一种葫芦巴中薯蓣皂苷的制备方法
1.本发明涉及分离纯化技术领域,尤其涉及一种葫芦巴中薯蓣皂苷的制备方法。
背景技术:
2.葫芦巴(trigonella foenum
‑
graeeum,l.),又名大叶芸香草、香苜蓿,是一种生存力极强的一年生草本植物,系豆科植物蝶形花亚科,它起源于南欧、西亚和地中海地区,自古以来被用作香料及调味剂。葫芦巴种子中含有丰富的化学成分,如半乳甘露聚糖、甾体皂苷类、黄酮类、生物碱类、三萜类和香豆素类等,药理作用非常广泛。甾体皂苷类可以降低血清总胆固醇的含量,抑制胆固醇的吸收,又是合成甾体激素药物的中间体,葫芦巴中甾体皂苷含量约为6.5%,而薯蓣皂苷是甾体皂苷的主要化学成分。因此,薯蓣皂苷作为葫芦巴甾体皂苷类的主要成分在全世界受到广泛关注。
3.目前,薯蓣皂苷的传统纯化方法主要有大孔树脂吸附法、膜分离法、高速逆流色谱法和柱层析法,但这些方法存在耗时长、分离纯化步骤繁琐的问题。
技术实现要素:
4.本发明的目的在于提供一种葫芦巴中薯蓣皂苷的制备方法,该方法具有操作简单、分离周期短和效率高等特点。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种葫芦巴中薯蓣皂苷的制备方法,包括以下步骤:
7.将葫芦巴粉与醇溶剂混合,进行提取,得到提取液;
8.将所述提取液进行泡沫分离,得到薯蓣皂苷粗产物;
9.采用制备型高效液相色谱将所述薯蓣皂苷粗产物进行分离纯化,得到薯蓣皂苷。
10.优选的,所述醇溶剂包括乙醇溶液;所述乙醇溶液的质量浓度为60~80%;所述葫芦巴粉的质量与醇溶剂的体积之比为1g:(20~40)ml。
11.优选的,所述提取的压力为300~400mpa,时间为6~12min。
12.优选的,所述提取后,还包括将提取所得料液依次进行减压抽滤和浓缩。
13.优选的,所述泡沫分离的条件为:上样浓度0.01~0.05mg/ml,温度25~45℃,气速350~550ml/min,装液量250~450ml。
14.优选的,所述泡沫分离的条件为:上样浓度0.03mg/ml,温度28℃,气速450ml/min,装液量400ml。
15.优选的,所述制备型高效液相色谱的色谱条件包括:流动相:水和乙腈;0~5min,体积分数5~15%乙腈;5~15min,体积分数15~45%乙腈;15~35min,体积分数45~100%乙腈;流速为10ml/min;进样量为5ml。
16.优选的,所述制备型高效液相色谱的色谱条件还包括:色谱柱:hederaods
‑
2柱,20
×
250mm,10μm;检测波长208nm。
17.本发明提供了一种葫芦巴中薯蓣皂苷的制备方法,包括以下步骤:将葫芦巴粉与
醇溶剂混合,进行提取,得到提取液;将所述提取液进行泡沫分离,得到薯蓣皂苷粗产物;采用制备型高效液相色谱将所述薯蓣皂苷粗产物进行分离纯化,得到薯蓣皂苷。本发明以葫芦巴为原料,采用泡沫分离法结合制备型高效液相色谱(p
‑
hplc)法对葫芦巴中的薯蓣皂苷进行分离纯化,薯蓣皂苷提取液中杂质较多,产品纯度低,而薯蓣皂苷属于螺甾烷型皂苷类,螺甾烷型甾体皂苷是由螺甾烷型的苷元和一个糖链构成,糖链具有较强的极性(水溶性),而苷元部分亲油性较好,因此甾体皂苷具有良好的起泡性能,本发明利用薯蓣皂苷提取液中具有起泡性能的物质,采用泡沫分离法通过鼓泡的形式将薯蓣皂苷醇提液中具有起泡性的物质吸附在气泡表面,利用气泡携带泡沫层的溶液排出达到分离的目的,能够缩短分离周期;然后采用制备型高效液相色谱将分离得到的薯蓣皂苷粗产物进一步纯化并收集,纯化效果好,具有操作简单、分离周期短和效率高等特点,对所得的单体化合物进行结构定性确定其为薯蓣皂苷适用于葫芦巴中薯蓣皂苷的分离制备,为葫芦巴中薯蓣皂苷的分离纯化以及规模化生产提供理论依据和技术参考。
附图说明
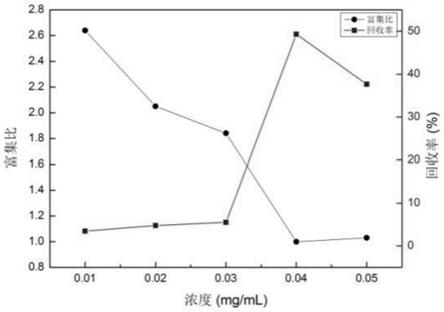
