环烯烃共聚物制备用催化剂、环烯烃共聚物的制备方法、环烯烃共聚物及其应用与流程
1.本技术实施例涉及工程塑料制备技术领域,特别是涉及一种环烯烃共聚物制备用催化剂、环烯烃共聚物的制备方法、环烯烃共聚物及其应用。
背景技术:
2.环烯烃聚合物是一类具有高附加值的热塑性工程塑料,由于其优异的光学透明性、耐热性、化学稳定性、熔体流动性、隔湿性、尺寸稳定性及低的介电常数等性能,已被广泛应用于各种电子产品、汽车头灯、眼镜、医药食品包装材料等领域。
3.环烯烃聚合物的合成主要有两种途径:一种方法为乙烯或α-烯烃(指双键在分子链端部的单烯烃)与降冰片烯类环烯烃单体的链式加成共聚合(如化学反应式(1)所示,m、n表示聚合度),由此方法制备的聚合物也称为环烯烃共聚物(cyclic olefin copolymer,coc);另一种方法为降冰片烯类等环烯烃单体的开环易位聚合(romp)和随后的氢化(如化学反应式(2)所示,n表示聚合度),由此得到的聚合物也称为环烯烃均聚物(cyclic olefin polymer,cop)。
[0004][0005]
环烯烃聚合物在合成、加工和实际应用过程中,其分子量和玻璃化转变温度(glass transition temperature,tg)是两个关键性能指标。分子量显著影响环烯烃聚合物的机械性能和加工性能。当环烯烃聚合物分子量较高(例如重均分子量大于10万)时,熔体流动指数(melt flow rate,mfr)很低,加工困难。而当环烯烃聚合物tg过高时,会造成环烯烃聚合物难以加工或较难注塑成型等;tg过低时,会限制环烯烃聚合物的使用环境和条件。因此,对于coc的实际应用而言,需要寻求有效方法制备同时具备低分子量(重均分子量小于或等于15万)和适中tg(110℃-180℃)的环烯烃共聚物。
技术实现要素:
[0006]
鉴于此,本技术实施例提供一种环烯烃共聚物制备用催化剂,采用该催化剂可在不额外引入氢气或丙烯等分子量调节剂的情况下制备获得低分子量(重均分子量小于或等于15万)环烯烃共聚物,同时保证环烯烃共聚物具有适中的玻璃化转变温度(110℃-180℃)。
[0007]
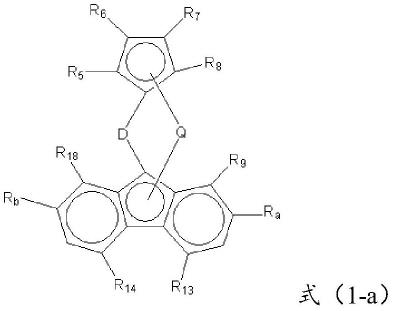
第一方面,本技术实施例提供一种环烯烃共聚物制备用催化剂,所述催化剂包括
结构式如式(1-a)所示的主催化剂:
[0008][0009]
式(1-a)中,d为桥联基团,q为金属中心;
[0010]
r5、r6、r7、r8独立地包括氢原子、烃基或含硅取代基,所述含硅取代基通过硅原子与对应取代位置的碳原子连接;
[0011]
ra、rb为含碳基团、含硅基团、含锗基团或含锡基团;
[0012]
所述r5、r6、r7、r8中至少一个为含硅取代基,和/或所述ra、rb中至少一个为含硅基团;
[0013]
r9、r
13
、r
14
、r
18
独立地包括氢原子、烃基或烃氧基。
[0014]
本技术实施例提供的环烯烃共聚物制备用催化剂,包括式(1-a)所示的主催化剂,该主催化剂为环戊二烯芴桥联的过渡金属催化剂,该主催化剂的环戊二烯基或芴基上引入有含硅杂原子基团,在乙烯、α-烯烃与环烯烃单体共聚的过程中,主催化剂的金属中心与环戊二烯基或芴基上引入的硅原子产生协同作用,可促进聚合过程的链转移,提升环烯烃单体的插入率,从而可在不额外引入氢气或丙烯等分子量调节剂的情况下通过链式加成共聚合方式得到具有低分子量(重均分子量小于或等于15万)和适中玻璃化转变温度的环烯烃共聚物。所得环烯烃共聚物由于具有较低分子量因而具有较低熔体流动指数,加工性能良好,且由于具有适中的玻璃化转变温度能够避免玻璃化转变温度太高导致难以加工难以注塑成型的问题,同时适中的玻璃化转变温度能够具有较好耐热性能,使得环烯烃共聚物适用于各种应用场景。
[0015]
本技术实施方式中,所述金属中心q表示为-m1(r1r2)-,所述m1表示钪、钛、钒、锆、铪、铌或钽,所述r1和r2独立地包括氢原子、卤原子、烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。
[0016]
本技术实施方式中,所述桥联基团d表示为-x(r3r4)-,所述x表示碳或硅,所述r3和r4独立地包括氢原子或烃基。
[0017]
本技术实施方式中,所述ra表示为-m2(r
10r11 r
12
),所述rb表示为-m3(r
15r16r17
),m2、m3独立地表示碳、硅、锗或锡,r
10
、r
11
、r
12
、r
15
、r
16
、r
17
独立地包括烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。
[0018]
本技术一实施方式中,式(1-a)所示的主催化剂具体结构式为式(1-b)所示:
[0019][0020]
本技术一些实施方式中,r5、r6、r7、r8中至少一个为含硅取代基,ra、rb不为含硅基团,ra、rb为含碳基团、含锗基团或含锡基团,即m2、m3独立地包括碳、锗或锡。本技术另一些实施方式中,ra、rb至少一个为含硅基团,即m2、m3中一者或两者为硅,r5、r6、r7、r8不为含硅取代基,r5、r6、r7、r8为氢原子或烃基。本技术另一些实施方式中,ra、rb至少一个为含硅基团,即m2、m3中一者或两者为硅,同时r5、r6、r7、r8中至少一个为含硅取代基。
[0021]
本技术一些实施方式中,所述r6、r7中至少一个为含硅取代基,和/或所述ra、rb中至少一个为含硅基团。含硅取代基位于环戊二烯3位和4位相比2位和5位离金属中心m1更远,在环戊二烯3位、4位引入含硅取代基,不仅能够实现与金属中心m1形成弱配位促进链转移,降低聚合物分子量,而且能够避免因较强配合作用而影响催化剂的聚合反应活性。
[0022]
本技术实施方式中,所述r5、r6、r7、r8中烃基和含硅取代基的碳原子数小于或等于6。
[0023]
本技术实施方式中,所述r
10
、r
11
、r
12
、r
15
、r
16
、r
17
的碳原子数小于或等于10。
[0024]
本技术实施方式中,所述环烯烃共聚物制备用催化剂还包括助催化剂,所述助催化剂包括但不限于甲基铝氧烷、改性的甲基铝氧烷、有机硼化合物中的一种或多种。本技术实施方式中,所述有机硼化合物包括三(五氟苯基)硼、三苯碳鎓四(五氟苯基)硼酸盐、n,n-二甲基苯铵四(五氟苯基)硼酸盐中的一种或多种。采用甲基铝氧烷、改性的甲基铝氧烷、有机硼化合物作为助催化剂,有利于保证环烯烃共聚物制备的共聚反应活性。
[0025]
本技术实施方式中,所述式(1-a)所示的主催化剂与所述助催化剂的摩尔比为1:(10-10000)。
[0026]
本技术实施方式中,所述催化剂的催化反应活性高于106g
·
mol-1
·
h-1
。本技术式(1-a)所示的主催化剂用于环烯烃共聚物制备具有较高的催化反应活性。
[0027]
本技术实施方式中,主催化剂和助催化剂可以是负载在载体上。载体例如可以是氧化硅、氧化铝、氧化钛等。
[0028]
第二方面,本技术实施例提供一种环烯烃共聚物的制备方法,包括:
[0029]
在第一方面所述的环烯烃共聚物制备用催化剂存在的条件下,使环烯烃单体与乙烯或α-烯烃发生共聚合反应,得到环烯烃共聚物。
[0030]
本技术实施方式中,所述共聚合的反应体系中包括惰性溶剂,所述惰性溶剂包括直链烷烃类化合物、环烃类化合物和芳烃类化合物中的一种或多种。
[0031]
本技术实施方式中,所述共聚合的反应体系中式(1-a)所示的主催化剂的用量为
0.001mmol/l-10mmol/l。
[0032]
本技术实施方式中,所述共聚合的反应体系中所述环烯烃单体的用量为0.01mol/l-10mol/l。
[0033]
本技术实施方式中,所述共聚合的反应体系中所述环烯烃单体与所述主催化剂的摩尔比为500-500000。本技术的主催化剂可适应较大范围的环烯烃单体使用量,催化活性高。
[0034]
本技术实施方式中,所述共聚合反应的温度为50℃-120℃;所述共聚合反应的时间为2min-10min。本技术环烯烃共聚物的制备方法,其共聚合反应温度要求低,时间短,效率高。
[0035]
