一种4-氮杂芴类化合物及其制备方法与应用
一种4
‑
氮杂芴类化合物及其制备方法与应用
技术领域
1.本发明属于有机合成化学技术领域,具体涉及一种4
‑
氮杂芴化合物及其制备方法与应用。
背景技术:
2.公开该背景技术部分的信息仅仅旨在增加对本发明的总体背景的理解,而不必然被视为承认或以任何形式暗示该信息构成已经成为本领域一般技术人员所公知的现有技术。
[0003]4‑
氮杂芴类化合物,核心骨架为茚并吡啶结构,是某些生物碱的母核结构,存在于多种天然活性产物、药物以及生物活性分子中,具有抗癌、抗过敏、抗炎、杀菌等活性,其骨架存在于抗疟药、血管扩张药、麻醉药、抗惊厥药和抗癫痫药等很多药物中。
[0004]
此外,氮杂芴类化合物还可以作为一种良好的发光配体,对于电子、发光材料等领域也有重要意义。如hwu等含有吡啶基部分的双芴类似物和有效的电子传输材料作为主体,与各种含重金属的红色(ir、ru、os和pt)和绿色(ir)荧光粉混合获得具有简单器件结构的高效磷光oled。wong等提供了新型杂芳烃嵌入和双极三芴类似物,在引入杂芳烃单元作为成分后,母体三芴的物理性质发生了显着变化,杂芳烃的存在及其与相邻亚苯基环的共面性为这些新型三芴类似物提供了用于发光器件的特性。
[0005]
目前,尽管化学工作者已经发展了很多4
‑
氮杂芴类化合物以及合成方法,4
‑
氮杂芴类化合物的种类还是比较少,选择应用时仍旧存在局限性,而且大多数现有的合成方法是基于2
‑
芳基吡啶或芳基(吡啶
‑3‑
基)甲酮衍生物底物的环合反应,需要预先构建三环骨架中五元环,还需要金属试剂,并在隔绝水或者氧气的条件下进行,步骤繁琐。
[0006]
因此,开发一种新的4
‑
氮杂芴类化合物,并提供一种相对简单、高效的合成方法尤为重要。
技术实现要素:
[0007]
为了解决现有技术的不足,本发明提供一种4
‑
氮杂芴类化合物及其制备方法,该4
‑
氮杂芴类化合物是重要的有机分子母核结构,可以应用于复杂的天然产物全合成、生物活性分子合成或者功能材料分子合成中;本技术采用烯炔酮类化合物与异氰基乙酸酯类化合物发生串联环合反应进行制备4
‑
氮杂芴类化合物,反应条件温和,操作简单、高效,原料和试剂稳定易得,无需金属试剂,实用性强。
[0008]
为了解决以上技术问题,本发明提供如下的技术方案:
[0009]
本发明第一方面提供一种4
‑
氮杂芴类化合物,该类化合物的结构通式如式ⅰ所示:
[0010][0011]
式ⅰ中,r1选自芳基或烷基中的一种,r2选自芳基或烷基中的一种,r3选自烷氧基、氟烷基或氢原子中的一种,r4选自烷基。
[0012]
本发明第二方面提供一种式ⅰ所示化合物的制备方法,所述方法为:烯炔酮类化合物与异氰基乙酸酯类化合物发生串联环合反应,得到式ⅰ所示化合物。
[0013]
反应路线如下所示:
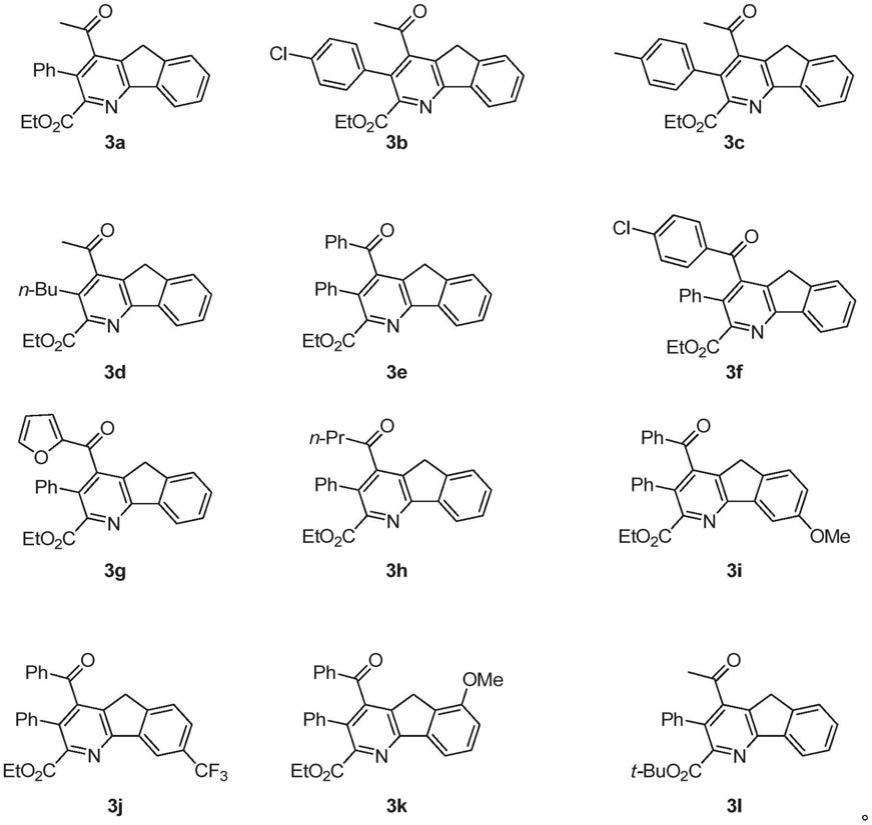
[0014][0015]
本发明第三方面提供一种药物组合物,包含如上所述式ⅰ所示化合物或其异构体或溶剂化物或可药用分子。
[0016]
本发明的第四方面提供一种式ⅰ所示4
‑
氮杂芴类化合物及其衍生物在制备宫颈癌细胞生长抑制剂中的应用。
[0017]
本发明的一个或多个实施方式至少具有以下有益效果:
[0018]
(1)本发明提供的多取代4
‑
氮杂芴类化合物,是重要的有机分子母核结构,可以应用于复杂的天然产物全合成、生物活性分子合成或者功能材料分子合成中,因而在有机合成和反应多样性领域上都占有非常重要的地位。此外,4
‑
氮杂芴类化合物也可作为重要的有机合成子中间体,用于重要的4
‑
氮杂芴酮衍生物的合成。
[0019]
(2)本发明提供的4
‑
