一类含水溶性取代基罗丹明荧光染料其制备方法和应用
1.本发明涉及一类含水溶性取代基的罗丹明荧光染料,具体涉及生物荧光分析中的荧光染料领域,特别是其在生物体系内的应用。
背景技术:
2.罗丹明类荧光染料具有较长的吸收和发射光谱、较高的亮度和较好的光稳定性,近年来被广泛应用于生物标记(蛋白质及核酸)、单分子示踪、离子检测等领域,逐渐成为生命科学研究不可替代的重要工具。但前人的工作往往更注重罗丹明类染料光物理性质的改造,而提高罗丹明类染料生物相容性的改造却很少被报道。近年来超分辨荧光显微镜、转盘共聚焦显微镜、全内反射显微镜、光片显微镜等先进成像技术的发展为荧光染料的发展和改造提出了新的要求:在调节荧光染料光物理性质的基础上更要重视提高染料的生物相容性以适应长时程活体荧光成像的需求。
3.锌离子(zn
2+
)在生物体内的生理及病理过程中扮演着重要角色。绝大多数胞内锌离子与蛋白质紧密结合,少部分则游离于细胞液中形成游离锌池。生物体中不同组织内锌离子浓度相差多达8个数量级,在细胞液中游离锌的浓度约为10
‑
10 m 而在胰岛囊泡中浓度则高达10
‑
2 m,这是由于胰岛素与锌离子以4:2的六聚体形式储存在胰岛囊泡中,生理状态的胰岛在受到葡萄糖刺激后通过分泌小泡的胞吐形式将胰岛素与锌离子共同释放到血液中。已经有很多荧光探针被用于锌离子的监测,但它们普遍亲和力过高(nm级别的k
d
)无法适用于监测生物体内高浓度锌离子的动态变化过程;此外具有远红发射的以硅罗丹明为母体的功能染料(锌、钙离子染料等)合成步骤较为繁琐且产率较低,极大地限制了远红及近红外功能染料的发展,无法与具有其他发射波长的探针(如绿色钙离子荧光探针gcamp6f)联用以指示组织内复杂的信号传递过程;目前已见文献报道的唯一一款低亲和力红色发射的锌离子探针rhodzin
‑
1也在监测胰岛素/锌离子共释放的过程中出现了明显的细胞内非特异性结合的现象并最终导致细胞凋亡。这充分说明锌离子荧光探针工具箱仍然需要进一步发展。
4.免疫荧光技术在蛋白质、细胞因子、细胞表面抗原、肿瘤标志物等多种生物活性物质的检测与鉴定中发挥着重要作用。免疫荧光根据抗原抗体反应的原理,将荧光染料与抗原或抗体共价偶联,与其相应的抗原或抗体结合后通过显微镜观察其荧光信号以确定抗原或抗体的定位及性质。一般认为水溶性更好的荧光染料具有更高的抗体标记效率。目前具有远红发射的抗体标记染料以磺酸cy5为主,但该染料标记效率较低、光稳定性较差、非特异性染色较为明显,难以适应长时间免疫荧光成像的要求。硅取代的罗丹明虽然也具有远红发射波长和较高的摩尔消光系数,但是水溶性和化学稳定性均较差,故未在免疫荧光标记中得到广泛应用。
5.综上所属,当前罗丹明类染料及其功能化衍生物存在的生物相容性差(光毒性高、非特异性标记、水溶性差)的问题严重限制了其在生物成像中的应用,但目前仍缺乏对该问题的普适性解决方案。因此,提出改善罗丹明生物相容性的普适方案并基于罗丹明母体构
建具有良好生物相容性的功能染料具有十分重要的意义。
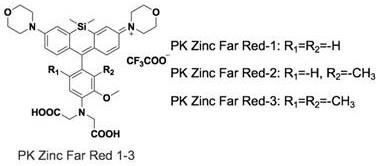
技术实现要素:
6.本发明的目的是针对当前罗丹明类染料及其功能化衍生物存在的生物相容性差(光毒性高、非特异性标记、水溶性差)的问题,提出了在罗丹明染料母核的n
‑
位连接不同水溶性取代基的普适解决方案并将其应用于具有红色和远红发射光谱的低亲和力不透膜锌离子探针和抗体标记染料的构建,并在活胰岛组织胰岛素和锌离子共释放的长时间监测和免疫荧光标记等领域得到了应用。
7.本发明首先提供了一类n
‑
水溶性基团取代的罗丹明功能染料,其通式为:
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(i)其中r1, r2, r3, r4, r5, r6, r7, r8彼此相互独立。
8.r1为 o, c(ch3)2, si(ch3)2, p(o)ch3, po2‑
, po2ch2ch3或so2。
9.r2为 o, c, p, s, nh, n
+
hch3, n
+
(ch3)2, p(o)ch3或so2。
10.r3为 ch2, chch3, c(ch3)2, chf, cf
2 , chcl, ccl2及它们之间可能的各种连接。
11.r4和r8为 h, ch3, ch2ch3, och3, och2ch3, coo
‑
, coome, cn, so2ch3, so2nh2或so2n(ch3)2。
12.r5和r7为 h, oh, och3, och2ch3, och2ch2och3, och2cooh, och2c6h5n, cooh 或以下结构。
13.其中,r9为 h, f, cl, br, ch3, och
3 或 no2。
14.r6为 h, cooh, n(ch2cooh)2或n(ch2cooch2ococh3)。
15.x
‑
为 卤素离子, clo4‑
, pf6‑
, bf4‑
, ch3coo
‑ 和cf3coo
‑
。
16.优选地,本发明在通式i的基础上提供了一系列具有红色发射的不透膜荧光染料pk zinc red 1
‑
5,其结构如通式ii所示;它们具有较低的亲和力(k
d
从190纳摩尔至74微摩尔)和良好的生物相容性。本发明还提供了该系列染料的合成方法并描述了其在体外样品中锌离子浓度的探测与定量和长时间监测活胰岛组织上胰岛素和锌离子共释放等应用。
17.(ii)本发明提供了上述染料pk zinc red 1
‑
5的制备方法,具体包括如下步骤。
18.优选地,本发明在通式i的基础上提供了一系列具有远红发射的不透膜荧光染料pk zinc red 1
‑
3,其结构如通式iii所示;它们同样具有较低的亲和力(k
d 30微摩尔)和良好的生物相容性,不仅适合于活胰岛组织胰岛素和锌离子共释放的长时间监测,还可以与蓝色细胞核染料hochest,绿色钙离子探针gcamp6f和红色线粒体染料pk mito red联用进行多色成像,解释它们之间复杂的信号传递关系。
19.(iii)本发明提供了上述染料pk zinc red 1
‑
3的制备方法,与前人所报道的合成路线相比(j. am. chem. soc. 133, 36: 14157
‑
14159),本文所报道的制备方法反应条件更加温和且产率更高,更适合与染料的克级制备,具体包括如下步骤:
检测锌离子方法的选择,是随着荧光探针的性质和体系、细胞和组织的性质而变化的,优选的检测技术方案是在酶标仪、荧光分光光度计和转盘共聚焦显微镜等设备上实现的。
20.优选地,本发明还在通式i的基础上提供了一款远红发射的荧光染料pk sir morpho,其结构如iv所示,该染料抗体标记效率较高(平均每个抗体标记4个染料)且与alexa 647、cy5等商用染料相比在免疫荧光成像中表现出了更高的光稳定性。