本技术实施方式中,所述环烯烃单体的结构式如式(2)所示:
[0036][0037]
式(2)中,r
19
为烃基或烃基硅基;r
20
和r
21
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团;
[0038]r22
和r
23
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团,或者r
22
和r
23
连接形成具有环状结构的基团;
[0039]
z为正整数。
[0040]
本技术实施方式中,所述α-烯烃可以是丙烯、1-丁烯、1-戊烯、2-甲基-1-丁烯、3-甲基-1-丁烯、1-己烯、2-甲基-1-戊烯、3-甲基-1-戊烯、4-甲基-1-戊烯或2-乙基-1-丁烯。
[0041]
本技术实施方式中,所述共聚合反应体系中不包含分子量调节剂。分子量调节剂例如为氢气、丙烯等。分子量调节剂的加入不仅使制备工艺变得更复杂,同时会影响聚合反应本身。例如氢气的引入会降低催化剂体系的活性,而丙烯的引入会在聚合物中引入丙烯分子,影响环烯烃共聚物的结构。
[0042]
本技术实施例提供的环烯烃共聚物的制备方法,通过采用本技术实施例第一方面提供的催化剂,不需要额外引入氢气或丙烯等分子量调节剂,即可制备获得具有低分子量和适中玻璃化转变温度的环烯烃共聚物,以满足光学透镜等各类光学制品、显示材料和包装材料等产品的加工性能、耐热性能等要求。该制备方法不仅大大简化了环烯烃共聚物材料的制备途径,还显著提高了聚合活性和能源经济效益,为大规模生产环烯烃共聚物材料开辟了一条新的路径。
[0043]
第三方面,本技术实施例提供一种环烯烃共聚物,根据第二方面所述的制备方法制得,所述环烯烃共聚物的结构式如式(3)所示:
[0044][0045]
式(3)中,r
19
为烃基或烃基硅基;r
20
和r
21
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团;
[0046]r22
、r
23
、r
24
、r
25
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团,或者r
22
和r
23
连接形成具有环状结构的基团,r
24
和r
25
连接形成具有环状结构的基团;
[0047]
x和y表示聚合度,x和y均为正数,1<x∶y<3,z为正整数。
[0048]
本技术实施方式中,所述环烯烃共聚物的重均分子量在5000至150000(g/mol)范围内;分子量分布指数在1.5至3.0范围内。
[0049]
本技术实施方式中,所述环烯烃共聚物的环烯烃单体的插入率在20%-60%范围内;所述环烯烃共聚物的玻璃化转变温度在110℃至180℃范围内。
[0050]
本技术实施方式中,所述环烯烃共聚物的成型体的可见光透过率大于90%。
[0051]
第四方面,本技术实施例提供一种组合物,包括本技术实施例第三方面所述的环烯烃共聚物,或包括第二方面所述的制备方法制得的环烯烃共聚物。
[0052]
本技术实施方式中,所述组合物还包括添加剂,所述添加剂包括填料、染料、抗氧化剂、光稳定剂、紫外线吸收剂、增塑剂、阻燃剂、抗静电剂、脱模剂中的一种或多种。
[0053]
第五方面,本技术实施例提供一种光学制品,所述光学制品包括本技术实施例第三方面所述的环烯烃共聚物,或包括第二方面所述的制备方法制得的环烯烃共聚物。
[0054]
本技术实施方式中,所述光学制品包括光学透镜、光学膜、光盘、导光板或显示面板。所述光学透镜包括眼镜透镜、相机透镜、传感器透镜、照明透镜、成像透镜。
[0055]
本技术实施例还提供一种设备,包括本技术实施例第五方面所述的光学制品。
[0056]
本技术实施例还提供一种电子设备,包括电子设备主体和装配于所述电子设备主体上的摄像头模组,所述摄像头模组包括镜头透镜,所述镜头透镜采用第三方面所述的环烯烃共聚物,或第四方面所述的组合物制备。
附图说明
[0057]
图1为本技术实施例提供的设备100的结构示意图;
[0058]
图2为本技术实施例1中催化剂a的核磁共振氢谱(1h nuclear magnetic resonance spectroscopy,1h nmr);
[0059]
图3为本技术实施例1中催化剂a的核磁共振碳谱(
13
c nuclear magnetic resonance spectroscopy,
13
c nmr);
[0060]
图4为本技术实施例1中环烯烃单体的核磁共振氢谱;
[0061]
图5为本技术实施例1的环烯烃单体的核磁共振碳谱;
[0062]
图6为本技术实施例1中环烯烃共聚物的核磁共振氢谱;
[0063]
图7为本技术实施例1的环烯烃共聚物的核磁共振碳谱;
[0064]
图8为本技术实施例1的环烯烃共聚物的dsc(differential scanning calorimetry,差示扫描量热)曲线;
[0065]
图9为本技术实施例1的环烯烃共聚物的可见光透过率测试曲线;
[0066]
图10为本技术实施例2的环烯烃共聚物的dsc曲线;
[0067]
图11为本技术实施例3的环烯烃共聚物的dsc曲线;
[0068]
图12为本技术实施例4的环烯烃共聚物的dsc曲线;
[0069]
图13为本技术实施例5的环烯烃共聚物的dsc曲线。
具体实施方式
[0070]
下面将结合本技术实施例中的附图,对本技术实施例进行说明。
[0071]
环烯烃共聚物coc的分子量和玻璃化转变温度是两个关键性能指标,分子量影响加工性能,玻璃化转变温度影响加工性能及使用环境和条件。目前,环烯烃共聚物的制备通常使用茂金属催化剂,然而采用现有的茂金属催化剂直接制备的环烯烃共聚物分子量通常在20万或以上,熔体流动指数低,加工困难,商用价值低。为了获得具有低分子量的环烯烃共聚物,现有传统的方法是在制备过程中额外引入氢气、丙烯等分子量调节剂,而额外引入分子量调节剂可能降低催化剂的催化活性,也可能使环烯烃共聚物中引入分子量调节剂,影响环烯烃共聚物的结构,而且额外引入分子量调节剂也会使制备工艺变得复杂。另外,在采用乙烯或α-烯烃与环烯烃单体共聚的过程中,通过调节环烯烃单体在共聚物中的插入率可以调节环烯烃共聚物coc的玻璃化转变温度。
[0072]
为了获得具有较低分子量和适合玻璃化转变温度的环烯烃共聚物coc,本技术实施例提供一种环烯烃共聚物制备用催化剂,该催化剂可在不额外引入氢气或丙烯等分子量调节剂的情况下,用于直接制备得到具有低分子量和适中玻璃化转变温度的环烯烃共聚物,该催化剂包括结构式如式(1-a)所示的主催化剂:
[0073][0074]
式(1-a)中,d为桥联基团,q为金属中心;
[0075]
r5、r6、r7、r8独立地包括氢原子、烃基或含硅取代基,所述含硅取代基通过硅原子与对应取代位置的碳原子连接;
[0076]
ra、rb为含碳基团、含硅基团、含锗基团或含锡基团;
[0077]
所述r5、r6、r7、r8中至少一个为含硅取代基,和/或所述ra、rb中至少一个为含硅基团;
[0078]
r9、r
13
、r
14
、r
18
独立地包括氢原子、烃基或烃氧基。
[0079]
本技术实施例提供的环烯烃共聚物制备用催化剂,式(1-a)所示的主催化剂为环戊二烯芴桥联的过渡金属催化剂,该环戊二烯芴桥联的过渡金属催化剂中的环戊二烯基或芴基的2位或7位上引入有含硅杂原子基团,在乙烯或α-烯烃与环烯烃单体共聚的过程中,主催化剂的金属中心m1与环戊二烯基或芴基上引入的硅原子产生协同作用,可促进聚合过程的链转移,提升环烯烃单体的插入率,从而可在不额外引入氢气或丙烯等分子量调节剂的情况下得到具有低分子量和适中玻璃化转变温度的环烯烃共聚物。具体地,环戊二烯基或芴基上引入的硅原子与金属中心m1空轨道产生弱的配位作用,与烯烃-金属中心m1两者之间的配位产生竞争,从而促进链转移,降低聚合物分子量使环烯烃共聚物具有低分子量;同时通过这种竞争作用,增加了乙烯或α-烯烃与金属中心m1配位的难度,提升了环烯烃单体的插入率,从而起到调节玻璃化转变温度的作用,使环烯烃共聚物具有适中的玻璃化转变温度,适用于各种应用场景。本技术实施例的环烯烃共聚物制备用催化剂能够使得低分子量环烯烃共聚物的制备更简便高效,大大简化聚合流程和聚合设备,利于规模化生产低分子量环烯烃共聚物。同时,该环烯烃聚合物的其他性能如光学性能、热性能和机械性能等都能达到传统方法制备的低分子量coc材料的水平,从而也能够适应传统方法制备的coc材料的应用场景。