氮杂芴类化合物的合成方法,无需预先构建三环骨架中五元环,同时无需金属试剂,无需隔绝水或者氧气,只需碱催化温和条件下,经历第一次分子内环和形成六元氮杂炔正离子中间体,并进一步发生分子内环合反应,然后发生质子迁移、氧化芳构化等得到最终产物。本制备方法条件温和,操作简单、高效,原料和试剂稳定易得,无需金属试剂,实用性强,适用于合成多种多取代4
‑
氮杂芴类化合物。经过实验数据表明,本发明提供的4
‑
氮杂芴类化合物衍生物对具有一定的hela抑制活性,可以用作宫颈癌细胞生长抑制剂。
附图说明
[0020]
构成本发明的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。
[0021]
图1为本发明实施例1得到的4
‑
氮杂芴类化合物的核磁氢谱图;
[0022]
图2为本发明实施例1得到的4
‑
氮杂芴类化合物的核磁碳谱图;
[0023]
图3为本发明实施例3得到的4
‑
氮杂芴类化合物的核磁氢谱图;
[0024]
图4为本发明实施例3得到的4
‑
氮杂芴类化合物的核磁碳谱图;
[0025]
图5为本发明实施例6得到的4
‑
氮杂芴类化合物的核磁氢谱图;
[0026]
图6为本发明实施例6得到的4
‑
氮杂芴类化合物的核磁碳谱图;
[0027]
图7为本发明实施例8得到的4
‑
氮杂芴类化合物的核磁氢谱图;
[0028]
图8为本发明实施例8得到的4
‑
氮杂芴类化合物的核磁碳谱图;
[0029]
图9为本发明实施例10得到的4
‑
氮杂芴类化合物的核磁氢谱图;
[0030]
图10为本发明实施例10得到的4
‑
氮杂芴类化合物的核磁碳谱图;
[0031]
图11为本发明实施例12得到的4
‑
氮杂芴类化合物的核磁氢谱图;
[0032]
图12为本发明实施例12得到的4
‑
氮杂芴类化合物的核磁碳谱图。
具体实施方式
[0033]
应该指出,以下详细说明都是示例性的,旨在对本发明提供进一步的说明。除非另有指明,本文使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
[0034]
需要注意的是,这里所使用的术语仅是为了描述具体实施方式,而非意图限制根据本发明的示例性实施方式。如在这里所使用的,除非上下文另外明确指出,否则单数形式也意图包括复数形式,此外,还应当理解的是,当在本说明书中使用术语“包含”和/或“包括”时,其指明存在特征、步骤、操作、器件、组件和/或它们的组合。
[0035]
正如背景技术所介绍的,现有技术中4
‑
氮杂芴类化合物的种类比较少,选择应用时仍旧存在局限性,而且大多数现有的合成方法是基于2
‑
芳基吡啶或芳基(吡啶
‑3‑
基)甲酮衍生物底物的环合反应,需要预先构建三环骨架中五元环,还需要金属试剂,并在隔绝水或者氧气的条件下进行,步骤繁琐。
[0036]
为了解决如上的技术问题,本发明第一方面提供一种4
‑
氮杂芴类化合物,该类化合物的结构通式如式ⅰ所示:
[0037][0038]
式ⅰ中,r1选自芳基或烷基中的一种,r2选自芳基或烷基中的一种,r3选自烷氧基、氟烷基或氢原子中的一种,r4选自烷基。
[0039]
本发明提供的多取代4
‑
氮杂芴类化合物,是重要的有机分子母核结构,可以应用于复杂的天然产物全合成、生物活性分子合成或者功能材料分子合成中,因而在有机合成和反应多样性领域上都占有非常重要的地位。此外,4
‑
氮杂芴类化合物也可作为重要的有机合成子中间体,用于重要的4
‑
氮杂芴酮衍生物的合成。
[0040]
在本发明的一些实施方式中,r1选自所述c1
‑
c5直链烷基、芳基、杂芳基、酯基或稠芳基;
[0041]
进一步,r1选自c1
‑
c5直链烷基、苯基、4
‑
氯苯基、4
‑
甲基苯基、2
‑
萘基、2
‑
噻吩基;
[0042]