21.(iv)本发明提供了上述染料pk sir morpho的制备方法,具体包括如下步骤:使用上述染料pk sir morpho进行免疫荧光成像时,优选的检测技术方案是在共聚焦显微镜和高内涵活细胞成像系统等设备上实现的。
22.附图说明
23.图1为本发明制备的化合物pk zinc red
‑
1 在各种锌离子浓度下的荧光及吸收光谱图。
24.图2为本发明制备的化合物pk zinc red
‑
1的锌离子浓度滴定试验数据图。
25.图3为本发明制备的化合物pk zinc red
‑
2 在各种锌离子浓度下的荧光及吸收光谱图。
26.图4为本发明制备的化合物pk zinc red
‑
2的锌离子浓度滴定试验数据图。
27.图5为本发明制备的化合物pk zinc red
‑
3 在各种锌离子浓度下的荧光及吸收光谱图。
28.图6为本发明制备的化合物pk zinc red
‑
3的锌离子浓度滴定试验数据图。
29.图7为本发明制备的化合物pk zinc red
‑
4 在各种锌离子浓度下的荧光及吸收光谱图。
30.图8为本发明制备的化合物pk zinc red
‑
4的锌离子浓度滴定试验数据图。
31.图9为本发明制备的化合物pk zinc red
‑
5 在各种锌离子浓度下的荧光及吸收光谱图。
32.图10为本发明制备的化合物pk zinc red
‑
5的锌离子浓度滴定试验数据图。
33.图11为本发明制备的化合物pk zinc farred
‑
1 在各种锌离子浓度下的荧光及吸收光谱图。
34.图12为本发明制备的化合物pk zinc farred
‑
1的锌离子浓度滴定试验数据图。
35.图13为本发明制备的化合物pk zinc farred
‑
2 在各种锌离子浓度下的荧光及吸收光谱图。
36.图14为本发明制备的化合物pk zinc farred
‑
2的锌离子浓度滴定试验数据图。
37.图15为本发明制备的化合物pk zinc farred
‑
3 在各种锌离子浓度下的荧光及吸收光谱图。
38.图16为本发明制备的化合物pk zinc farred
‑
3的锌离子浓度滴定试验数据图。
39.图17为本发明制备的化合物pk zinc red
‑
1与已报道的化合物rhodzin
‑
1在小鼠的完整离体活胰岛上进行胰岛素/锌离子共释放检测的实验结果对比图;其中,图a中的roi 1中的信号为细胞凋亡信号,roi 2中的信号为细胞内非特异性标记信号,图b中的roi 3 为细胞内背景:roi 4中的荧光点为胰岛素/锌离子共释放信号。
40.图18为本发明制备的化合物pk zinc red
‑
5在小鼠的胰岛细胞团上进行胰岛素/锌离子共释放检测的实验结果图;虚线显示细胞团的外轮廓,图中的荧光点为pk zinc red
‑
5检测到胰岛素/锌离子共释放信号;图19为本发明制备的化合物pk zinc red
‑
1在药物福斯克林刺激下在小鼠的完整离体活胰岛上进行胰岛素/锌离子共释放检测的实验结果图;其中使用荧光染料fm 4
‑
64标记了细胞膜,图中的荧光点为pk zinc red
‑
1检测到的胰岛素/锌离子共释放信号图20为本发明制备的化合物pk zinc far red
‑
3与蓝色细胞核染料hochest,绿色钙离子探针gcamp6f和红色线粒体染料pk mito red联用进行多色成像的实验结果图;
图21为本发明制备的化合物pk sir morpho与商用染料alexa 647和cy5进行免疫荧光成像时荧光信号变化的实验结果图。
41.具体实施方式:下面的实施例可以使本领域技术人员更全面地理解本发明,但不以任何方式限制本发明。
42.下面结合实例进一步描述本发明n
‑
水溶性基团取代罗丹明类荧光探针的合成方法。
43.实例1:化合物pk zinc red
‑
1的合成:(1) 化合物1b的合成:首先将化合物1a(1.00 g, 3.39 mmol, 1.0 e.q.)溶于10 ml干燥的dmf中,在
‑
20℃下搅拌30 min。然后,将pocl3(2.88 g, 18.8 mmol, 5.5 e.q.)滴加入上述溶液中,升温至60℃反应3 h。上述反应结束后,将所得溶液缓滴入20 ml冰
‑
水混合液中,随后用na2co3溶液中和反应液并用二氯甲烷萃取三次(每次40 ml),收集有机层,用na2so4固体干燥。真空/减压条件下浓缩所得溶液,采用硅胶快速柱层析法纯化(展开剂10% 乙酸乙酯/石油醚)得到黄色固体化合物1b(920 mg, 2.84 mmol, 产率84%)。
[0044]1h nmr (400 mhz, chloroform
‑
d) δ 9.78 (s, 1h), 7.36
ꢀ–ꢀ
7.32 (m, 2h), 6.74 (d, j = 8.6 hz, 1h), 4.23 (q, j = 7.2 hz, 4h), 4.18 (s, 4h), 3.83 (s, 3h), 1.29 (t, j = 7.1 hz, 6h)。
[0045]
13
c nmr (101 mhz, chloroform
‑
d) δ 190.79, 170.96, 150.67, 145.11, 130.10, 126.79, 116.46, 110.27, 61.18, 55.89, 54.43, 14.42。
[0046]
(esi) 计算值c
16
h
22
no
6+ [m+h]
+ 324.14, 实际值324.35。
[0047]
(2) 化合物1c的合成:室温下,在化合物1b(250 mg, 0.772 mmol, 1.0 e.q.)的三氟乙酸溶液中(6 ml)分别加入3
‑
吗啉苯酚(346 mg, 1.94 mmol, 2.5 e.q.)和对甲苯磺酸(13.3 mg, 0.0772 mmol, 0.1 e.q.),随后在80℃下搅拌12 h。反应结束后,将所得混合物溶于50 ml chcl3,随后用3 m naoac的水溶液洗涤三次。收集有机层,真空/减压条件下浓缩。随后将粗产物溶于1:1 dcm/meoh的混合液中(4 ml / 4 ml),分三次加入ddq(总量176 mg, 0.775 mmol, 1.0 e.q.),继续于室温下反应4 h。反应结束后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从50%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物1c的三氟乙
酸盐(80 mg, 0.12 mmol, 产率16%)。
[0048]1h nmr(400 mhz, methanol
‑