[0080]
本技术实施方式中,所述金属中心q表示为-m1(r1r2)-,所述m1表示钪、钛、钒、锆、铪、铌或钽,所述r1和r2独立地包括氢原子、卤原子、烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。
[0081]
本技术实施方式中,所述桥联基团d表示为-x(r3r4)-,所述x表示碳或硅,所述r3和r4独立地包括氢原子或烃基。
[0082]
本技术实施方式中,所述ra表示为-m2(r
10r11 r
12
),所述rb表示为-m3(r
15r16r17
),m2、m3独立地表示碳、硅、锗或锡,r
10
、r
11
、r
12
、r
15
、r
16
、r
17
独立地包括烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。ra通过m2与芴环上对应位置碳原子连接,rb通过m3与芴环上对应位置碳原子连接。
[0083]
本技术一实施方式中,式(1-a)所示的主催化剂具体结构式为式(1-b)所示:
[0084]
[0085]
本技术实施方式中,式(1-b)所示的主催化剂是一类桥联的双茂过渡金属化合物,m1为金属中心,表示钪、钛、钒、锆、铪、铌或钽等前过渡金属,r1和r2与金属中心m1连接,r1和r2独立地包括氢原子、卤原子、烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。环戊二烯和芴通过桥联基团-x(r3r4)-连接,且环戊二烯和芴与金属中心m1配位结合。x表示碳或硅,r3和r4独立地包括氢原子或烃基,烃基可以是烷基、烯基、芳基、芳烷基、烷芳基或芳烯基。具体地,烃基可以是碳原子数小于或等于10(即碳原子数为1-10)的烷基、烯基、芳基、芳烷基、烷芳基或芳烯基。本技术一些实施方式中,r3和r4也可以是连成环状结构。
[0086]
本技术实施方式中,式(1-b)中,r1和r2为卤原子时,卤原子可以是氟、氯、溴或碘。本技术实施方式中,烷基可以是直链、支链或具有环状结构的烷基。烷基可以是非取代烷基,也可以是取代烷基。烯基可以是直链或支链的烯基。烯基可以是非取代烯基,也可以是取代烯基。烷氧基可以是直链、支链或具有环状结构的烷氧基。本技术实施方式中,烷基、烷氧基、烯基的碳原子数可以是1、2、3、4、5、6、7、8、9、10。具体地,烷基例如可以是甲基、乙基、丙基、丁基等。本技术实施方式中,芳基可以是非取代芳基,也可以是取代芳基。芳基、芳氧基、芳烷基、烷芳基、芳烯基的碳原子数可以是6、7、8、9、10。本技术一些实施方式中,桥联基团-x(r3r4)-例如可以是但不限于是亚甲基、亚乙基、亚异丙基(-c(ch3)
2-)、亚二苯甲基(-c(c6h5)
2-)、双三甲基硅基亚甲基(-c(si(ch3)3)
2-)等。
[0087]
本技术实施方式中,式(1-b)中,r5、r6、r7、r8可独立地包括氢原子、烃基或含硅取代基。具体地,烃基可以是碳原子数小于或等于10(即碳原子数为1-10)的烷基、烯基、芳基、芳烷基、烷芳基或芳烯基。烷基可以是直链、支链或具有环状结构的烷基。烷基可以是非取代烷基,也可以是取代烷基。烯基可以是直链或支链的烯基。烯基可以是非取代烯基,也可以是取代烯基。烷基、烯基的碳原子数可以是1、2、3、4、5、6、7、8、9、10。具体地,烷基例如可以是甲基、乙基、丙基、丁基等。本技术实施方式中,芳基可以是非取代芳基,也可以是取代芳基。芳基、芳烷基、烷芳基、芳烯基的碳原子数可以是6、7、8、9、10。r5、r6、r7、r8分别对应环戊二烯的2位、3位、4位、5位的碳原子。本技术实施方式中,r5、r6、r7或r8为含硅取代基时,含硅取代基与环戊二烯2位、3位、4位或5位的碳原子形成碳硅键合。本技术实施方式中,可以是r5、r6、r7和r8中的一者为含硅取代基,也可以是r5、r6、r7和r8中的两者或三者或四者为含硅取代基。当r5、r6、r7和r8中部分为含硅取代基时,不为含硅取代基的基团可以是氢原子或烃基。当r5、r6、r7和r8中有多个为含硅取代基时,可以是相同含硅取代基,也可以是不同含硅取代基。当r5、r6、r7和r8中有多个为烃基时,可以是相同烃基,也可以是不同烃基。含硅取代基通过硅原子与对应取代位置的碳原子连接,即含硅取代基通过硅原子与对应位置环戊二烯环上的碳原子连接,含硅取代基可以表示为-si(r’r”r
”’
),r’、r”、r
”’
可以是烷基或芳基,即含硅取代基可以是烷基硅烷基或芳基硅烷基。烷基硅烷基具体例如可以是三甲基硅烷基(即r’、r”、r
”’
为甲基)、三乙基硅烷基(即r’、r”、r
”’
为乙基)等,芳基硅烷基具体例如可以是三苯基硅烷基(即r’、r”、r
”’
为苯基)。本技术一些实施方式中,含硅取代基可以是碳原子总数为1-10的烷基硅烷基。一些实施例中,r5、r6、r7、r8独立地包括碳原子数小于或等于6(即c
1-c6)的烃基或含硅取代基。较小的碳原子数的烃基或含硅取代基能够减小金属中心m1周围的空间位阻,有利于使催化剂的催化活性保持较高水平。
[0088]
环戊二烯碳位上引入的含硅取代基可以与金属中心m1产生协同作用,促进聚合过程的链转移,提升环烯烃单体的插入率,使环烯烃共聚物具有低分子量和适中的玻璃化转
变温度。本技术一些实施方式中,r6或r7中至少一个为含硅取代基。环戊二烯3位和4位的取代基相比2位和5位的取代基离金属中心m1更远,在环戊二烯3位、4位引入含硅取代基,不仅能够实现与金属中心m1形成弱配位促进链转移,降低聚合物分子量,而且能够避免因较强配合作用而影响催化剂的聚合反应活性。例如,一实施方式中,r6为含硅取代基,r5、r7和r8为氢或烃基。另一实施方式中,r7为含硅取代基,r5、r6和r8为氢或烃基。一些实施方式中,r6和r7为含硅取代基,r5和r8为氢或烃基。
[0089]
本技术实施方式中,式(1-b)中,m2表示碳、硅、锗或锡,m3表示碳、硅、锗或锡。m2、m3可以是相同的原子,也可以是不同的原子。m2或m3可以与金属中心m1产生协同作用,促进聚合过程的链转移,提升环烯烃单体的插入率,起到调节分子量和调节玻璃化转变温度的作用,从而使环烯烃共聚物具有低分子量和适中玻璃化转变温度。
[0090]
为使式(1-b)所示的主催化剂能够在环烯烃共聚物制备过程中实现调节分子量和调节玻璃化转变温度的作用,最终获得具有低分子量和适中玻璃化转变温度的环烯烃共聚物,本技术实施方式中,环戊二烯环上的取代基r5、r6、r7、r8中至少有一个基团为含硅取代基,或者芴环的2位或7位的取代基至少一个为含硅基团,即m2、m3中至少一个为硅。芴环的2位和7位离金属中心m1的距离适中,2位和7位的含硅基团与金属中心m1的协同配位作用既不会太强也不会太弱,从而有利于通过弱配位协同作用促进链转移,降低聚合物分子量,而且能够避免因较强配合作用而影响催化剂的聚合反应活性。当m2、m3中一者或两者为硅时,r5、r6、r7、r8均为氢原子或烃基,有利于保证聚合反应活性保持在更高水平,更好地平衡聚合物低分子量与聚合反应活性,且可以降低催化剂制备难度。当m2、m3中一者或两者为硅时,r5、r6、r7、r8中至少一个为含硅基团,通过硅与金属中心m1的配位作用能够有利于制备获得具有更低分子量的聚合物。
[0091]
本技术一些实施方式中,r5、r6、r7、r8中至少一个为含硅取代基,m2、m3独立地包括碳、锗或锡。本技术另一些实施方式中,r5、r6、r7、r8独立地包括氢原子或烃基,m2、m3中至少一个为硅。本技术另一些实施方式中,r5、r6、r7、r8中至少一个为含硅取代基,同时m2、m3中至少一个为硅。一实施方式中,r5、r6、r7、r8独立地包括氢原子或烃基,m2、m3为硅,r9、r
13
、r
14
、r
18