更进一步,r1选自苯基、4
‑
氯苯基、4
‑
甲基苯基或正丁基中的一种。
[0043]
在本发明的一些实施方式中,r2选所述c1
‑
c5直链烷基、芳基、杂芳基、酯基或稠芳基;
[0044]
进一步,r2选自c1
‑
c5直链烷基、苯基、4
‑
氯苯基、4
‑
甲基苯基、2
‑
萘基、2
‑
呋喃基;
[0045]
更进一步,r2选自苯基、4
‑
氯苯基、2
‑
呋喃基、正丙基或甲基中的一种。
[0046]
在本发明的一些实施方式中,r3选自8
‑
甲氧基、8
‑
三氟甲基、6
‑
甲氧基或氢原子中的一种。
[0047]
在本发明的一些实施方式中,r4选自甲酯基、乙酯基或叔丁酯基中的一种;
[0048]
进一步,r4选自乙基或叔丁基中的一种。
[0049]
在本发明的一些实施方式中,式ⅰ所示化合物包括以下结构:
[0050][0051]
本发明第二方面提供一种式ⅰ所示化合物的制备方法,所述方法为:烯炔酮类化合物与异氰基乙酸酯类化合物发生串联环合反应,得到式ⅰ所示化合物。
[0052]
现有的4
‑
氮杂芴类化合物的合成方法一般是基于2
‑
芳基吡啶或芳基(吡啶
‑3‑
基)甲酮衍生物底物的环合反应,该方法需要预先构建三环骨架中五元环,还需要金属试剂,并在隔绝水或者氧气的条件下进行,反应步骤较为繁琐,反应条件也较为苛刻。
[0053]
而本发明提供的4
‑
氮杂芴类化合物的合成方法,是将烯酮类化合物与异氰基乙酸酯类化合物在有机溶剂中进行反应,和现有技术相比,本发明无需预先构建三环骨架中五元环,同时无需金属试剂,无需隔绝水或者氧气,只需碱催化温和条件下,经历第一次分子内环和形成六元氮杂炔正离子中间体,并进一步发生分子内环合反应,然后发生质子迁移、氧化芳构化等得到最终产物。
[0054]
当异腈的α
‑
位引入酯基、氰基、磷酰基、磺酰基、或酰胺基等吸电子基团时,其α
‑
酸性得到显著增强,即:活泼亚甲基异腈化合物。该类化合物不仅具有异腈的反应性,而且α
‑
位也能够发生各类亲核反应,其中异氰基乙酸乙酯就是典型活泼亚甲基异腈。由于本身α
‑
酸性特征以及异氰基团特有的属性,导致异氰基乙酸乙酯常用作1,3
‑
偶极子,并与极化多重键形成一系列的杂环化环加成反应,而利用异氰基乙酸酯的1,3
‑
双亲核性实现杂环化合物的构建的相关报道至今没有出现。另一方面,共轭烯炔酮类化合物是一类具有高活性和多反应位点的反应合成剂,它的基本结构骨架中含有共轭的羰基、碳碳双键(或者苯环)和碳碳三键,能够构建多种环状化合物,具备新的反应模式、催化特性、多样性、高选择性等优点。
[0055]
因此,异氰基酸酯类化合物与共轭烯炔酮发生串联环合反应为多取代4
‑
氮杂芴类化合物的合成提供了新的方法。本制备方法条件温和,操作简单、高效,原料和试剂稳定易得,无需金属试剂,实用性强,适用于合成多种多取代4
‑
氮杂芴类化合物。
[0056]
进一步,反应路线如下所示:
[0057][0058]
其中,r1、r2、r3、r4的定义如上所述。
[0059]
进一步,上述反应路线包括以下步骤:
[0060]
1)在有机溶剂中,异氰基酸酯与共轭烯炔酮发生串联环合反应;
[0061]
2)反应后,将产物除去有机溶剂,然后经过硅胶柱层析,得到4
‑
氮杂芴类化合物。
[0062]
在本发明的一些实施方式中,共轭烯炔酮选自(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮、(e)
‑3‑
(4
‑
氯亚苄基)
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮、(e)
‑3‑
(4
‑
甲基亚苄基)