18.图1为上样浓度对泡沫分离薯蓣皂苷的影响图;
19.图2为温度对泡沫分离薯蓣皂苷的影响图;
20.图3为气速对泡沫分离薯蓣皂苷的影响图;
21.图4为装液量对泡沫分离薯蓣皂苷的影响图;
22.图5为上样浓度
‑
温度、上样浓度
‑
气速和上样浓度
‑
装液量两因素的交互作用对于薯蓣皂苷泡沫分离的影响图;
23.图6为温度
‑
气速、温度
‑
装液量和气速
‑
装液量两因素的交互作用对于薯蓣皂苷泡沫分离的影响图;
24.图7为温度
‑
上样浓度、气速
‑
上样浓度和装液量
‑
上样浓度两因素的交互作用对于薯蓣皂苷泡沫分离的影响图;
25.图8为气速
‑
温度、装液量
‑
温度和装液量
‑
气速两因素的交互作用对于薯蓣皂苷泡沫分离的影响图;
26.图9为薯蓣皂苷标品(a)、供试品(b)和阴性样品(c)的p
‑
hplc图;
27.图10为薯蓣皂苷标品(a)、供试品(b)和阴性样品(c)的hplc图;
28.图11为薯蓣皂苷标品和实施例1制备的薯蓣皂苷样品的红外谱图;
29.图12为实施例1制备的薯蓣皂苷样品的质谱图;
30.图13为实施例1制备的薯蓣皂苷样品的核磁共振氢谱图;
31.图14为实施例1制备的薯蓣皂苷样品的核磁共振碳谱图。
具体实施方式
32.本发明提供了一种葫芦巴中薯蓣皂苷的制备方法,包括以下步骤:
33.将葫芦巴粉与醇溶剂混合,进行提取,得到提取液;
34.将所述提取液进行泡沫分离,得到薯蓣皂苷粗产物;
35.采用制备型高效液相色谱将所述薯蓣皂苷粗产物进行分离纯化,得到薯蓣皂苷。
36.在本发明中,若无特殊说明,所需制备原料均为本领域技术人员熟知的市售商品。
37.本发明将葫芦巴粉与醇溶剂混合,进行提取,得到提取液。在本发明中,所述葫芦巴粉的制备方法优选为将葫芦巴种子依次进行粉碎和干燥,得到葫芦巴粉;本发明对所述粉碎的过程及粉碎后物料的粒径没有特殊的限定,按照本领域熟知的过程进行粉碎即可。在本发明中,所述干燥的方式优选为烘干,所述烘干的温度优选为50℃,所述烘干的时间优选为烘干至物料恒重。
38.在本发明中,所述醇溶剂优选包括乙醇溶液;所述乙醇溶液的质量浓度优选为60~80%,更优选为71%;所述葫芦巴粉的质量与醇溶剂的体积之比(即料液比)优选为1g:(20~40)ml,更优选为1g:30ml。
39.本发明对所述葫芦巴粉与醇溶剂混合的过程没有特殊的限定,按照本领域熟知的过程能够将物料混合均匀即可。
40.在本发明中,所述提取的压力优选为300~400mpa,更优选为360mpa,时间优选为6~12min,更优选为563s。在所述提取过程中,通过醇溶剂将葫芦巴粉中的薯蓣皂苷提取出来。
41.完成所述提取后,本发明优选还包括将所得料液依次进行减压抽滤和浓缩。本发明对所述减压抽滤的过程没有特殊的限定,按照本领域熟知的过程进行即可。在本发明中,所述浓缩优选通过旋转蒸发仪进行,优选将减压抽滤所得溶液浓缩至无醇味即可。
42.得到提取液后,本发明将所述提取液进行泡沫分离,得到薯蓣皂苷粗产物。在本发明对所述泡沫分离所用泡沫分离柱没有特殊的限定,本领域熟知的市售泡沫分离柱均可;在本发明的实施例中,具体为循环水恒温泡沫分离柱。
43.在本发明中,所述泡沫分离的条件优选为:上样浓度0.01~0.05mg/ml,温度25~45℃,气速350~550ml/min,装液量250~450ml,更优选为:上样浓度0.03mg/ml,温度28℃,气速450ml/min,装液量400ml。
44.完成所述泡沫分离后,本发明优选将所得薯蓣皂苷粗产物进行冷冻干燥,将所得粉末物质加甲醇溶解,进行后续色谱分离。本发明对所述甲醇的用量没有特殊的限定,能够将粉末物质完全溶解即可。
45.得到薯蓣皂苷粗产物后,本发明采用制备型高效液相色谱将所述薯蓣皂苷粗产物进行分离纯化,得到薯蓣皂苷。在本发明中,所述制备型高效液相色谱所用仪器优选购于江苏汉邦科技有限公司。