为氢原子,即芴环上2位和7位为含硅基团,其余取代位均为氢原子,该实施方式的主催化剂不仅能够通过协同作用调节共聚物分子量和玻璃化转变温度,而且结构简单易于制备。
[0092]
本技术实施方式中,r
10
、r
11
、r
12
、r
15
、r
16
、r
17
可独立地包括烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。具体地,r
10
、r
11
、r
12
、r
15
、r
16
、r
17
可以是独立地包括碳原子数小于或等于10的烷基、烷氧基、烯基、芳基、芳氧基、芳烷基、烷芳基或芳烯基。本技术实施方式中,烷基可以是直链、支链或具有环状结构的烷基。烷基可以是非取代烷基,也可以是取代烷基。烯基可以是直链或支链的烯基。烯基可以是非取代烯基,也可以是取代烯基。烷氧基可以是直链、支链或具有环状结构的烷氧基。本技术实施方式中,烷基、烷氧基、烯基的碳原子数可以是1、2、3、4、5、6、7、8、9、10。具体地,烷基例如可以是甲基、乙基、丙基、丁基等。本技术实施方式中,芳基可以是非取代芳基,也可以是取代芳基。芳基、芳氧基、芳烷基、烷芳基、芳烯基的碳原子数可以是6、7、8、9、10。
[0093]
本技术实施方式中,式(1-b)中,r9、r
13
、r
14
、r
18
独立地包括氢原子、烃基或烃氧基。具体地,烃基可以是碳原子数小于或等于10(即碳原子数为1-10)的烷基、烯基、芳基、芳烷基、烷芳基或芳烯基。烃氧基可以是碳原子数小于或等于10(即碳原子数为1-10)的烷氧基
或芳氧基。烷基可以是直链、支链或具有环状结构的烷基。烷基可以是非取代烷基,也可以是取代烷基。烯基可以是直链或支链的烯基。烯基可以是非取代烯基,也可以是取代烯基。烷氧基可以是直链、支链或具有环状结构的烷氧基。本技术实施方式中,烷基、烷氧基、烯基的碳原子数可以是1、2、3、4、5、6、7、8、9、10。具体地,烷基例如可以是甲基、乙基、丙基、丁基等。本技术实施方式中,芳基可以是非取代芳基,也可以是取代芳基。芳基、芳氧基、芳烷基、烷芳基、芳烯基的碳原子数可以是6、7、8、9、10。本技术一些实施方式中,r9、r
13
、r
14
、r
18
均为氢原子,即芴基上仅2位和7位碳上具有取代基,这样可以使芴基上的取代结构变得更简单,简化催化剂的制备工艺。
[0094]
本技术一些实施方式中,环烯烃共聚物制备用催化剂包括式(1-a)所示的主催化剂,还包括助催化剂,助催化剂可以是包括甲基铝氧烷(mao)、改性的甲基铝氧烷(mmao)、有机硼化合物中的一种或多种。其中,有机硼化合物可以是包括三(五氟苯基)硼、三苯碳鎓四(五氟苯基)硼酸盐、n,n-二甲基苯铵四(五氟苯基)硼酸盐中的一种或多种。助催化剂有利于提升主催化剂的活性。相比甲基铝氧烷(mao),采用改性的甲基铝氧烷(mmao)作为助催化剂可以有利于减少环烯烃单体的用量。采用甲基铝氧烷、改性的甲基铝氧烷、有机硼化合物作为助催化剂,有利于保证环烯烃共聚物制备的共聚反应活性。
[0095]
本技术实施方式中,环烯烃共聚物制备用催化剂包括式(1-a)所示的主催化剂和助催化剂,助催化剂用量越多,环烯烃聚合物的分子量越低,玻璃化转变温度越高,考虑环烯烃聚合物分子量和玻璃化转变温度,本技术中,主催化剂与助催化剂的摩尔比可以是1:(10-10000)。一些实施例中,主催化剂与助催化剂的摩尔比可以是1:(100-5000)。一些实施例中,主催化剂与助催化剂的摩尔比可以是1:(500-4000)。
[0096]
本技术实施方式中,环烯烃共聚物制备用催化剂用于制备环烯烃共聚物,具有较高的催化反应活性,具体地,催化反应活性高于1
×
106g
·
mol-1
·
h-1
。一些实施例中,催化反应活性高于1
×
107g
·
mol-1
·
h-1
。本技术式(1-a)所示的主催化剂用于环烯烃共聚物制备具有较高的催化反应活性,催化反应活性高,有利于提升共聚合反应的反应速度和转化率。
[0097]
本技术实施方式中,主催化剂和助催化剂可以是负载在载体上。载体例如可以是氧化硅、氧化铝、氧化钛等。
[0098]
本技术实施例上述的式(1-b)所示的化合物可以是采用如下方式制备:
[0099]
制备-x(r3r4)-桥联的环戊二烯芴配体,将环戊二烯芴配体采用锂化试剂锂化后,与m1金属盐发生配位反应,得到式(1-b)所示的化合物。反应过程如反应式(a)所示。
[0100]
[0101]
锂化试剂可以是但不限于是正丁基锂。m1金属盐可以是钪盐、钛盐、钒盐、锆盐、铪盐、铌盐或钽盐。锂化过程可以是在无水无氧、冰浴的条件下进行。环戊二烯芴配体锂化后进行抽滤,获得的产物移入手套箱加入有机溶剂和m1金属盐,搅拌过夜进行配位反应,有机溶剂可以是甲苯、己烷等能够溶解上述产物的溶剂。配位反应后获得的产物可以通过洗涤、萃取、重结晶获得最终产物。
[0102]
以催化剂a为例,催化剂a的具体的制备过程可以是包括如下步骤:
[0103]
(1)环戊二烯芴配体的制备:
[0104]
无水无氧条件下,-78℃反应温度下,在反应容器中加入2,7-二溴芴和无水四氢呋喃,再逐滴滴加含正丁基锂的己烷溶液,随后逐滴滴加含三甲基氯硅烷(me3sicl)的四氢呋喃溶液,反应过夜。然后再次逐滴滴加含正丁基锂的己烷溶液和含三苯基氯硅烷(ph3sicl)的四氢呋喃溶液,反应过夜。随后,在室温下加入氢氧化钠水溶液进行水解,用乙醚萃取分液,有机相用无水硫酸镁干燥后得到2,7-双(三苯基硅基)芴;
[0105]
无水无氧条件下,在反应容器中加入所制备的2,7-双(三苯基硅基)芴和无水四氢呋喃,在室温下逐滴滴加等摩尔量的甲基锂乙醚溶液,室温过夜反应,然后逐滴加入溶有6,6-二甲基富烯的无水四氢呋喃溶液,反应过夜,加入四丁基氯化铵水溶液,搅拌,萃取分液,水相用乙醚水洗三次,有机相用无水硫酸镁干燥后,干燥重结晶,得到环戊二烯芴配体前体。
[0106]
无水无氧条件下,在反应容器中加入上述制备的环戊二烯芴配体前体和无水四氢呋喃,在-78℃下加入等摩尔量的正丁基锂己烷溶液,随后逐滴滴加三甲基氯硅烷(me3sicl),搅拌过夜,抽干溶剂,用己烷洗涤,得到环戊二烯芴配体(即催化剂a前体)。
[0107]
上述步骤的反应过程可参见反应式(1-1)。
[0108][0109]
(2)催化剂a的制备:
[0110]
在无水无氧的条件下,向反应容器中加入环戊二烯芴配体(催化剂a前体),冰浴下加入正丁基锂,撤掉冰浴后反应2-12个小时后抽干溶剂,移入手套箱内加入己烷,充分搅拌下加四氯化锆,搅拌过夜。过夜反应后过滤,滤饼用己烷洗涤后溶于过量甲苯中,过滤出土黄色不溶物,然后浓缩甲苯溶液,重结晶得到桃红色固体催化剂a。该步骤的反应过程可参见反应式(1-2)。
[0111][0112]
本技术实施方式中,式(1-b)所示的其他结构催化剂的制备可参考催化剂a的制备,此处不再一一说明。
[0113]
本技术实施例还提供一种环烯烃共聚物的制备方法,采用本技术实施例上述的环
烯烃共聚物制备用催化剂,该制备方法包括:
[0114]
在上述的环烯烃共聚物制备用催化剂存在的条件下,使环烯烃单体与乙烯或α-烯烃发生共聚合反应,得到环烯烃共聚物。
[0115]
本技术实施方式中,共聚合的反应体系中包括惰性溶剂,惰性溶剂包括直链烷烃类化合物、环烃类化合物和芳烃类化合物中的一种或多种。直链烷烃类化合物具体可以是碳原子数为5-16的直链烷烃,例如戊烷、己烷、庚烷、辛烷等。环烃类化合物具体可以是碳原子数为5-11的环烃,例如环戊烷、环己烷等。芳烃类化合物具体可以是碳原子数为6-20的液态芳烃,例如甲苯。
[0116]
本技术实施方式中,共聚合的反应体系中式(1-a)所示的主催化剂的用量为0.001mmol/l-10mmol/l。一些实施方式中,共聚合的反应体系中式(1-a)所示的主催化剂的用量为0.01mmol/l-1mmol/l。一些实施方式中,共聚合的反应体系中式(1-a)所示的主催化剂的用量为0.01mmol/l-0.1mmol/l。
[0117]
本技术实施方式中,共聚合的反应体系中式(1-a)所示的主催化剂与助催化剂的摩尔比可以是1:(10-10000)。一些实施例中,主催化剂与助催化剂的摩尔比可以是1:(100-5000)。一些实施例中,主催化剂与助催化剂的摩尔比可以是1:(500-4000)。