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮、(e)
‑3‑
(苯乙炔基)辛
‑3‑
烯
‑2‑
酮、(e)
‑2‑
亚苄基
‑
1,4
‑
二苯基丁
‑3‑
炔
‑1‑
酮、(e)
‑2‑
亚苄基
‑1‑
(4
‑
氯苯基)
‑4‑
苯基丁
‑3‑
炔
‑1‑
酮、(e)
‑2‑
亚苄基
‑1‑
(呋喃
‑2‑
基)
‑4‑
苯基丁
‑3‑
炔
‑1‑
酮、(e)
‑3‑
亚苄基
‑1‑
苯基庚
‑1‑
炔
‑4‑
酮、(e)
‑2‑
亚苄基
‑4‑
(4
‑
甲氧基苯基)
‑1‑
苯基丁
‑3‑
炔
‑1‑
酮、(e)
‑2‑
亚苄基
‑4‑
(4
‑
三氟甲基苯基)
‑1‑
苯基丁
‑3‑
炔
‑1‑
酮、(e)
‑2‑
亚苄基
‑4‑
(2
‑
甲氧基苯基)
‑1‑
苯基丁
‑3‑
炔
‑1‑
酮。
[0063]
在本发明的一些实施方式中,异氰基乙酸酯选自异氰基乙酸乙酯、异氰基乙酸叔丁酯。
[0064]
在本发明的一些实施方式中,反应过程加入催化剂,在催化剂的作用下进行反应;
[0065]
进一步的,所述催化剂为叔丁醇钾;
[0066]
进一步,催化剂的摩尔百分含量为20
‑
50mol%;进一步优选为30mol%。
[0067]
在本发明的一些实施方式中,共轭烯炔酮与异氰基酸酯的摩尔比为1:1
‑
2;进一步优选为1:1.2。
[0068]
在本发明的一些实施方式中,有机溶剂选自1,4
‑
二氧六环、1,2
‑
二氯甲烷、四氢呋喃、乙酸乙酯、乙腈;优选为1,4
‑
二氧六环。
[0069]
在本发明的一些实施方式中,每进行0.2毫摩尔反应用1毫升溶剂。
[0070]
在本发明的一些实施方式中,反应温度为25
‑
80℃;进一步的,反应温度为65℃。
[0071]
在本发明的一些实施方式中,反应时间为1
‑
12h;进一步反应时间为8h。不同原料因活性差异而导致时间缩短或延长。
[0072]
本发明第三方面提供一种药物组合物,包含如上所述式ⅰ所示化合物或其异构体或溶剂化物或可药用分子。
[0073]
本发明的第四方面提供一种式ⅰ所示4
‑
氮杂芴类化合物及其衍生物在制备宫颈癌细胞生长抑制剂中的应用。
[0074]
为了使得本领域技术人员能够更加清楚地了解本发明的技术方案,以下将结合具体的实施例详细说明本发明的技术方案。
[0075]
实施例1
[0076]4‑
氮杂芴类化合物3a的制备
[0077][0078]
向15ml耐压管中加入(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a(0.2mmol),异氰基乙酸乙酯2a(0.24mmol),并用1ml1,4
‑
二氧六环溶解,加入催化剂叔丁醇钾(0.06mmol),并加入搅拌子,拧紧耐压管旋塞后放入预热至65℃的金属模块中进行搅拌,反应时间8h,此时tlc检测底物1a完全消失,停止反应并静置至室温,用饱和氯化铵溶液水洗,二氯甲烷溶液萃取三次合并有机相溶液,再用饱和氯化钠溶液水洗一次,减压蒸去溶剂,并通过硅胶柱层析进行分离以得到最终产物,经核磁氢谱、碳谱以及质谱检测证实其为多取代4
‑
氮杂芴类化合物3a,收率为73%。
[0079]
图1为本发明实施例1得到的4
‑
氮杂芴类化合物的核磁氢谱图,图2为其核磁碳谱图。
[0080]
谱图解析数据:
[0081]1h nmr(400mhz,cdcl3)δ0.97(t,j=6.8hz,3h),1.91(s,3h),3.95(s,2h),4.14(q,j=6.8hz,2h),7.33
‑
7.36(m,2h),7.42
‑
7.45(m,3h),7.45
‑
7.49(m,2h),7.57
‑
7.59(m,1h),8.19
‑
8.21(m,1h).