46.在本发明中,所述制备型高效液相色谱的色谱条件优选包括:流动相:水和乙腈;0~5min,体积分数5%~15%乙腈;5~15min,体积分数15%~45%乙腈;15~35min,体积分数45%~100%乙腈;流速为10ml/min;进样量为5ml;优选还包括:色谱柱:hedera ods
‑
2柱,20
×
250mm,10μm;检测波长208nm。
47.完成所述分离纯化后,本发明优选将所得纯化物进行蒸发浓缩,去除有机相蒸发浓缩,冷冻干燥,得到薯蓣皂苷。本发明对所述蒸发浓缩和冷冻干燥的过程没有特殊的限定,按照本领域熟知的过程进行即可。
48.下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
49.实施例1
50.将葫芦巴种子粉碎后,在50℃烘箱干燥至恒重,将所得葫芦巴粉1g与30ml质量浓度为71%的乙醇溶液放置于聚乙烯瓶中,在360mpa下处理563s,将所得料液进行减压抽滤,使用旋转蒸发仪将所得溶液浓缩至无醇味,将所得原液在上样浓度0.03mg/ml,温度28℃,气速450ml/min,装液量400ml条件下在循环水恒温泡沫分离柱中进行泡沫分离后,将所得薯蓣皂苷粗产物冷冻干燥,将所得粉末加甲醇溶解,摇匀,在制备型高效液相色谱上进行分离纯化,色谱条件为:色谱柱:hedera ods
‑
2柱,20
×
250mm,10μm;检测波长208nm;流动相:水和乙腈;0~5min,体积分数5~15%乙腈;5~15min,体积分数15~45%乙腈;15~35min,体积分数45~100%乙腈;流速为10ml/min;进样量为5ml,完成分离后,收集有机相进行蒸发浓缩,冷冻干燥,得到薯蓣皂苷。
51.实施例2
52.与实施例1的区别在于:在装液量为350ml,气速为500ml/min,温度为30℃条件下进行泡沫分离,上样浓度依次为0.01mg/ml、0.02mg/ml、0.03mg/ml、0.04mg/ml和0.05mg/ml。
53.实施例3
54.与实施例1的区别在于:在装液量为350ml,气速为500ml/min,上样浓度为0.04mg/ml条件下进行泡沫分离,温度依次为25℃、30℃、35℃、40℃、45℃。
55.实施例4
56.与实施例1的区别仅在于:在装液量为350ml,温度为30℃,上样浓度为0.04mg/ml条件下进行泡沫分离,气速依次为350ml/min、400ml/min、450ml/min、500ml/min和550ml/min。
57.实施例5
58.与实施例1的区别仅在于:在气速为500ml/min,温度为30℃,上样浓度为0.04mg/ml条件下进行泡沫分离,装液量依次为250ml、300ml、350ml、400ml和450ml。
59.评价与测试
60.1、葫芦巴中薯蓣皂苷泡沫分离的评价指标
61.采用香草醛
‑
高氯酸显色法对葫芦巴中薯蓣皂苷的浓度进行测定,分别按式(1)、(2)计算葫芦巴中薯蓣皂苷的回收率e和富集比r。
[0062][0063][0064]
式中:ρ
f
、ρ
s
‑
泡沫层、残留层中薯蓣皂苷的质量浓度,μg/ml,v0、v
s
、v
f
‑
依次为原液、残留层、泡沫层的体积,ml。
[0065]
2、根据上述式(1)、(2)计算实施例2~5制备的葫芦巴中薯蓣皂苷的回收率e和富集比r,所得结果分别见图1~4;
[0066]
图1为上样浓度对泡沫分离薯蓣皂苷的影响图,如图1所示,富集比随浓度的增大
而逐渐降低,而回收率随浓度的增大而逐渐升高。当溶液浓度较低时,泡沫稳定性较差,溶液黏度较小,泡沫持液率较低,因此呈现富集比高而回收率低的现象。随着溶液浓度的不断增大泡沫稳定性逐渐提高,同时溶液的黏度不断增大,泡沫夹带率变高,液体不易回流,因此回收率逐渐升高。
[0067]