[0118]
本技术实施方式中,共聚合的反应体系中环烯烃单体的用量可以是0.01mol/l-10mol/l。一些实施例中,共聚合的反应体系中环烯烃单体的用量可以是0.01mol/l-5mol/l。一些实施例中,共聚合的反应体系中环烯烃单体的用量可以是0.01mol/l-1mol/l。
[0119]
本技术实施方式中,共聚合的反应体系中环烯烃单体与主催化剂的摩尔比为500-500000。一些实施方式中,共聚合的反应体系中环烯烃单体与主催化剂的摩尔比为1000-100000。一些实施方式中,共聚合的反应体系中环烯烃单体与主催化剂的摩尔比为5000-100000。本技术的主催化剂可适应较大范围的环烯烃单体使用量,催化活性高。
[0120]
本技术实施方式中,共聚合反应的温度可以是50℃-120℃;共聚合反应的时间可以是2min-10min。本技术通过采用上述催化剂进行共聚合,反应温度较温和,时间短,可以优化聚合工艺。一些实施方式中,共聚合反应的温度可以是60℃-110℃。一些实施方式中,共聚合反应的温度可以是80℃-100℃。一些实施方式中,共聚合反应的时间可以是3min-6min。
[0121]
本技术实施方式中,环烯烃单体的结构式可以是如式(2)所示:
[0122][0123]
式(2)中,r
19
为烃基或烃基硅基;r
20
和r
21
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团;
[0124]r22
和r
23
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团,或者r
22
和r
23
连接形成具有环状结构的基团;
[0125]
z为正整数。
[0126]
本技术实施方式中,r
19
为烃基或烃基硅基,烃基可以是亚烷基、亚烯基、亚芳基、亚芳烷基、亚烷芳基或亚芳烯基。烃基的碳原子数可以是小于或等于10(即碳原子数为1-10)。具体地,亚烷基、亚烯基的碳原子数可以是1、2、3、4、5、6、7、8、9、10。亚芳基、亚芳烷基、亚烷芳基、亚芳烯基的碳原子数可以是6、7、8、9、10。烃基硅基可以是烷基亚硅基、芳基亚硅基。烃基硅基的碳原子数可以是小于或等于10(即碳原子数为1-10)。一些实施例中,烃基硅基具体可以是二甲基亚硅基(-si(ch3)
2-),二乙基亚硅基(-si(c2h5)
2-),二苯基亚硅基(-si(c6h5)
2-)等。
[0127]
本技术实施方式中,可取代上述基团的原子或原子团是指可以取代氢原子、卤原子、烷基、芳基、烷氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基的原子或原子团,具体例如可以是氢原子的同位素原子(氘等)、硼烷、金属配体等。
[0128]
本技术实施方式中,r
20
、r
21
、r
22
和r
23
中,卤原子可以是氟、氯、溴或碘。烷基可以是碳原子数为1-20的烷基。在一些实施方式中,烷基的碳原子数为2-10;在另一些实施方式中,烷基的碳原子数为8-20;在其他一些实施方式中,烷基的碳原子数为8-15。烷基可以是直链、支链或具有环状结构的烷基。具体地,烷基例如可以是甲基、乙基、丙基、丁基等。烷基可以是非取代烷基,也可以是取代烷基。本技术实施方式中,芳香基可以是碳原子数为6-20的芳香基团,进一步地,芳香基的碳原子数可以是6-10;更进一步地,芳香基的碳原子数可以是7-8。芳香基可以是非取代芳香基,也可以是取代芳香基。本技术实施方式中,烷氧基的碳原子数可以是1-20。在一些实施方式中,烷氧基的碳原子数为2-10;在另一些实施方式中,烷氧基的碳原子数为8-20;在其他一些实施方式中,烷氧基的碳原子数为8-15。烷氧基可以是直链、支链或具有环状结构的烷氧基。
[0129]
本技术实施方式中,r
22
、r
23
连接形成的环状结构可以是饱和或不饱和碳环、饱和或不饱和杂环,杂环中的杂原子可以是氮、硫、氧、硼、硅等。例如,r
22
、r
23
连接形成的环状结构上连接的基团可以是包括氢原子、卤原子、烷基、芳香基、烷氧基、羟基、酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团。
[0130]
本技术实施方式中,z为正整数,具体可以是1、2、3、4等。
[0131]
本技术实施方式中,α-烯烃是指双键在分子链端部的单烯烃,α-烯烃的碳原子数可以是2-20。具体地,α-烯烃可以是丙烯、1-丁烯、1-戊烯、2-甲基-1-丁烯、3-甲基-1-丁烯、1-己烯、2-甲基-1-戊烯、3-甲基-1-戊烯、4-甲基-1-戊烯或2-乙基-1-丁烯。
[0132]
本技术实施方式中,共聚合反应体系中不包含分子量调节剂。本技术实施例的环烯烃共聚物制备方法可以仅通过本技术实施例提供的环烯烃制备用催化剂实现调节分子量的效果,获得低分子量的环烯烃共聚物。
[0133]
本技术实施方式中,以环烯烃单体与乙烯发生共聚合反应为例,其制备得到环烯烃共聚物的反应过程可以如反应式(1-3)所示:
[0134][0135]
本技术实施例提供的环烯烃共聚物的制备方法,通过采用本技术实施例提供的催化剂,不需要额外引入氢气或丙烯等分子量调节剂,即可制备获得具有低分子量和适中玻璃化转变温度的环烯烃共聚物,以满足光学透镜等各类光学制品、显示材料和包装材料等产品的加工性能、耐热性能等要求,同时环烯烃共聚物的其他性能如光学性能、热性能和机械性能等与传统的低分子量环烯烃共聚物材料保持同等水平,因此由该方法生产出的低分子量环烯烃共聚物材料可适用传统的环烯烃共聚物材料应用场景。该制备方法不仅大大简化了环烯烃共聚物材料的制备途径,还显著提高了聚合活性和能源经济效益,为大规模生产环烯烃共聚物材料开辟了一条新的路径。本技术实施例环烯烃单体与α-烯烃的聚合反应具有较高的活性。实验结果表明,本技术实施例上述方法制备得到的环烯烃共聚物具有适中的tg(110-180℃)和较低的分子量(≤15万),环烯烃单体的插入率在20%-60%之间,分子量分布指数在1.5-3.0之间。
[0136]
本技术实施例还提供一种采用上述方法制备得到的环烯烃共聚物,其结构式如式(3)所示:
[0137][0138]
式(3)中,r
19
为烃基或烃基硅基;r
20
和r
21
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团;
[0139]r22
、r
23
、r
24
、r
25
分别独立地包括氢原子、卤原子、烷基、烷氧基、芳基、芳氧基、羟基、酯基、碳酸酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团,或者r
22
和r
23
连接形成具有环状结构的基团,r
24
和r
25
连接形成具有环状结构的基团;
[0140]
x和y表示聚合度,x和y均为正数,1<x∶y<3,z为正整数。
[0141]
可以理解地,本技术实施例式(3)所示的环烯烃聚合物,其中r
19
、r
20
、r
21
、r
22
、r
23
与式(2)所示的环烯烃单体中的r
19
、r
20
、r
21
、r
22
、r
23
具体选择一致,此处不再赘述。
[0142]
本技术实施方式中,r
24
、r
25
中,卤原子可以是氟、氯、溴或碘。烷基可以是碳原子数为1-20的烷基。在一些实施方式中,烷基的碳原子数为2-10;在另一些实施方式中,烷基的
碳原子数为8-20;在其他一些实施方式中,烷基的碳原子数为8-15。烷基可以是直链、支链或具有环状结构的烷基。具体地,烷基例如可以是甲基、乙基、丙基、丁基等。烷基可以是非取代烷基,也可以是取代烷基。本技术实施方式中,芳基可以是碳原子数为6-20的芳香基团,进一步地,芳基的碳原子数可以是6-10;更进一步地,芳基的碳原子数可以是7-8。芳基可以是非取代芳香基,也可以是取代芳香基。本技术实施方式中,烷氧基的碳原子数可以是1-20。在一些实施方式中,烷氧基的碳原子数为2-10;在另一些实施方式中,烷氧基的碳原子数为8-20;在其他一些实施方式中,烷氧基的碳原子数为8-15。烷氧基可以是直链、支链或具有环状结构的烷氧基。