13
c nmr(100mhz,cdcl3)δ13.5,30.7,33.8,61.6,121.8,125.1,127.4,128.5,128.6,129.3,129.4,129.6,134.3,136.2,139.4,144.0,145.4,150.1,160.5,167.2,203.1.hrms(esi
‑
tof)m/z calculated for c
23
h
20
no
3+
([m+h
+
)358.1443,found 358.1440.
[0082]
实施例2
[0083]4‑
氮杂芴类化合物3b的制备
[0084][0085]
用(e)
‑3‑
(4
‑
氯亚苄基)
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1b代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3b,收率为68%。
[0086]1h nmr(400mhz,cdcl3)δ1.06(t,j=6.8hz,3h),1.98(s,3h),3.94(s,2h),4.18(q,j=6.8hz,2h),7.28(d,j=8.4hz,2h),7.42(d,j=8.4hz,2h),7.46
‑
7.49(m,2h),7.57
‑
7.59(m,1h),8.18
‑
8.21(m,1h).
13
c nmr(100mhz,cdcl3)δ13.6,30.9,33.8,61.7,121.9,125.1,127.5,128.1,128.9,129.8,130.7,134.3,134.7,134.9,139.2,144.0,145.5,149.9,160.9,166.8,202.7.hrms(esi
‑
tof)m/z calculated for c
23
h
19
clno
3+
([m+h]
+
)392.1053,found 392.1047.
[0087]
实施例3
[0088]4‑
氮杂芴类化合物3c的制备
[0089][0090]
用(e)
‑3‑
(4
‑
甲基亚苄基)
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1c代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3c,收率为68%。
[0091]
图3为本发明实施例3得到的4
‑
氮杂芴类化合物的核磁氢谱图,图4为其核磁碳谱图。
[0092]
谱图解析数据:
[0093]1h nmr(400mhz,cdcl3)δ1.03(t,j=7.2hz,3h),1.92(s,3h),2.41(s,3h),3.94(s,2h),4.17(q,j=7.2hz,2h),7.20
‑
7.25(m,4h),7.45
‑
7.48(m,2h),7.56
‑
7.58(m,1h),8.18
‑
8.20(m,1h).
13
c nmr(100mhz,cdcl3)δ13.6,21.2,30.7,33.8,61.5,121.8,125.1,127.4,129.2,129.3,129.4,129.5,133.1,134.3,138.5,139.5,144.0,145.4,150.2,160.3,167.3,203.2.hrms(esi
‑
tof)m/z calculated for c
24
h
22
no
3+
([m+h]
+
)372.1600,found 372.1602.
[0094]
实施例4
[0095]4‑
氮杂芴类化合物3d的制备
[0096][0097]
用(e)
‑3‑
(苯乙炔基)辛
‑3‑
烯
‑2‑
酮1d代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3d,收率为62%。
[0098]
谱图解析数据:
[0099]1h nmr(400mhz,cdcl3)δ0.91(t,j=7.6hz,3h),1.37(q,j=7.6hz,2h),1.45(t,j=7.2hz,3h),1.51
‑
1.57(m,2h),2.61(s,3h),2.79(t,j=8.4hz,2h),3.83(s,2h),4.51(q,j=7.2hz,2h),7.41
‑
7.46(m,2h).,7.54(dd,j1=6.4hz,j2=2.0hz,1h),8.11
‑
8.13(m,1h).
13
c nmr(100mhz,cdcl3)δ13.7,14.2,22.9,29.5,31.5,33.4,34.1,61.8,121.6,125.0,127.4,129.3,129.7,133.1,139.4,143.0,147.0,149.6,158.8,167.1,203.4.hrms(esi
‑
tof)m/z calculated for c
21
h
24
no
3+
([m+h]
+
)338.1756,found 338.1738
[0100]
实施例5
[0101]4‑
氮杂芴类化合物3e的制备
[0102][0103]
用(e)
‑2‑
亚苄基
‑
1,4
‑
二苯基丁
‑3‑
炔
‑1‑
酮1e代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3e,收率为81%。
[0104]
谱图解析数据:
[0105]1h nmr(400mhz,cdcl3)δ0.95(t,j=7.2hz,3h),3.80(s,2h),4.13(q,j=7.2hz,2h),7.17
‑
7.21(m,5h),7.31(t,j=7.6hz,2h),7.44
‑
7.46(m,1h),7.49(td,j1=7.2hz,j2=2.0hz,3h),7.59
‑
7.61(m,2h),8.25(dd,j1=7.6hz,j2=1.6hz,1h).