图2为温度对泡沫分离薯蓣皂苷的影响图,如图2所示,富集比随温度的升高先增大后减小,回收率不断减小。当温度较低时,溶液黏度及泡沫稳定性高,泡沫具有较高持液率,因此回收率较高。当温度较高时,泡沫持液率降低,泡沫层夹带的薯蓣皂苷含量降低,因此回收率降低。
[0068]
图3为气速对泡沫分离薯蓣皂苷的影响图;如图3所示,随着气速的增加,富集比不断降低,回收率先升高后降低。当气速较低时,泡沫在分离柱中停留的时间较长,气泡并聚、排液较充分,可携带出的薯蓣皂苷较少,因此富集比高,回收率低。当气速增大时,泡沫的生成速度及上升速度加快,持液率较高,因此,回收率升高。
[0069]
图4为装液量对泡沫分离薯蓣皂苷的影响图,如图4所示,当装液量较少时,泡沫在分离柱中停留的时间较长,泡沫中携带的薯蓣皂苷较大程度上回流至原液中,因此,富集比升高。而当装液量增加时,泡沫的持液率较高,泡沫中的夹带液在回流前被鼓出来,因此,回收率升高。
[0070]
3、box
‑
behnken响应面优化试验
[0071]
1)根据图1~4的结果,以上样浓度(a)、温度(b)、气速(c)和装液量(d)为因素,薯蓣皂苷提取得率为指标,采用响应面box
‑
behnken中心组合试验设计优化方案,四因素三水平的选择见表1。
[0072]
表1 因素水平设计表
[0073][0074]
选取浓度(a)、温度(b)、气速(c)、装液量(d)进行四因素三水平的box
‑
behnken响应面实验设计,响应面实验设计及结果见表2。
[0075]
表2 响应面实验设计及结果
[0076]
[0077][0078]
2)回归模型的建立与检验
[0079]
对表2进行多元线性回归拟合后,得到回归方程并对此方程进行方差分析,方差分析结果见表3和表4。
[0080]
富集比(e)=+1.85
‑
0.83a+0.15b
‑
0.52c
‑
0.21d
‑
0.040ab+0.32ac+0.21ad
‑
0.017bc+0.070bd+0.080cd
‑
1.083e
‑
003a2‑
0.050b2+0.23c2+0.50d2;
[0081]
回收率(r)=+89.13+2.69a+1.96b+0.87c+1.19d
‑
0.54ab
‑
3.02ac
‑
4.24ad+2.62bc
‑
6.70bd
‑
5.33cd
‑
1.50a2‑
6.80b2‑
1.89c2‑
3.11d2。
[0082]
表3 富集比的线性回归表
[0083][0084]
注:“**”表示极显著(p<0.01);“*”表示显著(0.01<p<0.05);不显著(p>0.1)
[0085]
表4 回收率的线性回归表
[0086][0087]
由表3和表4可知,富集比和回收率模型中f值分别达42.34、11.27,均达到显著水平,而富集比的r2为0.9769,回收率的r2为0.9185,校正后富集比和回收率的校正决定系数r
2adj
分别为0.9539、0.8370,此结果表明该模型与实际实验拟合较好,对于富集比能解释94.92%的响应值,对于回收率能解释91.85%的响应值。富集比的失拟项f值为1.56,p值为0.3544,说明由于误差造成失拟项f值的可能性为35.44%,回收率的失拟项f值为1.33,p值为0.4218,说明由于误差造成失拟项f值的可能性为42.18%,e和r的模型都说明了失拟项对于纯误差来说并不显著,表明预测值与实际情况下测得的值间相关性相对较好,试验误差小,故可用于设计范围内的预测。由表3和表4还可看出,对于富集比模型:ad、c2均为显著,a、b、c、d、ac、d2为极显著,根据f值大小可知,因素主效应关系为:a>c>d>b;对于回收率模型:a、ad、bd、cd、b2、d2为极显著,b、ac、bc为显著,根据f值大小可知,因素主效应关系为:a>b>d>c。
[0088]
3)响应曲面分析及最佳工艺条件的确定
[0089]
利用design