[0143]
本技术实施方式中,r
24
、r
25
连接形成的环状结构可以是饱和或不饱和碳环、饱和或不饱和杂环,杂环中的杂原子可以是氮、硫、氧、硼、硅等。例如,r
24
、r
25
连接形成的环状结构上连接的基团可以是包括氢原子、卤原子、烷基、芳香基、烷氧基、羟基、酯基、氰基、氨基、硫醇基、可取代上述基团的原子或原子团。
[0144]
本技术实施方式中,z为正整数,具体可以是1、2、3、4等。x和y的比值可以是,1.5<x∶y<2.5。
[0145]
本技术实施方式中,环烯烃共聚物的重均分子量小于或等于150000;分子量分布指数在1.5至3.0范围内。一些实施方式中,环烯烃共聚物的重均分子量在5000至150000范围内;一些实施方式中,环烯烃共聚物的重均分子量在10000至120000范围内。一些实施方式中,环烯烃共聚物的重均分子量在20000至100000范围内。环烯烃共聚物具有相对较低的重均分子量,加工性能更好,有利于提升商用价值。一些实施方式中,分子量分布指数在1.7-2.4的范围内。
[0146]
本技术一些实施方式中,环烯烃单体的插入率在20%-60%之间;一些实施方式中,环烯烃单体的插入率在20%-50%之间;一些实施方式中,环烯烃单体的插入率在30%-40%之间。适合的环烯烃单体插入率能够使得环烯烃聚合物具有适合的玻璃化转变温度。本技术一些实施方式中,环烯烃共聚物的玻璃化转变温度在110℃至180℃范围内。一些实施方式中,环烯烃共聚物的玻璃化转变温度在120℃至160℃范围内。一些实施方式中,环烯烃共聚物的玻璃化转变温度在130℃至150℃范围内。玻璃化转变温度在120℃至160℃范围内既能够获得较优异的加工成型性能,又能保证共聚物制品在较高的环境温度条件下使用,从而更好地兼顾加工性能和使用条件。
[0147]
本技术实施方式中,环烯烃共聚物的成型体的可见光透过率大于90%。成型体可以是通过热压形成的片状成型体;也可以是涂覆形成的膜状成型体;成型体的厚度可以是在0.1mm-1mm范围内。
[0148]
本技术中,
“‑”
表示范围,包括两个端点值。例如,共聚合的反应体系中主催化剂的用量为0.001mmol/l-10mmol/l,表示主催化剂的用量为0.001mmol/l至10mmol/l范围内的任意值,包括端点值0.001mmol/l和10mmol/l。
[0149]
本技术实施例还提供一种组合物,包括本技术实施例上述的环烯烃共聚物。该组合物可以作为光学材料。
[0150]
本技术实施方式中,所述组合物还包括添加剂,添加剂可以是包括填料、染料、抗氧化剂、光稳定剂、紫外线吸收剂、增塑剂、阻燃剂、抗静电剂、脱模剂中的一种或多种。组合物还可以包括其他聚合物,其他聚合物可以是与本技术实施例不同的其他的环烯烃聚合
物、也可以是非环烯烃聚合物,具体可根据需要适量加入。组合物中,本技术实施例上述的环烯烃聚合物的质量含量可以是大于或等于60%。一些实施例中,本技术实施例上述的环烯烃聚合物的质量含量可以是60%、70%、80%、90%、95%、98%。
[0151]
本技术实施例还提供一种光学制品,光学制品包括本技术实施例上述的环烯烃共聚物。可以通过各种已知的成型方法将上述环烯烃共聚物或组合物加工成光学制品。光学制品可以是局部采用上述环烯烃共聚物或组合物加工制成,也可以是整体均采用上述环烯烃聚合物或光学材料进行加工获得。
[0152]
本技术实施方式中,光学制品具体可包括光学透镜、光学膜、光盘、导光板或显示面板。
[0153]
本技术实施方式中,光学透镜具体可以包括眼镜透镜、相机透镜、传感器透镜、照明透镜、成像透镜等。相机透镜具体可以是手机相机透镜、笔记本电脑相机透镜、台式相机透镜、汽车相机透镜等。其中,眼镜透镜可以包括近视眼镜透镜、老花镜透镜、太阳镜透镜、隐形眼镜矫正透镜、护目镜透镜等。其中,传感器透镜可以是运动检测器透镜、接近传感器透镜、姿态控制透镜、红外传感器透镜等。其中,照明透镜可以是室内照明透镜、室外照明透镜、车辆前照灯透镜、车辆雾灯透镜、车辆后照灯透镜、车辆行车灯透镜、车辆雾灯透镜、车辆内部透镜、发光二极管(led)透镜或有机发光二极管(oled)透镜等。其中,成像透镜可以是扫描仪透镜、投影仪透镜、望远镜透镜、显微镜透镜、放大镜透镜等。
[0154]
本技术实施方式中,光学膜可以包括导光膜、反射膜、增透膜、扩散膜、滤光膜、偏振膜、分光膜和位相膜等。光学膜可以用于显示领域、照明领域等,例如可以用于液晶基板用膜。
[0155]
参见图1,本技术实施例还提供一种设备100,包括本技术实施例上述的光学制品。该设备100可以是电子设备,具体可以是包括移动终端、眼镜、相机、车辆(例如汽车、摩托车、火车等)、照明设备(例如台灯、天花板灯、路灯等)、成像设备(例如内窥镜、显微镜、望远镜、投影仪、扫描仪等)、安防设备等。其中,移动终端可以具体包括各种具有无线通信功能的手持设备(如各类手机、平板电脑、移动笔记本、上网本)、可穿戴设备(如智能手表)、或连接到无线调制解调器的其他处理设备,以及各种形式的用户设备(user equipment,ue),移动台(mobile station,ms),终端设备(terminal device)等。设备100包括摄像头模组2,摄像头模组2包括摄像头镜片,摄像头镜片采用本技术实施例上述环烯烃共聚物制备。设备100还包括盖设在摄像头镜片上的摄像头保护盖板103。
[0156]
本技术一具体实施例中,设备100为移动终端,移动终端包括摄像头模组,摄像头模组包括摄像头镜片,摄像头镜片采用本技术实施例上述环烯烃共聚物制备。
[0157]
本技术一具体实施例中,设备100为内窥镜,内窥镜包括摄像头模组,摄像头模组包括摄像头镜片,摄像头镜片采用本技术实施例上述环烯烃共聚物制备。
[0158]
本技术一具体实施例中,设备100为车辆,车辆包括摄像头模组,摄像头模组包括摄像头镜片,摄像头镜片采用本技术实施例上述环烯烃共聚物制备。
[0159]
本技术一具体实施例中,设备100为安防设备,安防设备包括摄像头模组,摄像头模组包括摄像头镜片,摄像头镜片采用本技术实施例上述环烯烃共聚物制备。
[0160]
下面分多个实施例对本技术实施例进行进一步的说明。
[0161]
实施例1
[0162]
催化剂制备:
[0163]
(1a)无水无氧条件下,在100ml反应容器中,-78℃反应温度下,加入5mmol的2,7-二溴芴和50ml无水四氢呋喃,逐滴滴加含10mmol正丁基锂的己烷溶液,随后逐滴滴加含10mmol三甲基氯硅烷(me3sicl)的四氢呋喃溶液,反应过夜。然后再次逐滴滴加含10mmol正丁基锂的己烷溶液和含10mmol三苯基氯硅烷(ph3sicl)的四氢呋喃溶液,反应过夜。随后,在室温下加入50ml的0.5mol/l氢氧化钠水溶液进行水解,用乙醚萃取分液,有机相用无水硫酸镁干燥后得到2,7-双(三苯基硅基)芴。
[0164]
(1b)无水无氧条件下,在100ml反应容器中,加入5mmol所制备的2,7-双(三苯基硅基)芴和50ml无水四氢呋喃,在室温下逐滴滴加等摩尔量的甲基锂乙醚溶液(1.4mol/l),室温过夜反应,然后逐滴加入20ml溶有5mmol 6,6-二甲基富烯的无水四氢呋喃溶液,反应过夜,加入100ml四丁基氯化铵水溶液,搅拌10min,萃取分液,水相用50ml乙醚水洗三次,有机相用无水硫酸镁干燥后,干燥重结晶,得到环戊二烯芴配体前体。
[0165]
(1c)无水无氧条件下,在100ml反应容器中,加入5mmol上述制备的环戊二烯芴配体前体和50ml无水四氢呋喃,在-78℃下加入等摩尔量的正丁基锂己烷溶液,随后逐滴滴加三甲基氯硅烷(me3sicl)5ml,搅拌过夜,抽干溶剂,用己烷洗涤1-3次,得到催化剂a前体。
[0166]