d4) δ 7.67 (d, j = 9.6 hz, 2h), 7.30 (dd, j = 9.6, 2.6 hz, 2h), 7.18 (d, j = 2.5 hz, 2h), 7.09 (d, j = 1.9 hz, 1h), 7.03 (dd, j = 8.2, 1.9 hz, 1h), 6.96 (d, j = 8.2 hz, 1h), 4.29
ꢀ–ꢀ
4.21 (m, 8h), 3.88
ꢀ–ꢀ
3.83 (m, 8h), 3.81 (s, 3h), 3.78
ꢀ–ꢀ
3.73 (m, 8h), 1.32 (t, j = 7.1 hz, 6h)。
[0049]
13
c nmr(101 mhz, methanol
‑
d4) δ 173.04, 160.37, 159.96, 158.73, 151.77, 142.88, 133.64, 130.85, 125.16, 124.85, 118.27, 115.81, 115.58, 98.55, 67.45, 62.10, 56.62, 55.42, 48.36, 14.61.hrms (esi) 计算值 c
36
h
42
n3o
8+ [m]
+ 644.2966, 实际值644.2986。
[0050]
(3) 化合物pk zinc red
‑
1的合成:在化合物1c(50 mg, 77 μmol, 1.0 e.q.)的meoh/h2o(4 ml / 1 ml)混合溶液中加入2 m lioh的水溶液(388 μl, 10.0 e.q.),所得混合液在室温下搅拌2 h。反应结束后,用2 m盐酸溶液酸化反应液,随后在真空/减压条件下浓缩。所得粗产物以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从35%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物pk zinc red
‑
1的三氟乙酸盐(15 mg, 26 μmol, 产率34%)。
[0051]1h nmr (400 mhz, methanol
‑
d4) δ 7.68 (d, j = 9.6 hz, 2h), 7.29 (dd, j = 9.6, 2.3 hz, 2h), 7.18 (d, j = 2.5 hz, 2h), 7.09 (d, j = 1.9 hz, 1h), 7.04
ꢀ–ꢀ
6.97 (m, 2h), 4.24 (s, 4h), 3.87
ꢀ–ꢀ
3.85 (m, 8h), 3.82 (s, 3h), 3.76
ꢀ–ꢀ
3.73 (m, 8h)。
[0052]
13
c nmr (101 mhz, methanol
‑
d4) δ 175.04, 160.41, 159.96, 158.72, 151.71, 142.95, 133.69, 130.85, 125.03, 124.92, 118.03, 115.79, 115.57, 98.55, 67.45, 56.54, 55.45, 48.36。
[0053]
hrms (esi) 计算值c
32
h
36
n3o
8+ [m]
+ 588.2340, 实际值 588.2362。
[0054]
实例2:化合物pk zinc red
‑
2的合成:(1) 化合物2b的合成:首先将化合物2a(730 mg, 2.36 mmol, 1.0 e.q.)溶于10 ml干燥的dmf中,在
‑
20℃下搅拌30 min。然后,将pocl3(3.00 g, 19.6 mmol, 8.3 e.q.)滴加入上述溶液中,将所得混合液升温至60℃ 反应3 h。上述反应结束后,将所得溶液缓滴入20 ml冰
‑
水混合液中,
随后将混合液用na2co3溶液中和并用二氯甲烷萃取三次(每次40 ml),收集有机层,用na2so4固体干燥。真空/减压条件下浓缩所得溶液,采用硅胶快速柱层析法纯化(展开剂10% 乙酸乙酯/石油醚)得到黄色固体化合物2b(680 mg, 2.02 mmol, 产率86%)。
[0055]1h nmr (400 mhz, chloroform
‑
d) δ 9.77 (s, 1h), 7.35
ꢀ–ꢀ
7.31 (m, 2h), 6.71 (d, j = 8.5 hz, 1h), 4.28
ꢀ–ꢀ
4.17 (m, 8h), 4.07 (q, j = 6.9 hz, 2h), 1.38 (t, j = 6.9 hz, 3h), 1.30 (t, j = 7.1 hz, 6h)。
[0056]
13
c nmr (101 mhz, chloroform
‑
d) δ 190.79, 171.02, 149.89, 145.20, 129.99, 126.57, 116.23, 110.76, 64.42, 61.16, 54.07, 14.50, 14.34。
[0057]
ms (esi) 计算值c
17
h
24
no
6+ [m+h]
+ 338.16, 实际值 338.21。
[0058]
(2) 化合物2c的合成:室温下,在化合物2b(50 mg, 0.148 mmol, 1.0 e.q.)的三氟乙酸溶液中(3 ml)分别加入3
‑
吗啉苯酚(66.0 mg, 0.371 mmol, 2.5 e.q.)和对甲苯磺酸(2.6 mg, 14.8 μmol, 0.1 e.q.),随后在80℃下搅拌12 h。反应结束后,将所得混合物溶于25 ml chcl3,随后用3 m naoac的水溶液洗涤。收集有机层,真空/减压条件下浓缩。随后将粗产物溶于1:1 dcm/meoh的混合液中(2 ml / 2 ml),分三次加入ddq(总量34.0 mg, 0.148 mmol, 1.0 e.q.),继续于室温下反应4 h。反应结束后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从50%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物2c的三氟乙酸盐(11 mg, 17 μmol, 产率11%)。
[0059]1h nmr(400 mhz, methanol
‑
d4) δ 7.66 (dd, j = 9.5, 2.3 hz, 2h), 7.29 (d, j = 9.4 hz, 2h), 7.17 (d, j = 2.1 hz, 2h), 7.05 (d, j = 1.6 hz, 1h), 7.02
ꢀ–ꢀ
6.92 (m, 2h), 4.30
ꢀ–ꢀ
4.20 (m, 8h), 4.04 (q, j = 6.9 hz, 2h), 3.89
ꢀ–ꢀ
3.81 (m, 8h), 3.77
ꢀ–ꢀ
3.71 (m, 8h), 1.40
ꢀ–ꢀ
1.29 (m, 9h).