‑
experter 8.0.6软件对葫芦巴中薯蓣皂苷的制备参数进行优化,分
别对两因素的交互作用进行研究,所得结果分别如图5~8所示,由图5~8计算得到薯蓣皂苷泡沫分离的最佳纯化工艺参数为:上样浓度0.03mg/ml,温度28.36℃,气速450ml/min,装液量400ml,在此条件下,薯蓣皂苷的回收率为88.31%,富集比为3.65。
[0090]
为验证预测上述富集比和回收率回归模型的可靠性,从实际情况出发,将最佳条件调整为:上样浓度0.03mg/ml,温度28℃,气速450ml/min,装液量400ml。在此条件下(即实施例1的条件),按照上述式(1)、(2)计算所得薯蓣皂苷的回收率为86.57%,富集比为3.82,接近其采用design
‑
experter 8.0.6软件计算的预测值(回收率为88.31%,富集比为3.65),说明由design
‑
expert 8.0.6设计软件得到的最佳工艺条件可行。
[0091]
4、色谱分析
[0092]
1)用甲醇将0.0062g薯蓣皂苷标准品配制成124μg/ml的标品溶液;取适量标准品溶液于紫外分光光度计进行全波段扫描,结果表明,208nm可作为最佳色谱分离波长。
[0093]
将实施例1制备的薯蓣皂苷溶于甲醇,得到供试品溶液(浓度为62μg/ml);
[0094]
将上述供试品溶液、标品溶液和阴性样品(纯甲醇)分别进样制备型高效液相色谱图,所得p
‑
hplc谱图见图9;其中a为标品、b为供试品,c为阴性样品;如图9所示,标品的出峰时间为15.62min,供试品的出峰时间为15.57min,阴性样品仅含有溶剂峰,说明本发明经过制备型高效液相色谱分离得到的纯品为薯蓣皂苷。
[0095]
2)用容器接收上述第4节中标品和供试品出峰时间段流出的样品组分,依次经冷冻干燥和甲醇复溶后,利用高效液相色谱仪分别对样品进行测定,同时以纯甲醇作为阴性样品,高效液相色谱的条件:色谱柱:hedera ods
‑
2柱(4.6
×
250mm,10μm),流动相:水(a)和乙腈(b);进行梯度洗脱:0~5min,5~15%b;5~15min,15~45%b;15~35min,45~100%b,检测波长208nm;进样量为20μl;流速为0.8ml/min;柱温28℃;所得结果见图10,其中a为标品、b为供试品,c为阴性样品;如图10所示,标品的出峰时间为23.26min,样品的出峰时间为23.03min,由于出峰时间相差不大,说明经制备型高效液相色谱纯化得到的样品为薯蓣皂苷。
[0096]
5、傅里叶红外光谱(fi
‑
ir)测定
[0097]
分别取适量实施例1制备的薯蓣皂苷样品及薯蓣皂苷标品与kbr混合,研磨成粉末后压成薄片,随后用nicolet is50型ft
‑
ir作全波段(4000
‑
400cm
‑1)扫描,所得结果见图11;从图11中可以看出,薯蓣皂苷的提取样品和标品图基本一致,标品和样品在3859
‑
3738cm
‑1处都有吸收,该吸收峰是
‑
oh伸缩振动吸收峰,3355
‑
3428cm
‑1处标品和样品都有较宽的吸收峰,在2930cm
‑1处有较强的吸收峰,该吸收峰是六元环上c
‑
h伸缩振动峰。通过图中两个峰比较得出:1380cm
‑1处为
‑
ch3吸收峰;1600
‑
1450cm
‑1处的吸收峰为c
‑
h的面外弯曲振动,1300
‑
1000cm
‑1是c
‑
o的强吸收振动峰。通过红外光谱图分析得出实施例1制备的薯蓣皂苷样品样品与标品的图谱基本一致,说明纯化方法好,提取物纯度高。
[0098]
6、高分辨质谱(hrms)测定
[0099]
用甲醇溶解实施例1制备的薯蓣皂苷样品后,用aglient 7250&jeol
‑
jms
‑