13
c nmr(100mhz,cdcl3)δ13.5,33.5,61.6,121.9,125.1,127.5,127.9,128.0,128.6,129.3,129.4,129.6,130.7,133.9,135.2,135.7,135.8,139.4,143.8,144.4,150.0,160.0,167.3,195.5.hrms(esi
‑
tof)m/z calculated for c
28
h
22
no
3+
([m+h]
+
)420.1600,found 420.1623.
[0106]
实施例6
[0107]4‑
氮杂芴类化合物3f的制备
[0108]
[0109]
用(e)
‑2‑
亚苄基
‑1‑
(4
‑
氯苯基)
‑4‑
苯基丁
‑3‑
炔
‑1‑
酮1f代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3f,收率为76%。
[0110]
图5为本发明实施例6得到的4
‑
氮杂芴类化合物的核磁氢谱图,图6为其核磁碳谱图。
[0111]
谱图解析数据:
[0112]1h nmr(400mhz,cdcl3)δ0.95(t,j=7.2hz,3h),3.79(s,2h),4.13(q,j=7.2hz,2h),7.19(s,5h),7.26(dt,j1=8.8hz,j2=2.0hz,2h),7.43
‑
7.47(m,1h),7.49
‑
7.53(m,4h),8.24(dt,j1=7.6hz,j2=1.2hz,1h).
13
c nmr(100mhz,cdcl3)13.5,33.5,61.6,122.0,125.1,127.6,128.1(2c),129.0,129.4,129.8,130.5,130.6,134.1,135.2,135.5,139.3,140.5,143.8(2c),150.1,160.2,167.2,194.3.hrms(esi
‑
tof)m/z calculated for c
28
h
21
clno
3+
([m+h]
+
)454.1210,found 454.1195.
[0113]
实施例7
[0114]4‑
氮杂芴类化合物3g的制备
[0115][0116]
用(e)
‑2‑
亚苄基
‑1‑
(呋喃
‑2‑
基)
‑4‑
苯基丁
‑3‑
炔
‑1‑
酮1g代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3g,收率为70%。
[0117]
谱图解析数据:
[0118]1h nmr(400mhz,cdcl3)δ0.96(t,j=7.2hz,3h),3.90(s,2h),4.14(q,j=7.2hz,2h),6.41(dd,j1=3.6hz,j2=1.6hz,1h),6.87(d,j=3.6hz,1h),7.25
‑
7.29(m,5h),7.46
‑
7.50(m,3h),7.55(d,j=6.4hz,1h),8.23
‑
8.26(m,1h).
13
c nmr(100mhz,cdcl3)δ13.5,33.4,61.6,112.6,120.8,121.9,125.1,127.5,128.0,128.1,129.3,129.7,130.7,135.5,135.7,139.3,143.1,143.8,147.8,150.0,151.6,160.1,167.3,182.3.hrms(esi
‑
tof)m/z calculated for c
26
h
20
no
4+
([m+h]
+
)410.1392,found 410.1387.
[0119]
实施例8
[0120]4‑
氮杂芴类化合物3h的制备
[0121][0122]
用(e)
‑3‑
亚苄基
‑1‑
苯基庚
‑1‑
炔
‑4‑
酮1h代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3h,收率为68%。
[0123]
图7为本发明实施例8得到的4
‑
氮杂芴类化合物的核磁氢谱图,图8为其核磁碳谱图。
[0124]
谱图解析数据:
[0125]1h nmr(400mhz,cdcl3)δ0.65(t,j=7.2hz,3h),0.96(t,j=7.6hz,3h),1.40(f,j=7.2hz,2h),2.08(t,j=7.2hz,2h),3.91(s,2h),4.13(q,j=7.6hz,2h),7.31
‑
7.35(m,2h),7.40
‑
7.44(m,3h),7.48(td,j1=7.2hz,j2=1.6hz,2h),7.56
‑
7.58(m,1h),8.19
‑
8.21(m,1h).
13
c nmr(100mhz,cdcl3)δ13.4,13.5,16.9,33.6,45.1,61.5,121.8,125.1,127.4,128.4,128.5,129.3,129.4,129.6,134.3,136.1,139.4,143.9,145.7,150.0,160.3,167.2,205.9.hrms(esi
‑
tof)m/z calculated for c
25
h
24
no
3+
([m+h]
+
)386.1756,found 386.1742.