13
c nmr(101 mhz, methanol
‑
d4) δ 173.04, 160.35, 159.91, 158.69, 150.97, 142.97, 133.63, 125.12, 124.72, 118.26, 116.25, 115.81, 115.53, 98.59, 67.45, 65.91, 62.13, 55.12, 48.39, 14.97, 14.55。
[0060]
hrms (esi) 计算值c
37
h
44
n3o
8+ [m]
+ 658.3123, 实际值658.3134。
[0061]
(3) 化合物pk zinc red
‑
2的合成:在化合物2c(10.0 mg, 15.2 μmol, 1.0 e.q.)的meoh/h2o(2 ml / 0.5 ml)混合
溶液中加入2 m lioh的水溶液(78 μl, 10.0 e.q.),所得混合液在室温下搅拌2 h。反应结束后,用2 m盐酸溶液酸化反应液,随后在真空/减压条件下浓缩。所得粗产物以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物pk zinc red
‑
2的三氟乙酸盐(2.1 mg, 3.3 μmol, 产率22%)。
[0062]1h nmr (400 mhz, methanol
‑
d4) δ 7.66 (d, j = 9.5 hz, 2h), 7.28 (dd, j = 9.6, 2.1 hz, 2h), 7.17 (d, j = 2.2 hz, 2h), 7.05 (d, j = 1.6 hz, 1h), 7.02
ꢀ–ꢀ
6.93 (m, 2h), 4.24 (s, 4h), 4.05 (q, j = 6.9 hz, 2h), 3.88
ꢀ–ꢀ
3.84 (m, 8h), 3.76
ꢀ–ꢀ
3.73 (m, 8h), 1.42 (t, j = 6.9 hz, 3h)。
[0063]
13
c nmr (101 mhz, methanol
‑
d4) δ 175.10, 160.36, 159.90, 158.67, 150.95, 143.07, 133.66, 124.95, 124.77, 117.95, 116.24, 115.78, 115.50, 98.58, 67.45, 66.07, 55.25, 48.38, 14.86。
[0064]
hrms (esi) 理论值c
33
h
36
n3o
8+ [m]
+ 602.2497, 实际值602.2488。
[0065]
实例3:化合物pk zinc red
‑
3的合成:(1) 化合物3b的合成:首先将化合物3a(700 mg, 2.06 mmol, 1.0 e.q.)溶于10 ml干燥的dmf中,在
‑
20℃下搅拌30 min。然后,将pocl3(2.52 g, 16.5 mmol, 8.0 e.q.)滴加入上述溶液中,将所得混合液升温至60℃反应3 h。上述反应结束后,将所得溶液缓滴入20 ml冰
‑
水混合液中,随后将混合液用na2co3溶液中和并用二氯甲烷萃取三次(每次40 ml),收集有机层,用na2so4固体干燥。真空/减压条件下浓缩所得溶液,采用硅胶快速柱层析法纯化(展开剂10% etoac/hexane)得到黄色油状化合物3b(500 mg, 1.36 mmol, 产率66%)。
[0066]1h nmr (400 mhz, chloroform
‑
d) δ 9.76 (s, 1h), 7.36
ꢀ–ꢀ
7.34 (m, 2h), 6.73 (d, j = 8.6 hz, 1h), 4.27
ꢀ–ꢀ
4.19 (m, 8h), 4.17
ꢀ–ꢀ
4.14 (m, 2h), 3.71
ꢀ–ꢀ
3.67 (m, 2h), 3.39 (s, 3h), 1.29 (t, j = 7.2 hz, 6h)。
[0067]
13
c nmr (101 mhz, chloroform
‑
d) δ 190.70, 170.86, 149.77, 145.23, 129.95, 126.79, 116.67, 111.64, 70.61, 68.07, 61.15, 58.94, 54.00, 14.34。
[0068]
ms (esi) 理论值c
18
h
26
no
7+ [m+h]
+ 368.17, 实际值368.58。
[0069]
(2) 化合物3c的合成:室温下,在化合物3b(50 mg, 0.136 mmol, 1.0 e.q.)的三氟乙酸溶液中(3 ml)分别加入3
‑
吗啉苯酚(61.0 mg, 0.340 mmol, 2.5 e.q.)和对甲苯磺酸(2.3 mg, 13.6 μ
mol, 0.1 e.q.),随后在80℃下搅拌12 h。反应结束后,将所得混合物溶于20 ml chcl3,随后用3 m naoac的水溶液洗涤。收集有机层,真空/减压条件下浓缩。随后将粗产物溶于1:1 dcm/meoh的混合液中(2 ml / 2 ml),分三次加入ddq(总量31.0 mg, 0.136 mmol, 1.0 e.q.),继续于室温下反应4 h。反应结束后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从50%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物3c的三氟乙酸盐(5.0 mg, 7.2 μmol, 产率5%)。
[0070]1h nmr (400 mhz, methanol
‑
d4) δ 7.65 (d, j = 9.4 hz, 2h), 7.28 (dd, j = 9.7, 2.2 hz, 2h), 7.15 (d, j = 2.2 hz, 2h), 7.12 (d, j = 1.7 hz, 1h), 7.06
ꢀ–ꢀ
6.93 (m, 2h), 4.31 (s, 4h), 4.25 (q, j = 7.1 hz, 4h), 4.16
ꢀ–ꢀ
4.12 (m, 2h), 3.87
ꢀ–ꢀ
3.84 (m, 8h), 3.76
ꢀ–ꢀ
3.72 (m, 8h), 3.70
ꢀ–ꢀ
3.68 (m, 2h), 3.39 (s, 3h), 1.32 (t, j = 7.1 hz, 6h)。
[0071]
13
c nmr (101 mhz, methanol
‑
d4) δ 172.85, 160.17, 159.86, 158.67, 151.01, 142.97, 133.64, 125.13, 125.11, 118.70, 117.08, 115.82, 115.49, 98.58, 72.00, 69.77, 67.45, 62.13, 59.19, 55.00, 48.39, 14.57。
[0072]
hrms (esi) 理论值c
38
h
46
n3o
9+ [m]
+ 688.3229, 实际值688.3262。
[0073]
(3) 化合物pk zinc red
‑
3的合成:在化合物3c(5.0 mg, 7.25 μmol, 1.0 e.q.)的meoh/h2o(1.2 ml / 0.3 ml)混合溶液中加入2 m lioh的水溶液(40 μl, 10.0 e.q.),所得混合液在室温下搅拌2 h。反应结束后,用2 m盐酸溶液酸化反应液,随后在真空/减压条件下浓缩。所得粗产物以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物pk zinc red
‑
3的三氟乙酸盐(2.0 mg, 3.1 μmol, 产率44%)。
[0074]1h nmr (400 mhz, methanol
‑
d4) δ 7.64 (d, j = 9.5 hz, 2h), 7.26 (dd, j = 9.5, 2.2 hz, 2h), 7.13 (d, j = 2.2 hz, 2h), 7.09 (d, j = 1.7 hz, 1h), 7.03
ꢀ–ꢀ
6.96 (m, 2h), 4.27 (s, 4h), 4.16
ꢀ–ꢀ
4.11 (m, 2h), 3.92
ꢀ–ꢀ
3.82 (m, 10h), 3.74
ꢀ–ꢀ
3.71 (m, 8h), 3.40 (s, 3h)。
[0075]
13
c nmr (101 mhz, methanol
‑
