6,6-二烷基哌啶-2-羧酸化合物的合成方法与流程
6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的合成方法
技术领域
1.本发明实施例属于化工领域,特别涉及一种6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的合成方法。
背景技术:
2.选择性n型电压激活钙通道(vacc)阻滞剂已在几种中风和疼痛模型中显示出效果,在寻找小分子作为n型钙通道阻滞剂的过程中,有文献报道一系列n,n
‑
二烷基肽胺在n型vacc上具有很强的功能活性,6,6
‑
二烷基哌啶
‑2‑
羧酸化合物是合成n,n
‑
二烷基肽胺类化合物的重要中间体。然而,关于6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的制备方法,并没有相关的现有技术,一些类似的化合物如1,2,3,4
‑
四氢
‑
1,1
‑
二甲基
‑3‑
异喹啉羧酸、6,6
‑
二甲基
‑3‑
氧代
‑1‑
(2,2,2
‑
三氟乙酰基)哌啶
‑2‑
羧酸乙酯、6
‑
甲基哌啶
‑2‑
羧酸等的合成技术,与6,6
‑
二烷基哌啶
‑2‑
羧酸在合成机理方面完全不同。因此,有必要提供一种6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的制备方法以满足需要。
技术实现要素:
3.本发明实施例提供一种6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的全新合成方法,以化合物i、三氟醋酸、氢氧化钠等为原料,经过两步反应得到目标产物6,6
‑
二烷基哌啶
‑2‑
羧酸。本发明实施例操作简便、反应条件温和、成本较低、环境友好,综合收率为70%左右,适合工业化规模生产。
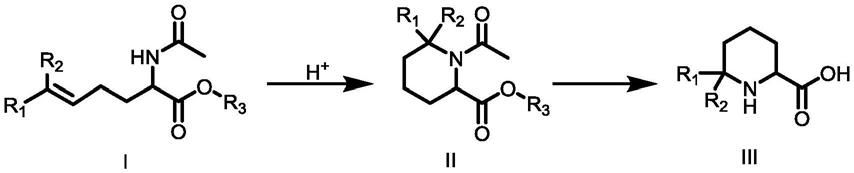
4.本发明实施例提供一种化合物iii的合成方法,
[0005][0006]
其包括以下步骤:
[0007]
(1)通过化合物i进行环化反应,得到化合物ii;
[0008]
(2)在碱性水溶液中,对化合物ii进行水解反应,得到化合物iii;
[0009][0010]
其中,r1和r2为c1‑
c3烷基,优选为甲基或乙基,更优选为甲基;r3为甲基或乙基。
[0011]
在一些实施方案中,步骤(1)的反应过程为将化合物i和酸液在第一反应温度下进行反应,待反应完成后,降温,加入到冰水中,将体系调节至弱碱性,然后依次通过有机溶剂萃取,干燥,柱层析洗脱得化合物ii,直接用于步骤(2)中。
[0012]
在一些实施方案中,所述酸液选自三氟醋酸或冰乙酸,优选为三氟醋酸。
[0013]
在一些实施方案中,所述第一反应温度为25~70℃,优选为50℃。
[0014]
在一些实施方案中,步骤(2)的反应过程为将化合物ii和碱性水溶液在第二反应温度下反应完全后,降温,将反应体系调节至酸性,而后通过有机溶剂萃取,干燥,减压蒸馏后得到化合物iii。
[0015]
在一些实施方案中,所述碱性水溶液为氢氧化钠水溶液。
[0016]
在一些实施方案中,化合物ii和氢氧化钠的摩尔比为1:(3~5),优选1:4。
[0017]
在一些实施方案中,第二反应温度为25
‑
~100℃,优选为100℃。
[0018]
本技术提供的6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的全新合成方法,其具有以下优点:
[0019]
1、本技术首次提出了6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的合成方法,促进了该化合物在医疗领域中的应用,并有望降低中风及疼痛等疾病的治疗成本。
[0020]
2、本技术采用价廉易得的2
‑
(乙酰氨基)
‑6‑
甲基