(1d)在无水无氧的条件下,向100ml反应容器中加入1g催化剂a前体,0℃反应温度下加入3.4ml正丁基锂,缓慢升温至室温后反应2-12小时后抽干溶剂,移入手套箱内加入己烷,充分搅拌下加四氯化锆0.6g,搅拌过夜。过夜反应后过滤,滤饼用己烷洗涤后溶于过量甲苯中,过滤出土黄色不溶物,然后浓缩甲苯溶液,重结晶得到桃红色固体催化剂a。本实施例得到的桃红色固体催化剂a0.3g,产率为19.7%,纯度为93.5%,图2是催化剂a的核磁共振氢谱。图3是催化剂a的核磁共振碳谱。图2的核磁共振氢谱和图3的核磁共振碳谱表明催化剂a成功制备。
[0167][0168]
环烯烃单体制备:在220ml高压釜中,依次加入78g双环戊二烯、110g降冰片烯和少量的2,6-二甲氧基苯酚(bht),在氮气的氛围下220℃加热反应24小时,反应完成后将反应体系的温度降至室温,直接减压蒸馏,先前馏分为未反应的双环戊二烯,之后馏分为环烯烃单体目标产物,反应过程如反应式(1-4)所示。本实施例得到的环烯烃单体为无色液体128g,收率67%,图4为本实施例环烯烃单体的核磁共振氢谱;图5为本技术实施例1的环烯烃单体的核磁共振碳谱。图4的核磁共振氢谱和图5的核磁共振碳谱表明环烯烃单体成功制备。
[0169][0170]
环烯烃共聚物制备:将装有2.5g上述环烯烃单体、2.5ml的mao溶液(1.5mol/l,溶于甲苯中)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入2.0mg本实施例制备的催化剂a的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倒入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物材料。聚合反应的过程如反应式(1-5)所示。图6为本技术实施例1的环烯烃共聚物的核磁共振氢谱;图7为本技术实施例1的环烯烃共聚物的核磁共振碳谱。图7中a)为虚线区域的放大图。图6的核磁共振氢谱和图7的核磁共振碳谱表明环烯烃聚合物成功制备。
[0171][0172]
本实施例制得的环烯烃共聚物材料的质量为2.4g,催化剂的聚合活性为1.4*107g mol-1
h-1
。采用高温凝胶色谱法检测环烯烃共聚物材料的相对数均分子量为26kg/mol(重均分子量为62.4kg/mol),分子量分布指数为2.4(分子量分布指数等于重均分子量除以数均分子量)。采用高温核磁碳谱对得到的环烯烃共聚物进行检测,结果表明,本实施例制备的coc材料的环烯烃单体的插入率为33%,插入率=(y/(x+y))*100%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,图8为本技术实施例1的环烯烃共聚物的dsc曲线,结果表明,本实施例制备的环烯烃共聚物的玻璃化转变温度为141.02℃,由图8也可以看出dsc曲线没有结晶峰出现,这是由于采用本技术实施例的主催化剂提升了coc材料的环烯烃单体插入率,从而有利于避免结晶性聚乙烯链段的生成。将本技术实施例的环烯烃共聚物成型成厚度为0.1mm-1mm的膜或片样品,经检测,本实施例制得的环烯烃共聚物膜或片样品的可见光透过率大于90%,图9为本技术实施例1的环烯烃共聚物的可见光透过率测试曲线。
[0173]
相比于现有技术,本技术实施例无需外部分子量调节剂,通过采用催化剂a即可直接实现低分子量环烯烃共聚物材料的制备,同时保证了环烯烃共聚物材料其他性能优异。
[0174]
实施例2
[0175]
催化剂和环烯烃单体的合成步骤与实施例1相同。
[0176]
环烯烃共聚物制备:将装有3.5g环烯烃单体、2.5ml的mao(1.5mol/l,溶于甲苯中)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入2.0mg催化剂a的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物材料。
[0177]
本实施例制得的环烯烃共聚物的质量为3.28g,催化剂的聚合活性为1.9*107g mol-1
h-1
。采用高温凝胶色谱法检测环烯烃共聚物的相对数均分子量为58kg/mol(重均分子量为98.6kg/mol),分子量分布指数为1.7。采用高温核磁碳谱对得到的环烯烃共聚物进行检测,结果表明,本实施例制备的环烯烃共聚物材料的环烯烃单体的插入率为38%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,图10为本技术实施例2的环烯烃共聚物的dsc曲线,结果表明,环烯烃共聚物的玻璃化转变温度为141.43℃。经检测,本实施例制得的环烯烃共聚物的可见光透过率大于90%。
[0178]
相比实施例1,实施例2增加了环烯烃单体的量,通过实施例1和实施例2可知,采用本技术实施例的主催化剂在一个较宽的单体浓度范围内,均可实现低分子量、适合玻璃化转变温度coc的制备。
[0179]
实施例3
[0180]
催化剂和环烯烃单体的合成步骤与实施例1相同。
[0181]
环烯烃共聚物制备:将装有2.5g环烯烃单体、2.6ml的mmao溶液(8%wt,溶于庚烷中)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入2.0mg催化剂a的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物。
[0182]
本实施例制得的环烯烃共聚物的质量为2.97g,催化剂的聚合活性为1.8*107g mol-1
h-1
。采用高温凝胶色谱法去检测本实施例得到的环烯烃共聚物材料的相对数均分子量为35kg/mol(重均分子量为84kg/mol),分子量分布指数为2.4。采用高温核磁碳谱对得到的环烯烃共聚物进行检测,结果表明,本实施例制备的环烯烃共聚物材料的环烯烃单体的插入率为30%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,图11为本技术实施例3的环烯烃共聚物的dsc曲线,结果表明,本实施例制备的环烯烃共聚物的玻璃化转变温度为129.81℃。经检测,本实施例制得的环烯烃共聚物的可见光透过率大于90%。
[0183]
实施例3相比实施例1改变了助催化剂,由实施例1和实施例3可知,以本技术实施例催化剂a作为主催化剂,在采用不同的助催化剂时,均可实现低分子量、适合玻璃化转变温度coc的制备。
[0184]
实施例4
[0185]
催化剂和环烯烃单体的合成步骤与实施例1相同。
[0186]
环烯烃共聚物制备:将装有3.5g环烯烃单体、2.6ml的mmao(8%wt庚烷)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入1.2mg本发明制备的催化剂的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物材料。
[0187]
本实施例制得的环烯烃共聚物的质量为1.96g,催化剂的聚合活性为1.2*107g mol-1
h-1
。采用高温凝胶色谱法检测本实施例得到的环烯烃共聚物材料的相对数均分子量为26kg/mol(重均分子量为62.4kg/mol),分子量分布指数为2.4。采用高温核磁碳谱对得到的环烯烃共聚物进行检测得到环烯烃单体的插入率为39%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,图12为本技术实施例4的环烯烃共聚物的dsc曲线,结果表明,本实施例制备的环烯烃共聚物材料的玻璃化转变温度为145.67℃。经检测,本实施例制得的环烯烃共聚物的可见光透过率大于90%。
[0188]
实施例4与实施例1相比改变了助催化剂和环烯烃单体用量,结果表明,采用本技术实施例的主催化剂,同时更换助催化剂和环烯烃单体用量,均可实现低分子量、适合玻璃化转变温度coc的制备,本技术实施例提供的主催化剂适用范围大,效果好。
[0189]
实施例5
[0190]
催化剂制备:
[0191]
(1a)无水无氧条件下,在100ml反应容器中,-78℃反应温度下,加入5mmol的2,7-二溴芴和50ml无水四氢呋喃,逐滴滴加含10mmol正丁基锂的己烷溶液,随后逐滴滴加含10mmol三甲基氯硅烷(me3sicl)的四氢呋喃溶液,反应过夜。然后再次逐滴滴加含10mmol正丁基锂的己烷溶液和含10mmol三甲基氯硅烷(me3sicl)的四氢呋喃溶液,反应过夜。随后,在室温下加入50ml的0.5mol/l氢氧化钠水溶液进行水解,用乙醚萃取分液,有机相用无水硫酸镁干燥后得到2,7-双(三甲基硅基)芴。