d4) δ 174.88, 160.19, 159.86, 158.65, 150.92, 143.04, 133.65, 125.13, 124.98, 118.28, 116.5, 115.76, 115.46, 98.56, 71.96, 69.83, 67.43, 59.23, 55.01, 48.36。
[0076]
hrms (esi) 理论值 c
34
h
38
n3o
9+ [m]
+ 632.2603, 计算值 632.2616。
[0077]
实例4:化合物pk zinc red
‑
4的合成:(1) 化合物4b的合成:
首先将化合物4a(800 mg, 2.18 mmol, 1.0 e.q.)溶于10 ml干燥的dmf中,在
‑
20℃下搅拌30 min。然后,将pocl3(2.66 g, 17.4 mmol, 8.0 e.q.)滴加入上述溶液中,将所得混合液升温至60℃反应3 h。上述反应结束后,将所得溶液缓滴入20 ml冰
‑
水混合液中,随后将混合液用na2co3溶液中和并用二氯甲烷萃取三次(每次40 ml),收集有机层,用na2so4固体干燥。真空/减压条件下浓缩所得溶液,采用硅胶快速柱层析法纯化(展开剂10% 乙酸乙酯/石油醚)得到黄色固体化合物4b(518 mg, 1.31 mmol, 产率60%)。
[0078]1h nmr (400 mhz, chloroform
‑
d) δ 9.76 (s, 1h), 7.40 (dd, j = 8.2, 1.8 hz, 1h), 7.26 (d, j = 1.6 hz, 1h), 6.81 (d, j = 8.2 hz, 1h), 4.65 (s, 2h), 4.31
ꢀ–ꢀ
4.17 (m, 10h), 1.33
ꢀ–ꢀ
1.23 (m, 9h)。
[0079]
13
c nmr (101 mhz, chloroform
‑
d) δ 190.33, 170.67, 168.06, 162.54, 148.66, 145.29, 129.79, 127.23, 117.17, 111.73, 65.62, 61.41, 61.04, 54.07, 36.46, 31.39, 14.21, 14.11。
[0080]
ms (esi) 理论值c
19
h
27
no
8+ [m+h]
+ 396.17, 计算值396.26。
[0081]
(2) 化合物4c的合成:室温下,在化合物4b(50 mg, 0.127 mmol, 1.0 e.q.)的三氟乙酸溶液中(3 ml)分别加入3
‑
吗啉苯酚(57.0 mg, 0.316 mmol, 2.5 e.q.)和对甲苯磺酸(2.2 mg, 12.7 μmol, 0.1 e.q.),随后在80℃下搅拌12 h。反应结束后,将所得混合物溶于20 ml chcl3,随后用3 m naoac的水溶液洗涤。收集有机层,真空/减压条件下浓缩。随后将粗产物溶于1:1 dcm/meoh的混合液中(2 ml / 2 ml),分三次加入ddq(总量29.0 mg, 0.127 mmol, 1.0 e.q.),继续于室温下反应4 h。反应结束后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从50%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物4c的三氟乙酸盐(5.2 mg, 7.3 μmol, 产率6%)。
[0082]1h nmr (400 mhz, methanol
‑
d4) δ 7.60 (d,j = 9.6 hz, 2h), 7.26 (dd,j = 9.6, 2.3 hz, 2h), 7.14 (d,j = 2.4 hz, 2h), 7.11
ꢀ–ꢀ
7.03 (m, 3h), 4.75 (s, 2h), 4.33 (s, 4h), 4.28
ꢀ–ꢀ
4.17 (m, 6h), 3.89
ꢀ–ꢀ
3.82 (m, 8h), 3.76
ꢀ–ꢀ
3.70 (m, 8h), 1.29 (t,j = 7.1 hz, 6h), 1.23 (t,j = 7.1 hz, 3h)。
[0083]
13
c nmr (101 mhz, methanol
‑
d4) δ 172.77, 170.19, 159.66, 159.49, 158.54, 149.85, 143.20, 133.58, 130.80, 126.12, 124.95, 119.28, 115.92, 115.30, 98.79, 67.52, 62.54, 62.16, 55.13, 53.31, 48.57, 14.63, 14.43。
hz, 2h), 7.27
ꢀ–ꢀ
7.21 (m, 3h), 7.18
ꢀ–ꢀ
7.12 (m, 4h), 5.48 (s, 2h), 4.31 (s, 4h), 4.13 (q,j = 7.1 hz, 4h), 3.89
ꢀ–ꢀ
3.83 (m, 8h), 3.77
ꢀ–ꢀ
3.71 (m, 8h), 1.25 (t,j = 7.1 hz, 6h)。
[0091]
13
c nmr (101 mhz, methanol
‑
d4) δ 172.74, 159.88, 159.43, 158.71, 154.59, 150.46, 145.92, 144.85, 143.66, 133.36, 130.84, 126.72, 126.34, 126.17, 120.18, 118.91, 115.88, 115.47, 98.59, 70.44, 67.43, 62.20, 55.19, 48.40, 14.50。
[0092]
hrms (esi) 理论值c
41
h
45
n4o
8+ [m]
+ 721.3232, 实际值721.3212。
[0093]
(2) 化合物pk zinc red
‑
5的合成:在化合物5c(24.0 mg, 33 μmol, 1.0 e.q.)的meoh/h2o(4 ml / 1 ml)混合溶液中加入2 m lioh的水溶液(168 μl, 10.0 e.q.),所得混合液在室温下搅拌2 h。反应结束后,用2 m盐酸溶液酸化反应液,随后在真空/减压条件下浓缩。所得粗产物以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长545 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到紫黑色固体化合物pk zinc red
‑
5的三氟乙酸盐(10.4 mg, 15.6 μmol, 产率48%)。
[0094]1h nmr (400 mhz, methanol
‑
d4) δ 8.77
ꢀ–ꢀ
8.70 (m, 1h), 8.33 (td,j = 7.8, 1.5 hz, 1h), 7.96 (d,j = 7.9 hz, 1h), 7.79 (dd,j = 7.6, 5.4 hz, 1h), 7.54 (d,j = 9.4 hz, 2h), 7.27
ꢀ–ꢀ
7.20 (m, 3h), 7.19
ꢀ–ꢀ
7.10 (m, 4h), 5.45 (s, 2h), 4.28 (s, 4h), 3.88
ꢀ–ꢀ
3.84 (m, 8h), 3.78
ꢀ–ꢀ
3.73 (m, 8h)。
[0095]
13
c nmr (101 mhz, methanol
‑
d4) δ 174.64, 159.92, 159.58, 158.71, 154.65, 150.27, 146.24, 144.36, 143.73, 133.44, 130.85, 126.55, 126.30, 126.08, 119.64, 118.66, 115.84, 115.48, 98.59, 70.63, 67.44, 55.17, 48.40。
[0096]
hrms (esi) 理论值 c
37
h
37
n4o
8+ [m]
+ 665.2606, 计算值665.2606。
[0097]
实例6:化合物pk zinc farred
‑
1的合成: (1) 化合物二(2
‑
溴
‑4‑
吗啉苯基)甲烷的合成:在4
‑
(3
‑
溴苯基)吗啉(3.08 g, 12.7 mmol, 2.0 e.q.)的acoh溶液(30 ml)中加入37%的福尔马林(0.475 ml, 6.34 mmol, 1.0 e.q.),随后将反应液升温至90℃搅拌3 h。反应结束待反应液冷却至室温后,用饱和的nahco3水溶液中和反应液并用ch2cl2萃取有机层。有机层用盐水洗涤后,以na2so4干燥,蒸干溶剂。以柱层析法(5% etoac/hexane)纯化所得产物,最终得到白色固体二(2