‑5‑
庚酸乙酯作为原料,于酸性溶剂中,历经ritter反应关环,得到化合物6,6
‑
二甲基哌啶
‑2‑
羧酸;该原料在反应过程中产生唯一碳翁离子,避免了分子的重排,从而进一步避免了副产物的生成;使反应不仅能够高效进行,而且简化了反应后处理,所得产物纯度及反应收率也较为理想。
[0021]
3、本技术中化合物6,6
‑
二甲基哌啶
‑2‑
羧酸在碱性溶剂中直接进行水解,该过程反应副产物少,产物纯度高,操作简便的同时,所用辅料均为市售价廉易得品,从而不仅适合实验室小规模制备,也适于大规模工业化生产。
[0022]
4.本技术实施例优化反应工艺和反应条件,提供了6,6
‑
二烷基哌啶
‑2‑
羧酸合成的优惠条件,使整个合成过程获得最优反应收率的同时,原料和辅料处于最佳配比,从而进一步降低合成成本,获得较高的经济效益,更适合工业化的扩大生成。
具体实施方式
[0023]
为了使本技术所解决的技术问题、技术方案及有益效果更加清楚明白,以下结合具体实施例,对本技术作进一步的说明。下述实施例中,除非另有说明,所述的试验方法具体条件通常按照常规条件或制造厂商建议的条件实施;原料、试剂均通过市售获得或者使用公开信息制备。
[0024]
本发明的合成路线如下:
[0025][0026]
反应机理如下:
[0027][0028]
反应步骤(1),化合物1在h
+
进攻下,遵照马氏规则,使烯烃部分质子化,形成稳定的碳翁离子,而后氮上的孤对电子进攻该碳翁离子,进行亲电加成反应,关环得到化合物2。反应步骤(2)中,碱性条件下,羟基负离子进攻缺电子羰基碳,酰胺端形成氧负离子,而后进行电子转移,失去一分子乙酸,形成氨负离子;酯基端经羟基进攻羰基碳,形成一个四面体中间体,消除乙酯基团,形成甲酸,再经质子转移,形成羧酸负离子;氨负离子及羧酸负离子在酸性条件中质子化,得到目标化合物3。
[0029]
实施例1
[0030]
第一步:化合物1
‑
乙酰基
‑
6,6
‑
二甲基哌啶
‑2‑
羧酸乙酯的合成
[0031]
将2
‑
(乙酰氨基)
‑6‑
甲基
‑5‑
庚酸乙酯(10g,44mmol,1eq)加入到100ml的三氟醋酸中,在50℃下,反应2小时,直到薄层色谱tlc检测达到反应终点,冷却至室温,加入到冰水中,用碳酸氢钠调节ph至8
‑
9,用600ml乙酸乙酯萃取2次,合并有机相用饱和食盐水反洗,加入无水硫酸钠干燥,减压蒸馏,经柱层析洗脱,柱层析所用的洗脱剂为体积比为5:1的石油醚和乙酸乙酯的混合溶剂,得到8.1g化合物1
‑
乙酰基
‑
6,6
‑
二甲基哌啶
‑2‑
羧酸乙酯,收率为81.00%,纯度98%,1hnmr(500mhz,cdcl3)δ4.66(t,j=8.0hz,1h,nch),4.14(q,j=6.0hz,2h,ch2ch3),2.22(ddd,j=18.2hz,7.9hz,5.6hz,1h,nchch2),2.05(s,3h,coch3),1.95(m,1h,nchch2),1.72(m,3h,ch2ch2),1.51(m,1h,ch2ch2),1.28(t,j=6.0hz,3h,ch2ch3),1.18(s,6h,ch3)。
[0032]
第二步:化合物6,6
‑
二甲基哌啶
‑2‑
羧酸的合成
[0033]
将氢氧化钠(5.7g,142.5mmol,4eq)溶于80ml水中,加入化合物1
‑
乙酰基
‑
6,6
‑
二甲基哌啶
‑2‑
羧酸乙酯(8.1g,35.6mmol,1eq),加热至100℃,搅拌反应2小时,直到薄层色谱tlc检测达到反应终点,冷却至室温,用盐酸调节ph值至3
‑
4,然后加入400ml二氯甲烷萃取5次,有机相合并后加入无水硫酸钠干燥,减压蒸馏除去二氯甲烷,得到4.8g化合物6,6
‑
二甲基哌啶
‑2‑