[0192]
(1b)至(1d)同实施例1。
[0193][0194]
环烯烃单体的合成步骤与实施例1相同。
[0195]
环烯烃共聚物制备:将装有2.5g环烯烃单体、2.5ml的mao(1.5mol/l的甲苯溶液)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅
拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入1.3mg催化剂b的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物材料。
[0196]
本实施例制得的环烯烃共聚物的质量为1.99g,催化剂的聚合活性为1.2*107g mol-1
h-1
。采用高温凝胶色谱法检测本实施例得到的环烯烃共聚物材料的相对数均分子量为35kg/mol(重均分子量为70kg/mol),分子量分布指数为2.0。采用高温核磁碳谱对得到的环烯烃共聚物进行检测得到环烯烃单体的插入率为31%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,图13为本技术实施例5的环烯烃共聚物的dsc曲线,结果表明,本实施例制备的环烯烃共聚物材料的玻璃化转变温度为131.51℃。经检测,本实施例制得的环烯烃共聚物的可见光透过率大于90%。
[0197]
实施例6
[0198]
催化剂制备:
[0199]
(1a)无水无氧条件下,在100ml反应容器中,-78℃反应温度下,加入5mmol的2,7-二溴芴和50ml无水四氢呋喃,逐滴滴加含10mmol正丁基锂的己烷溶液,随后逐滴滴加含10mmol三甲基氯硅烷(me3sicl)的四氢呋喃溶液,反应过夜。然后再次逐滴滴加含10mmol正丁基锂的己烷溶液和含10mmol三乙基氯硅烷(et3sicl)的四氢呋喃溶液,反应过夜。随后,在室温下加入50ml的0.5mol/l氢氧化钠水溶液进行水解,用乙醚萃取分液,有机相用无水硫酸镁干燥后得到2,7-双(三乙基硅基)芴。
[0200]
(1b)至(1d)同实施例1。
[0201][0202]
环烯烃单体的合成步骤与实施例1相同。
[0203]
环烯烃共聚物制备:将装有2.5g环烯烃单体、2.5ml的mao(1.5mol/l的甲苯溶液)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入1.5mg催化剂c的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物材料。
[0204]
本实施例制得的环烯烃共聚物的质量为0.62g,催化剂的聚合活性为3.7*106g mol-1
h-1
。采用高温凝胶色谱法检测本实施例得到的环烯烃共聚物材料的相对数均分子量为44kg/mol(重均分子量为70.4kg/mol),分子量分布指数为1.6。采用高温核磁碳谱对得到的环烯烃共聚物进行检测得到环烯烃单体的插入率为37%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,结果表明,本实施例制备的环烯烃共聚物材料的玻璃化转变温度为145.7℃。经检测,本实施例制得的环烯烃共聚物的可见光透过率大于90%。
[0205]
实施例7
[0206]
催化剂制备:
[0207]
与实施例1的区别仅在于不需要进行步骤(1c),直接将步骤(1b)得到的环戊二烯芴配体前体作为催化剂d前体进行步骤(1d)。
[0208][0209]
环烯烃单体的合成步骤与实施例1相同。
[0210]
环烯烃共聚物制备:将装有2.5g环烯烃单体、2.5ml的mao(1.5mol/l的甲苯溶液)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,加入1.9mg催化剂d的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流2小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色coc材料。
[0211]
本实施例制得的环烯烃共聚物的质量为1.3g,催化剂的聚合活性为7.8*106g mol-1
h-1
。采用高温凝胶色谱法检测本实施例得到的环烯烃共聚物材料的相对数均分子量为11.3kg/mol(重均分子量为21.5kg/mol),分子量分布指数为1.9。采用高温核磁碳谱对得到的环烯烃共聚物进行检测得到环烯烃单体的插入率为35%。采用示差扫描量热法(dsc)对得到的环烯烃共聚物的玻璃化转变温度进行检测,结果表明,本实施例制备的环烯烃共聚物材料的玻璃化转变温度为142℃。经检测,本实施例制得的环烯烃共聚物的可见光透过率大于90%。
[0212]
对比例1
[0213]
采用现有催化剂i(亚异丙基桥联的环戊二烯芴二氯化锆)进行环烯烃共聚物制备。
[0214][0215]
环烯烃共聚物制备:在将装有4.5g环烯烃单体、3.4ml的mao(1.5mol/l的甲苯溶液)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为50℃,在通乙烯的条件下加入0.9mg催化剂i的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流俩小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物。
[0216]
该对比例制得的环烯烃共聚物的质量为1.78g,催化剂的聚合活性为1.1*107g mol-1
h-1
。采用与上述实施例相同的方法测得环烯烃共聚物的数均分子量为124kg/mol(重均分子量为248kg/mol),分子量分布指数为2.0。环烯烃单体的插入率为33%。环烯烃共聚物的玻璃化转变温度为136.98℃。
[0217]
对比例2
[0218]
环烯烃共聚物制备:将装有4.5g环烯烃单体、1.7ml的mao(1.5m的甲苯溶液)和45ml甲苯的玻璃反应釜接入乙烯管道中,用氮气置换三次乙烯管道后开通乙烯气体和搅拌使玻璃釜内甲苯溶液对乙烯饱和,调节聚合温度为90℃,在通乙烯的条件下加入0.9mg催化剂i的2ml甲苯溶液,调节并保持乙烯的压力为一个大气压,聚合反应5分钟。聚合完成后,将得到的反应液倾入10%的盐酸水溶液中,充分搅拌后分液,有机层用水再洗两遍;将得到的有机层用丙酮充分搅拌下沉降,过滤后加入适量的丙酮回流俩小时,最后聚合物过滤并用丙酮洗三次,将产品置于真空干燥箱内,130℃干燥18小时,得到白色环烯烃共聚物。
[0219]
该对比例制得的环烯烃共聚物的质量为1.15g,催化剂的聚合活性为0.7*107g mol-1
h-1
。采用与上述实施例相同的方法测得环烯烃共聚物的数均分子量为117kg/mol(重均分子量为199kg/mol),分子量分布指数为1.7。环烯烃单体的插入率为35%。环烯烃共聚物的玻璃化转变温度为141.24℃。
[0220]
由对比例1和对比例2可知,采用对比例1的催化剂i进行环烯烃共聚物制备,在不同助催化剂使用量和聚合反应温度下,制得的环烯烃共聚物的重均分子量均远大于150kg/mol(即15万),分子量较大。而实施例1-7通过采用本技术实施例提供的催化剂,可以获得重均分子量小于15万的具有较小分子量的环烯烃共聚物,这主要是由于主催化剂环戊二烯基或芴基上的含硅基团能够通过与金属中心空轨道产生弱的配位作用,与烯烃/金属中心之间的配位产生竞争,从而促进链转移,降低聚合物分子量;同时,通过这种竞争作用,增加了烯烃与金属中心配位的难度,提升了环烯烃单体的插入率,从而有利于调节环烯烃共聚物的玻璃化转变温度。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1