‑
溴
‑4‑
吗啉苯基)甲烷(1.36 g, 2.74 mmol, 产率43% )。
[0098]1h nmr (400 mhz, chloroform
‑
d) δ 7.12 (d,j = 2.6 hz, 2h), 6.88 (d,j = 8.6 hz, 2h), 6.76 (dd,j = 8.5, 2.6 hz, 2h), 4.03 (s, 2h), 3.86
ꢀ–ꢀ
3.80 (m, 8h), 3.16
ꢀ–ꢀ
3.08 (m, 8h)。
[0099]
13
c nmr (101 mhz, chloroform
‑
d) δ 150.84, 130.97, 130.42, 125.67, 119.59, 114.90, 66.88, 49.15, 40.31。
[0100]
hrms (esi) 理论值c
21
h
25
br2n2o
2+ [m+h]
+ 497.0264, 实际值497.0234。
[0101]
(2)化合物吗啉基硅杂蒽酮的合成:在经热烘干并鼓满氩气的烧瓶中加入二(2
‑
溴
‑4‑
吗啉苯基)甲烷(600 mg, 1.22 mmol, 1.0 e.q.)和无水thf(9 ml)。将该溶液冷却至
‑
78℃后用注射器快速滴射入n
‑
buli(1.6 m的正己烷溶液1.70 ml,2.72 mmol, 2.2 e.q.),反应在
‑
78℃下搅拌10 min。相同温度下,用注射器缓滴入sime2cl
2 (0.165 ml, 9.35 mmol, 1.2 e.q.),随后将反应液升至室温并搅拌30 min。反应结束后,加入2 m盐酸淬灭反应,用饱和的nahco3水溶液中和反应液后,用ch2cl2萃取有机层。有机层用盐水洗涤后,以na2so4干燥,蒸干溶剂。所得粗产物(硅
‑
派洛宁)无需后处理,直接用于下步反应。将产物溶于15 ml ch3coch3中,并将溶液冷却至0℃。将kmno4(578 mg, 3.66 mmol, 3.0 e.q.)研磨成细密粉末,并在搅拌下1 h内少量多次投入反应液中。室温下,继续反应1 h后,加入20 ml ch2cl2稀释反应液,用滤纸过滤后蒸干溶剂(过程中可以加入硅胶粉末以吸附细密mno2粉末,利于过滤)。所得产物以柱层析法(10% 乙酸乙酯/石油醚)纯化,得到淡黄色固体吗啉基硅杂蒽酮(114 mg, 0.278 mmol, 产率23%)。
[0102]1h nmr (400 mhz, chloroform
‑
d) δ 8.39 (d,j = 8.7 hz, 2h), 7.06
ꢀ–ꢀ
6.99 (m, 4h), 3.92
ꢀ–ꢀ
3.85 (m, 8h), 3.38
ꢀ–ꢀ
3.32 (m, 8h), 0.47 (s, 6h)。
[0103]
13
c nmr (101 mhz, chloroform
‑
d) δ 185.47, 152.62, 140.65, 132.42, 131.76, 117.21, 115.92, 66.82, 47.79,
ꢀ‑
0.99。
[0104]
hrms (esi) 理论值c
23
h
29
n2o3si
+ [m+h]
+ 409.1947, 实际值409.1916。
[0105] (3)化合物1e的合成:室温下,将化合物1d(1.00 g, 4.95 mmol, 1.0 e.q.)、三乙胺(1.45 ml, 10.4 mmol, 10.0 e.q.)和4
‑
(二甲氨基)吡啶(60.0 mg, 0.50 mmol, 0.1 e.q.)溶于20 ml ch2cl2中,随后将5 ml 1,2
‑
二(氯代二甲基硅基)乙烷(1.17 g, 5.45 mmol, 1.1 e.q.)的ch2cl2溶液缓滴入上述混合液中,搅拌3 h。反应结束后,加入30 ml正己烷以沉淀氯化三乙胺盐,随后将其过滤除净。所得鹅黄色油状物以柱层析法(氧化铝基底,5% 乙酸乙酯/石油
ch2cl2中,随后将5 ml 1,2
‑
二(氯代二甲基硅基)乙烷(1.17 g, 5.45 mmol, 1.1 e.q.)的ch2cl2溶液缓滴入上述混合液中,搅拌3 h。反应结束后,加入30 ml正己烷以沉淀氯化三乙胺盐,随后将其过滤除净。所得鹅黄色油状物以柱层析法(氧化铝基底,5% 乙酸乙酯/石油醚)纯化,最终得到无色油状化合物2e(1.27 g, 3.57 mmol, 产率72%)。
[0120]1h nmr (400 mhz, chloroform
‑
d) δ 6.95 (s, 1h), 6.75 (s, 1h), 3.70 (s, 3h), 2.27 (s, 3h), 0.84 (s, 4h), 0.06 (s, 12h)。
[0121]
13
c nmr (101 mhz, chloroform
‑
d) δ 154.24, 134.76, 130.10, 129.24, 117.05, 114.88, 55.11, 22.11, 8.87, 0.14。
[0122]
(2) 化合物2f的合成:在经热烘干并鼓满氩气的烧瓶中加入化合物2e(100 mg, 0.280 mmol, 4.8 e.q.)和无水thf(3 ml)。将该溶液冷却至
‑
20℃后用注射器快速滴射入n
‑
buli(1.6 m的正己烷溶液0.19 ml,0.31 mmol, 5.3 e.q.),反应在
‑
20℃下搅拌15 min。相同温度下,用注射器缓滴入1 ml硅杂蒽酮 (25.0 mg, 57.8 μmol, 1.0 e.q.)的无水thf溶液,并继续搅拌20 min。反应结束后,加入2 m盐酸淬灭反应并升至室温,用饱和的nahco3水溶液中和反应液后,用etoac萃取有机层。有机层用盐水洗涤后,以na2so4干燥,随后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物2f的三氟乙酸盐(19.0 mg, 36.0 μmol, 产率62%)。
[0123]1h nmr (400 mhz, methanol
‑
d4) δ 7.56 (d, j = 2.9 hz, 2h), 7.37 (s, 1h), 7.15 (d, j = 9.6 hz, 2h), 7.02 (s, 1h), 6.96 (dd, j = 9.7, 2.8 hz, 2h), 3.91 (s, 3h), 3.89
ꢀ–ꢀ
3.80 (m, 16h), 2.00 (s, 3h), 0.62 (s, 3h), 0.61 (s, 3h)。
[0124]
13
c nmr (101 mhz, methanol
‑
d4) δ 168.80, 155.38, 151.58, 150.19, 142.65, 140.19, 129.95, 129.14, 125.71, 123.14, 122.63, 115.79, 114.15, 67.58, 57.05, 48.46, 18.52,
ꢀ‑
1.34,
ꢀ‑
1.49。
[0125]
hrms (esi) 理论值 c
31
h
38
n3o3si
+ [m]
+ 528.2677, 实际值 528.2645。
[0126]
(3) 化合物2g的合成:室温下,在1 ml化合物2f(15.0 mg, 28 μmol, 1.0 e.q.)的dmf溶液中分别加入
k2co3(38.0 mg, 0.28 mmol, 10.0 e.q.)、ki(2.4 mg, 14 μmol, 0.5 e.q.)和溴乙酸乙酯(46 mg, 0.28 mmol, 10.0 e.q.),反应在95℃下搅拌12 h。反应结束后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从50%升至95%;流速5.0 ml/min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物2g的三氟乙酸盐(13.3 mg, 18.9 μmol, 产率68%)。
[0127]1h nmr (400 mhz, methanol
‑
d4) δ 7.52 (d,j = 2.8 hz, 2h), 7.26 (d,j = 9.7 hz, 2h), 6.95 (dd,j = 9.7, 2.9 hz, 2h), 6.71 (s, 1h), 6.69 (s, 1h), 4.24 (q,j = 7.1 hz, 4h), 4.19 (s, 4h), 3.88
ꢀ–ꢀ