羧酸,收率为85.70%,纯度99%,1hnmr(600mhz,dmso)δ3.30(t,j=7.5hz,1h,nhch),2.29(s,1h,nh),1.93(m,1h,chch2),1.83(m,1h,chch2),1.69(dd,j=12.0hz,6.0hz,1h,ch2ch2),1.63(dd,j=12.0hz,6.1hz,1h,ch2ch2),1.54(dt,j=11.8hz,5.8hz,1h,ch2ch2),1.35(m,1h,ch2ch2),1.32(s,6h,ch3)。
[0034]
在本技术中,申请人还尝试在步骤(1)中的使用盐酸作为酸液,但其反应速度缓慢,升温反应生成大量杂质,收率大大降低。此外,还尝试了步骤(2)中使用氢氧化钾代替氢
氧化钠,其反应效果类似氢氧化钠,但是氢氧化钾腐蚀性更强。
[0035]
申请人还在以下不同反应条件下进行了试验,其余步骤与实施例1相同,在此不在赘述。
[0036]
表1
[0037]
序号步骤(1)反应条件化合物ii的收率实施例1三氟醋酸,50℃,反应2h81.00%实施例2冰乙酸,50℃,反应2h70.00%实施例3盐酸,50℃,反应2h23.01%实施例4三氟醋酸,25℃,反应2h9.30%实施例5三氟醋酸,70℃,反应2h75.00%序号步骤(2)反应条件化合物iii的收率实施例14.0eq氢氧化钠,100℃,反应2h85.70%实施例63.0eq氢氧化钠,100℃,反应2h72.70%实施例75.0eq氢氧化钠,100℃,反应2h80.00%实施例84.0eq氢氧化钠,75℃,反应2h75.33%实施例94.0eq氢氧化钠,50℃,反应2h67.03%实施例104.0eq氢氧化钠,110℃,反应2h81.70%
[0038]
实施例2至实施例3中的步骤(1)采用与实施例1不同的酸液进行反应,实施例2采用冰乙酸进行反应,相对于实施例1,收率有所下降,为70.00%。特别是,实施例3中使用盐酸进行反应,不仅反应速率明显缓慢,且升温过程中生成较多的副产物,使反应收率相比于实施例1明显下降,仅23.01%。
[0039]
实施例4至实施例5中的步骤(1)采用与实施例1不同的反应温度。相对于实施例1,实施例4降低反应温度至25℃,反应速率明显降低,相同反应时间内的反应收率有所降低,仅9.30%;实施例5升高反应温度至70℃,促进了副反应的发生,使反应收率相比于实施例1降低至75.00%。
[0040]
实施例6至实施例7中的步骤(2)采用与实施例1不同用量的氢氧化钠进行反应,相对于实施例1,实施例6和实施例7分别相对于实施例1减少氢氧化钠的用量至1
‑
乙酰基
‑
6,6
‑
二甲基哌啶
‑2‑
羧酸乙酯的3当量和增加氢氧化钠的用量至1
‑
乙酰基
‑
6,6
‑
二甲基哌啶
‑2‑
羧酸乙酯的5当量,反应收率相比于实施例1略有下降,分别为72.70%和80.00%。
[0041]
实施例8至实施例10的步骤(2)采用与实施例1不同的反应温度进行反应,实施例8和实施例9降低反应温度至75℃和50℃,使得反应速率有所降低,从而使相同反应时间内的反应收率降低,分别为75.33%和67.03%;实施例10升高反应温度至110℃,促进了副反应的发生,反应收率相比实施例1降低至81.70%。
[0042]
综上可知,本技术的6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的合成方法,采用式i所示化合物为原料,经过两步反应得到目标产物6,6
‑
二烷基哌啶
‑2‑
羧酸化合物,为6,6
‑
二烷基哌啶
‑2‑
羧酸化合物的制备提供了优良的合成路线,本发明操作简便、反应条件温和、成本较低、环境友好,综合收率70%左右,适合工业化规模生产。
[0043]
上述实施例的作用在于说明本技术的实质性内容,但并不以此限定本技术的保护范围。本领域的普通技术人员应当理解,可以对本技术的技术方案进行修改或者等同替换,
而不脱离本技术技术方案的实质和保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1