t100lpaccutof高分辨质谱仪对其进行测定,所得结果见图12;由图12可知,样品中出现了m/z 891.47128的分子离子峰,这与前人的实验结果近乎一致(姚志红等的研究结果表明薯蓣皂苷经一级全扫描后会得到m/z 891.9的准分子离子峰,姚志红,林舒颖,曹秀珍,等.甲基原薯蓣皂苷静脉注射给予大鼠后体内代谢物的lc
‑
ms~n分析[j].分析测试学报,2014,
33(09):1010
‑
1018.)。将薯蓣皂苷结构式画在chemdraw软件后,对其进行模拟分析,得到质荷比为868.48,而由样品的质谱图12中可知,与之最接近的质荷比为869.48952,根据主峰及旁边的峰计算该质荷比对应的分子电荷量为1,因此与目标化合物[m+h]
+
的分子量对应。
[0100]
7、核磁共振碳谱(13c
‑
nmr)、氢谱(1h
‑
nmr)测定
[0101]
将实施例1制备的薯蓣皂苷样品溶于氘带甲醇中,用brukeravance400核磁共振波谱仪对其进行测定,所得结果见图13~14;
[0102]
如图13所示,薯蓣皂苷核磁氢谱数据:除溶剂峰外,在5.41,5.23,4.59,4.53,4.51,4.44,4.43,4.41,4.39,4.17,4.15,4.14,4.13,3.95,3.94,3.94,3.91,3.85,3.83,3.80,3.69,3.67,3.65,3.64,3.61,3.59,3.57,3.54,3.52,3.48,3.46,3.45,3.44,3.43,3.42,3.40,3.39,3.37,3.34,3.33,3.33,2.48,2.46,2.34,2.32,2.29,2.05,2.01,1.94,1.93,1.88,1.81,1.79,1.77,1.72,1.70,1.69,1.67,1.66,1.64,1.61,1.58,1.56,1.52,1.49,1.48,1.45,1.42,1.35,1.33,1.32,1.29,1.27,1.26,1.22,1.19,1.18,1.14,1.13,1.10,1.08,1.07,0.99,0.98,0.83,0.82,0.81ppm处有信号,分子结构中大部分质子都能找到对应的信号,其中,糖苷上的羟基在cd3od中不显信号。
[0103]
如图14所示,薯蓣皂苷核磁碳谱数据:141.89(5),122.63(6),110.58(22),103.00(rh2
‑
1),102.31(rh1
‑
1),100.46(glc
‑
1),82.20(16),79.96(glc
‑
2),79.30(glc
‑
3),78.02(3/glc
‑
5),76.57(glc
‑
4),73.91(rh2
‑
4),73.72(rh1
‑
4),72.44(rh2
‑
2/3),72.36(rh1
‑
3),72.17(rh1
‑
2),70.66(rh2
‑
5),69.77(rh1
‑
5),67.85(26),63.73(glc
‑
6),61.94(17),57.80(14),51.70(9),42.90(20),41.42(13),40.93(12),39.50(4),38.56(1),38.04(10),33.18(23),32.80(7),32.74(15),32.42(8),31.44(25),30.75(2),29.88(24),21.98(11),19.84(19),17.99(27),17.87(rh2
‑
6),17.50(rh1
‑
6),16.78(21),14.90(18)。分子结构中基本上所有的碳均可以在图谱中找到对应信号。
[0104]
由图13~14可知,实施例1制备的薯蓣皂苷中的碳原子和氢原子均能在样品的碳谱图(图14)和氢谱图(图13)中找到相对应的信号,结合质谱图(图12)说明本发明的方法提取纯化后得到的样品为薯蓣皂苷。
[0105]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1