3.79 (m, 16h), 3.70 (s, 3h), 1.89 (s, 3h), 1.31 (t,j = 7.2 hz, 6h), 0.60 (s, 3h), 0.59 (s, 3h)。
[0128]
13
c nmr (101 mhz, methanol
‑
d4) δ 173.25, 171.76, 155.38, 150.12, 149.88, 143.37, 140.82, 132.06, 129.96, 129.31, 122.28, 120.31, 115.58, 114.78, 67.59, 61.96, 56.59, 55.29, 48.36, 19.01, 14.64,
ꢀ‑
1.30,
ꢀ‑
1.52。
[0129]
hrms (esi) 理论值c
39
h
50
n3o7si
+ [m]
+ 700.3413, 计算值 700.3396。
[0130]
(4) 化合物pk zinc farred
‑
2的合成:将化合物2g(13.3 mg, 18.9 μmol)溶于1 ml 12 m的盐酸溶液中,回流反应30 min。反应结束后在真空/减压条件下浓缩反应液,所得粗产物以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物pk zinc farred
‑
2的三氟乙酸盐(8.3 mg, 13 μmol, 产率68%)。
[0131]1h nmr (400 mhz, methanol
‑
d4) δ 7.51 (d, j = 2.9 hz, 2h), 7.27 (d, j = 9.6 hz, 2h), 6.95 (dd, j = 9.7, 2.9 hz, 2h), 6.75 (s, 1h), 6.69 (s, 1h), 4.17 (s, 4h), 3.87
ꢀ–ꢀ
3.79 (m, 16h), 3.72 (s, 3h), 1.90 (s, 3h), 0.60 (s, 3h), 0.58 (s, 3h)。
[0132]
13
c nmr (101 mhz, methanol
‑
d4) δ 175.41, 171.84, 155.39, 150.12, 149.87, 143.42, 140.80, 132.06, 129.98, 129.34, 122.27, 120.11, 115.59, 114.81, 67.59, 56.49, 55.41, 48.36, 19.03,
ꢀ‑
1.30,
ꢀ‑
1.53。
[0133]
hrms (esi) 理论值 c
35
h
42
n3o7si
+ [m]
+ 644.2787, 实际值 644.2761。
[0134]
实例8:化合物pk zinc farred
‑
3的合成: (1) 化合物3d的合成:
在10 ml 2
‑
甲氧基
‑
3,5
‑
二甲基苯胺(1.80 g, 11.9 mmol, 1.0 e.q.)的ch2cl2溶液中经30 min缓滴入经10 ml ch2cl2稀释的溴化四丁铵(5.74 g, 11.9 mmol, 1.0 e.q.),所得混合液在室温下继续搅拌反应15 min。反应结束后,用饱和的nahco3水溶液中和反应液,再用ch2cl2萃取有机层。所收集有机层用盐水洗涤后,以na2so4干燥,随后蒸干溶剂。用柱层析法(洗脱溶5% chcl3/et2o)纯化粗产物后得到纯净的无色油状化合物3d(1.75 g, 7.81 mmol, 产率66%)。
[0135]1h nmr (400 mhz, dmso
‑
d6) δ 6.55 (s, 1h), 4.92 (s, 2h), 3.57 (s, 3h), 2.21 (s, 3h), 2.17 (s, 3h)。
[0136]
13
c nmr (101 mhz, dmso
‑
d6) δ 143.10, 140.23, 132.60, 129.97, 114.49, 111.97, 58.85, 23.12, 16.39。
[0137]
(2) 化合物3e的合成:室温下,将化合物3d(1.13 g, 4.95 mmol, 1.0 e.q.)、三乙胺(1.45 ml, 10.4 mmol, 10.0 e.q.)和4
‑
(二甲氨基)吡啶(60.0 mg, 0.50 mmol, 0.1 e.q.)溶于20 ml ch2cl2中,随后将5 ml 1,2
‑
二(氯代二甲基硅基)乙烷(1.17 g, 5.45 mmol, 1.1 e.q.)的ch2cl2溶液缓滴入上述混合液中,搅拌3 h。反应结束后,加入30 ml正己烷以沉淀氯化三乙胺盐,随后将其过滤除净。所得鹅黄色油状物以柱层析法(氧化铝基底,5% 乙酸乙酯/石油醚)纯化,最终得到无色油状化合物3e(1.03 g, 2.77 mmol, 产率56%)。
[0138]1h nmr (400 mhz, chloroform
‑
d) δ 6.74 (s, 1h), 3.56 (s, 3h), 2.35 (s, 3h), 2.30 (s, 3h), 0.83 (s, 4h), 0.15 (s, 12h)。
[0139]
13
c nmr (101 mhz, chloroform
‑
d) δ 152.60, 139.36, 133.34, 132.01, 126.52, 120.55, 59.84, 23.78, 17.40, 9.19, 0.50。
[0140]
(3) 化合物3f的合成:在经热烘干并鼓满氩气的烧瓶中加入化合物3e(100 mg, 0.270 mmol, 4.7 e.q.)和无水thf(3 ml)。将该溶液冷却至
‑
20℃后用注射器快速滴射入n
‑
buli(1.6 m的正己烷溶液0.18 ml,0.30 mmol, 5.2 e.q.),反应在
‑
20℃下搅拌15 min。相同温度下,用注射器缓滴入1 ml硅杂蒽酮 (25.0 mg, 57.8 μmol, 1.0 e.q.)的无水thf溶液,并继续搅拌20 min。反应结束后,加入2 m盐酸淬灭反应并升至室温,用饱和的nahco3水溶液中和反应液后,用etoac萃取有机层。有机层用盐水洗涤后,以na2so4干燥,随后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/
min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物3f的三氟乙酸盐(20.4 mg, 37.6 μmol, 产率66%)。
[0141]1h nmr (400 mhz, methanol
‑
d4) δ 7.53 (d, j = 2.8 hz, 2h), 7.20 (d, j = 9.7 hz, 2h), 6.96 (dd, j = 9.7, 2.9 hz, 2h), 6.93 (s, 1h), 3.86
ꢀ–ꢀ
3.82 (m, 16h), 3.80 (s, 3h), 1.91 (s, 3h), 1.89 (s, 3h), 0.61 (s, 3h), 0.60 (s, 3h)。
[0142]
13
c nmr (101 mhz, methanol
‑
d4) δ 170.52, 155.48, 150.06, 147.55, 141.97, 136.04, 133.93, 133.11, 130.72, 129.45, 122.41, 119.23, 116.00, 67.58, 60.81, 48.42, 19.48, 13.38,
ꢀ‑
1.52,
ꢀ‑
1.56。
[0143]
hrms (esi) 理论值 c
32
h
40
n3o3si
+ [m]
+ 542.2833, 实际值 542.2814。
[0144]
(4) 化合物3g的合成:室温下,在2 ml化合物3f(20.0 mg, 37 μmol, 1.0 e.q.)的dmf溶液中分别加入k2co3(50.0 mg, 0.37 mmol, 10.0 e.q.)、ki(3.2. mg, 19 μmol, 0.5 e.q.)和溴乙酸乙酯(61 mg, 0.37 mmol, 10.0 e.q.),反应在95℃下搅拌12 h。反应结束后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从50%升至95%;流速5.0 ml/min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物3g的三氟乙酸盐(15.6 mg, 21.8 μmol, 产率59%)。
[0145]1h nmr (400 mhz, methanol
‑
