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸576.0g,收率为72.5%,滴定法检测含量为89.0%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0081]
实施例6
[0082]
(1)将三正丁胺39.3g(0.21mol)和正丁胺11.6g(0.16mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制40℃预热;
[0083]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0084]
(3)取1600ml 28%氨水,转移至氨水滴加装置;
[0085]
(4)控制氯乙酸溶液及氨水滴加速度分别为16.0g/min、12.0g/min,温度控制在40℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2小时滴加完毕;
[0086]
(5)滴加完毕后升温至60℃,保温反应4小时;
[0087]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1236.7g,其中副产物氯化铵含量为42.0%;
[0088]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸725.4g,收率为91.3%,滴定法检测含量为96.7%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0089]
对比例
[0090]
(1)将乌洛托品74.3g(0.53mol)加入装有温度计、搅拌、加热、循环水降温、滴加装置的反应容器中,控制40℃预热;
[0091]
(2)称量1000.0g(10.6mol)氯乙酸,加入600g水溶解,转移至氯乙酸滴加装置;
[0092]
(3)取1600ml 25%氨水,转移至氨水滴加装置;
[0093]
(4)控制氯乙酸溶液及氨水滴加速度分别为16.0g/min、12.0g/min,温度控制在40℃,氯乙酸溶液约1.5小时滴加完毕,氨水约2小时滴加完毕;
[0094]
(5)滴加完毕后升温至60℃,保温反应2小时;
[0095]
(6)反应结束后,将上述反应液减压蒸馏除水、过滤处理,得到氨基乙酸
‑
氯化铵混晶湿品;将上述湿品于65℃真空干燥至恒重,其质量为1073.5g,其中副产物氯化铵含量为46.7%;
[0096]
(7)粗品使用98%乙醇
‑
水混合液50℃下重结晶,经过滤、干燥得到氨基乙酸630.0g,收率为79.3%,滴定法检测含量为96.5%。将滤液缓慢降温至10℃,过滤分离得到氯化铵。
[0097]
尽管通过参考附图并结合优选实施例的方式对本发明进行了详细描述,但本发明并不限于此。在不脱离本发明的精神和实质的前提下,本领域普通技术人员可以对本发明的实施例进行各种等效的修改或替换,而这些修改或替换都应在本发明的涵盖范围内/任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以权利要求所述的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1