[0126]
实施例9
[0127]4‑
氮杂芴类化合物3i的制备
[0128][0129]
用(e)
‑2‑
亚苄基
‑4‑
(4
‑
甲氧基苯基)
‑1‑
苯基丁
‑3‑
炔
‑1‑
酮1i代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3i,收率为51%。
[0130]
谱图解析数据:
[0131]1h nmr(400mhz,cdcl3)δ0.93(t,j=7.6hz,3h),3.71(s,2h),3.93(s,3h),4.12(q,j=7.6hz,2h),7.02(dd,j1=8.4hz,j2=2.4hz,1h),7.16
‑
7.21(m,5h),7.30(td,j1=8.0hz,j2=2.0hz,2h),7.38(d,j=8.0hz,1h),7.47(tt,j1=8.0hz,j2=1.2hz,1h),7.58
‑
.7.61(m,2h),7.75(d,j=2.4hz,1h).
13
c nmr(100mhz,cdcl3)δ13.5,32.8,55.7,61.6,104.9,118.2,125.8,127.9,128.0,128.6,129.3,129.4,130.7,133.9,135.7,135.8,136.1,136.3,140.6,144.3,149.8,159.7,160.0,167.3,195.4.hrms(esi
‑
tof)m/z calculated for c
29
h
24
no
4+
([m+h]
+
)450.1705,found 450.1701.
[0132]
实施例10
[0133]4‑
氮杂芴类化合物3j的制备
[0134][0135]
用(e)
‑2‑
亚苄基
‑4‑
(4
‑
三氟甲基苯基)
‑1‑
苯基丁
‑3‑
炔
‑1‑
酮1j代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3j,收率为49%。
[0136]
图9为本发明实施例10得到的4
‑
氮杂芴类化合物的核磁氢谱图,图10为其核磁碳谱图。
[0137]
谱图解析数据:
[0138]1h nmr(400mhz,cdcl3)δ0.99(t,j=7.2hz,3h),3.88(s,2h),4.16(q,j=7.2hz,2h),7.18
‑
7.23(m,5h),7.32(td,j1=8.0hz,j2=1.6hz,2h),7.48(tt,j1=8.0hz,j2=1.6hz,1h),7.60(dd,j1=8.4hz,j2=1.6hz,2h),7.61(d,j=8.0hz,1h),7.71(dd,j1=8.0hz,j2=1.6hz,1h),8.54(s,1h).
13
c nmr(100mhz,cdcl3)δ13.5,33.7,61.8,119.0(q,j=3.9hz),124.1(q,j=270.7hz),125.6,126.3(q,j=3.6hz),128.1(2c),128.6,129.3,129.4,130.2(q,j=32.3hz),131.5,134.1,135.3,135.4,135.6,140.0,144.7,147.1,150.6,158.6,167.0,195.2.hrms(esi
‑
tof)m/z calculated for c
29
h
21
f3no
3+
([m+h]
+
)488.1474,found 488.1461.
[0139]
实施例11
[0140]4‑
氮杂芴类化合物3k的制备
[0141][0142]
用(e)
‑2‑
亚苄基
‑4‑
(2
‑
甲氧基苯基)
‑1‑
苯基丁
‑3‑
炔
‑1‑
酮。1k代替实施例1中的(e)
‑3‑
亚苄基
‑5‑
苯基戊
‑4‑
炔
‑2‑
酮1a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3k,收率为72%。
[0143]
谱图解析数据:
[0144]1h nmr(400mhz,cdcl3)δ0.94(t,j=7.2hz,3h),3.71(s,2h),3.87(s,3h),4.12(q,j=7.2hz,2h),6.95(d,j=8.0hz,1h),7.17
‑
7.23(m,5h),7.31(t,j=8.0hz,2h),7.45
‑
7.50(m,2h),7.60
‑
7.62(m,2h),7.86(d,j=7.6hz,1h).
13
c nmr(100mhz,cdcl3)δ13.6,30.9,55.2,61.5,111.0,114.4,127.9,128.0,128.6,129.2,129.3,129.4,130.7,131.6,134.0,135.2,135.7(2c),140.8,144.5,149.9,156.0,160.0,167.3,195.3.hrms(esi
‑
tof)m/z calculated for c
29
h
24
no
4+
([m+h]
+
)450.1705,found 450.1700.
[0145]
实施例12
[0146]4‑
氮杂芴类化合物3l的制备
[0147][0148]
用异氰基乙酸叔丁酯2b代替实施例1中的异氰基乙酸乙酯2a,其他条件同实施例1,得到4
‑
氮杂芴类化合物3l,收率为62%。
[0149]
图11为本发明实施例12得到的4
‑
氮杂芴类化合物的核磁氢谱图,图12为其核磁碳
谱图。
[0150]
谱图解析数据:
[0151]1h nmr(400mhz,cdcl3)δ1.23(s,9h),1.90(s,3h),3.93(s,2h),7.34
‑
7.36(m,2h),7.42
‑
7.47(m,5h),7.56
‑
7.57(m,1h),8.22
‑
8.24(m,1h).