d4) δ 7.52 (d, j = 2.9 hz, 2h), 7.18 (d, j = 9.6 hz, 2h), 6.95 (dd, j = 9.7, 2.9 hz, 2h), 6.80 (s, 1h), 4.27 (s, 4h), 4.21 (q, j = 7.1 hz, 4h), 3.86
ꢀ–ꢀ
3.80 (m, 16h), 3.76 (s, 3h), 1.88 (s, 3h), 1.88 (s, 3h), 1.28 (t, j = 7.1 hz, 6h), 0.60 (s, 3h), 0.59 (s, 3h)。
[0146]
13
c nmr (101 mhz, methanol
‑
d4) δ 172.64, 171.16, 155.51, 150.05, 149.34, 143.70, 142.13, 133.46, 131.99, 131.21, 129.51, 122.35, 119.88, 115.96, 67.60, 61.88, 60.15, 54.55, 48.40, 19.69, 14.60, 13.46,
ꢀ‑
1.51,
ꢀ‑
1.57。
[0147]
hrms (esi) 理论值c
40
h
52
n3o7si
+ [m]
+ 714.3569, 实际值 715.3566。
[0148] (5) 化合物pk zinc farred
‑
3的合成:将化合物3g(15.6 mg, 21.8 μmol)溶于2 ml 12 m的盐酸溶液中,回流反应30 min。反应结束后在真空/减压条件下浓缩反应液,所得粗产物以hplc纯化(洗脱剂,20 min
线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物pk zinc farred
‑
3的三氟乙酸盐(10.1 mg, 15.3 μmol, 产率68%)。
[0149]1h nmr (400 mhz, methanol
‑
d4) δ 7.51 (d,j = 2.9 hz, 2h), 7.21 (d,j = 9.6 hz, 2h), 6.96 (dd,j = 9.7, 2.8 hz, 2h), 6.80 (s, 1h), 4.18 (s, 4h), 3.87
ꢀ–ꢀ
3.78 (m, 16h), 3.67 (s, 3h), 1.86 (s, 6h), 0.60 (s, 3h), 0.59 (s, 3h)。
[0150]
13
c nmr (101 mhz, methanol
‑
d4) δ 178.65, 171.59, 155.51, 150.00, 148.63, 143.05, 142.31, 132.53, 132.18, 131.33, 129.68, 122.25, 118.91, 116.01, 67.59, 60.74, 58.95, 48.39, 19.74, 13.62,
ꢀ‑
1.52,
ꢀ‑
1.54。
[0151]
hrms (esi) 理论值c
36
h
44
n3o7si
+ [m]
+ 658.2943, 实际值 658.2947。
[0152]
实例9 化合物pk sir morpho的合成:在经热烘干并鼓满氩气的烧瓶中加入化合物1h(76 mg, 0.270 mmol, 4.7 e.q.)和无水thf(3 ml)。将该溶液冷却至
‑
20℃后用注射器快速滴射入n
‑
buli(1.6 m的正己烷溶液0.18 ml,0.30 mmol, 5.2 e.q.),反应在
‑
100℃下搅拌15 min。相同温度下,用注射器缓滴入1 ml硅杂蒽酮 (25.0 mg, 57.8 μmol, 1.0 e.q.)的无水thf溶液,并继续搅拌20 min。反应结束后,加入2 m盐酸淬灭反应并升至室温,用饱和的nahco3水溶液中和反应液后,用etoac萃取有机层。有机层用盐水洗涤后,以na2so4干燥,随后在真空/减压条件下浓缩反应液,以hplc纯化(洗脱剂,20 min线性梯度,溶剂b组分从30%升至95%;流速5.0 ml/min;检测波长650 nm;洗脱剂a:含0.1% (v/v) tfa的ddh2o;洗脱剂b:ch3cn)后得到深蓝色固体化合物pk sir morpho的三氟乙酸盐(23.0 mg, 39.9 μmol, 产率70%)。
[0153]1h nmr (400 mhz, methanol
‑
d4) δ 7.88 (s, 2h), 7.57 (d, j = 2.8 hz, 2h), 7.13 (d, j = 9.6 hz, 2h), 6.97 (dd, j = 9.6, 2.7 hz, 2h), 3.89
ꢀ–ꢀ
3.83 (m, 16h), 2.06 (s, 6h)。
[0154]
实例10 锌离子探针pk zinc red 1
‑
5 和pk zinc farred 1
‑
3 锌离子滴定和解离常数(k
d
)测试方法。
[0155]
(1)测试溶液的制备:以“零锌离子缓冲液”和“高浓度锌离子缓冲液”相互稀释的方法制备包含 1 μm pk zinc 染料的各种测试溶液。首先需要制备的零锌离子溶液包含 100 mm ph 7.4 hepes,100 mm nano
3 ;“高浓度锌离子缓冲液”包含100 mm ph 7.4 hepes,100 mm nano3和 10 mm znso4。对于游离锌离子浓度大于 100 nm的缓冲体系来说,直接使用两种缓冲液进行相互稀释。对于游离锌离子浓度小于 100 nm的缓冲体系来说,在溶液中再加入10 mm nta以螯合高浓度锌离子,并通过文献(org. lett., 2011, 13(17): 4558
‑
4561)所述公式计算出缓冲体系内实际的游离锌离子浓度。
[0156]
(2)光谱测试:荧光光谱是在shimadzu rf
‑
5301pc上在室温下进行测试的,吸收光
谱是在uv
‑
1780上在室温下进行测试的。本发明所述所有锌离子染料的荧光光谱和吸收光谱使用分别制备的溶液在每种锌离子浓度下分别重复进行了3次实验。
[0157]
不同染料在不同锌离子浓度下的荧光光谱测试结果如图1,3,5,7,9,11,13,15所示(右上角内嵌图为吸收光谱),浓度滴定测试结果如图2,4,6,8,10,12,14,16所示。k
d
是根据方程来计算的。其中f是给定锌离子浓度下的荧光强度,f
min
是在=0时的荧光强度;f
max
是在染料被锌离子饱和时的荧光强度。本发明所述化合物pk zinc red 1
‑
5 和pk zinc farred 1
‑
3的重要光物理性质在表1下方列出:表1实例11 使用锌离子探针在胰岛beta细胞或离体胰岛组织上观测胰岛素/锌离子共释放的测试方法。
[0158]
胰岛在35 mm玻璃底共聚焦培养皿(cellvis,d35
‑
14
‑1‑
n)上培养24小时,然后洗涤两次并在含有125 mm nacl、5.9 mm kcl、2.4 mm cacl2、1.2 mm mgcl2、1 mm l
‑
谷氨酰胺、25 mm hepes、3 mm葡萄糖、0.1%牛血清白蛋白、10%葡萄糖和1 μm 锌离子染料的krbb溶液中预热约15分钟。接下来,用含有指定葡萄糖浓度或/和药物的krbb溶液刺激胰岛并进行成像。荧光图像17,18,19,20都是通过安置csu
‑
x1转盘的奥林巴斯倒置ix
‑
81 激光共聚焦显微镜获得的。图像由60x(na1.35,olympus)或100x(na1.30,olympus)油镜采集,单色采样率~2hz,双通道采样率~1hz,四色成像采样率~0.3hz。
[0159]
实例12 使用pk sir morpho和商用染料alexa 647和cy5进行免疫荧光成像的测试方法。
[0160]
将染料与二抗以浓度比为20:1 的比例在pbs 7.4缓冲液中进行共孵育,使用葡聚糖凝胶g
‑
25柱或脱盐柱分离被染料标记的二抗和游离的染料并使用nanodrop 2000通过a
260
/a
650
确定二抗的染料平均标记个数。而后在固定hela细胞上进行免疫荧光染色,并在高内涵活细胞成像系统上进行成像。采用短时间拍摄+1 s 650 nm激光照射的方式进行荧光信号的记录,每一个循环算一次,具体拍摄时间及激光光强为:pk sir morpho 100 ms,50%;alexa 647 50 ms,50%;cy5 20 ms,50%。记录的荧光信号随时间变化的曲线如图21所示。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1