13
c nmr(100mhz,cdcl3)δ27.5,30.8,33.8,82.6,121.9,125.0,127.4,128.4,128.6,128.7,129.5,129.7,133.7,136.5,139.6,144.0,145.4,151.3,160.4,166.3,203.3.hrms(esi
‑
tof)m/z calculated for c
25
h
24
no
3+
([m+h]
+
)386.1756,found 386.1748.
[0152]
实施例13
[0153]4‑
氮杂芴酮化合物4a的制备
[0154][0155]
向15ml耐压管中加入4
‑
乙酰基
‑3‑
苯基
‑
5h
‑
茚并[1,2
‑
b]吡啶
‑2‑
乙酸乙酯3a(0.2mmol),用1ml1,4
‑
二氧六环溶解,加入催化剂叔丁醇钾(0.06mmol)、bht(0.4mmol)并加入搅拌子,拧紧耐压管旋塞后放入预热至65℃的金属模块中进行搅拌,反应时间1h,此时tlc检测底物3a完全消失,停止反应并静置至室温,用饱和氯化铵溶液水洗,二氯甲烷溶液萃取三次合并有机相溶液,再用饱和氯化钠溶液水洗一次,减压蒸去溶剂,并通过硅胶柱层析进行分离以得到最终产物,经核磁氢谱、碳谱以及质谱检测证实其为4
‑
乙酰基
‑5‑
氧代
‑3‑
苯基
‑
5h
‑
茚并[1,2
‑
b]吡啶
‑2‑
乙酸乙酯4a,收率为92%。
[0156]
谱图解析数据:
[0157]1h nmr(400mhz,cdcl3)δ0.94(t,j=7.2hz,3h),2.22(s,3h),4.10(q,j=7.2hz,2h),7.26
‑
7.28(m,2h),7.39
‑
7.47(m,3h),7.49(td,j1=7.6hz,j2=1.2hz,1h),7.65(td,j1=7.6hz,j2=1.2hz,1h),7.74(d,j=7.6hz,1h),7.97(d,j=7.6hz,1h).
13
c nmr(100mhz,cdcl3)δ13.5,31.2,61.9,121.9,123.6,124.4,128.5,128.9,129.2,131.5,131.7,133.8,134.8,135.8,142.4,147.5,154.7,163.7,166.0,189.9,200.5.hrms(esi
‑
tof)m/z calculated for c
23
h
18
no
4+
([m+h]
+
)372.1236,found 372.1223.
[0158]
采用制备的化合物3a
‑
3l、4a对人宫颈癌细胞(hela)的生长抑制活性测定。
[0159]
实验材料:olympus细胞培养用显微镜,forma
tm
·
310细胞培养箱,thermo scientific heraguard eco超净台,eppendorf 5910r冷冻离心机,spectra max m5酶标仪,松下mvs
‑
83高压灭菌锅,人宫颈癌细胞hela,96孔细胞培养板,dmem培养基,pbs缓冲液,gibco胎牛血清,0.25%胰蛋白酶
‑
edta消化液,青霉素
‑
链霉素混合液,mtt。
[0160]
实验方法:
[0161]
(1)接种细胞:用含10%胎小牛血清得培养液配成单个细胞悬液,以每孔1000个细胞接种到96孔板,每孔体积100μl.
[0162]
(2)培养细胞:于37℃、5%co2细胞培养箱中培养。
[0163]
(3)呈色:待细胞贴壁后,每孔加入不同浓度(0.1μμ、0.5μμ、1.0μμ)的药物3a
‑
3l、4a,培养24h后,吸掉细胞培养液,用pbs洗板3次,加入mtt溶液(0.5mg/ml pbs配)100μl/孔,同时设立空白对照为200μl空白培养基.
[0164]
(4)继续37℃孵育4小时,每孔加150μl dmso终止培养,振荡10分钟,使结晶物充分融解。
[0165]
(5)在酶联免疫监测仪上,选择490nm波长,测定并记录各孔光吸收值。
[0166][0167]
计算获得的抑制率如表1所示。
[0168]
表1.化合物在0.1μm、0.5μm和1.0μm浓度下对宫颈癌细胞hela的抑制率
[0169][0170][0171]
由表1可知,化合物3a
‑
3l、4a对人宫颈癌细胞的生长均有抑制活性。其中,化合物3a、3b、3c、3e、3f、4j、4a均对子宫颈癌细胞的生长有较强抑制活性。
[0172]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1