一种瑞德西韦重要中间体制备方法与流程
1.本发明属于医药合成领域,特别涉及一种瑞德西韦重要中间体制备方法及其两种新的化合物。
背景技术:
2.瑞德西韦(remdesivir)是由美国吉利德公司开发,最早的研发初衷是抗埃博拉病毒。但随着研究的深入,人们发现瑞德西韦的抗病毒效果并不仅限于埃博拉病毒这类丝状病毒,其对于冠状病毒等多种病毒也有抑制效果。2020年5月1日,fda为瑞德西韦发放了治疗新冠肺炎的紧急使用授权。
[0003][0004]
文献jmed.chem.2017,60,1648-1661.报道了瑞德西韦的合成路线。
[0005]
第一代的路线如下
[0006][0007]
第二代路线如下
[0008]
[0009]
通过这两代反应,我们发现杂环化合物上面的氨基对糖环的加成都存在收率偏低的问题,这个就造成了对后续路线的制约。而收率偏低主要是由于杂环上面裸露的氨基的影响。包括后续的氰化反应,脱苄反应均因为氨基的活泼氢这个因素导致收率偏低。采用新的反应底物就势在必行。
技术实现要素:
[0010]
本发明一方面提供了瑞德西韦重要中间体化合物i(3ar,4r,6r,6ar)-4-(4-氨基吡咯并[2,1-f][1,2,4]三嗪-7-基)-6-(羟甲基)-2,2-二甲基四氢呋喃并[3,4-d][1,3]二氧杂环戊烷-4-甲腈的一种新的合成路线。
[0011]
本发明提供了一种瑞德西韦中间体i的制备方法,其包括如下步骤:
[0012][0013]
本发明还提供了一种瑞德西韦中间体ii的制备方法,其包括如下步骤:
[0014][0015]
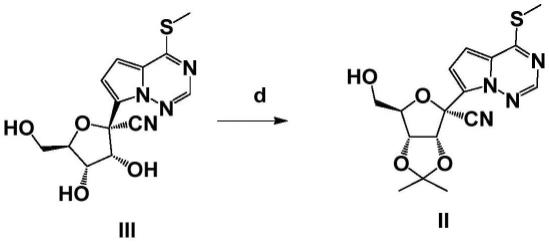
本发明还提供了一种瑞德西韦中间体iii的制备方法,其包括如下步骤:
[0016][0017]
作为本发明一种可选实施方案,本发明还提供了如下的瑞德西韦中间体i的制备方法,其包括如下步骤:
[0018][0019]
具体方案如下:
[0020]
首先步骤a化合物vi在大位阻强碱作用下与化合物vii反应生成化合物v,
[0021]
优选地,步骤a中,所述大位阻强碱为二异丙基氨基锂、二(三甲基硅基)氨基钠、双(三甲基硅基)氨基锂、双(三甲基硅基)氨基钠或四甲基哌啶锂等。
[0022]
接着步骤b在路易斯酸作用下,将化合物v中的羟基取代成氰基得到化合物iv,
[0023]
优选地,步骤b中,所述路易斯酸为三氟化硼乙醚,tmsotf,tbsotf
[0024]
步骤c脱保护反应得到化合物iii,
[0025]
优选地,步骤c中,所述的脱保护试剂为钯碳和氢氧化钯碳联用,其比例在于钯碳:氢氧化钯碳质量比=1.0:0.5~1.0,所用溶剂为甲醇,乙醇。
[0026]
步骤d上保护生成化合物ii,
[0027]
优选地,步骤d中,所述的催化剂为三氟化硼乙醚,对甲苯磺酸,苯磺酸,硫酸铜,保护试剂为丙酮,2,2-二甲氧基丙烷。
[0028]
步骤e最后经取代反应得到化合物i。
[0029]
优选地,步骤e中,所述的氨源为醋酸铵,氨甲醇溶液,硫酸铵,碳酸氢铵。
[0030]
进一步地,步骤b中,所述路易斯酸为三氟化硼乙醚,tmsotf,tbsotf。
[0031]
进一步地,步骤c中,所述的脱保护试剂为钯碳和氢氧化钯碳联用,其比例在于钯碳:氢氧化钯碳质量比=1.0:0.5~1.0,所用溶剂为甲醇,乙醇。
[0032]
进一步地,步骤d中,所述的催化剂为三氟化硼乙醚,对甲苯磺酸,苯磺酸,硫酸铜,保护试剂为丙酮,2,2-二甲氧基丙烷。
[0033]
进一步地,步骤e中,所述的氨源为醋酸铵,氨甲醇溶液,硫酸铵,碳酸氢铵。
[0034]
另一方面,本发明还提供新的瑞德西韦中间体化合物iii与化合物ii:
[0035]
[0036]
综上,本发明提供了一种新的合成瑞德西韦中间体的方法,其以甲硫基取代的杂环为起始物料,经位阻强碱拔氢和糖环反应,氰化反应,脱苄基反应,上丙酮叉保护,和最后的取代氨解反应,可以高效率的拿到瑞德西韦的中间体i。因为不再使用含有活泼氢的氨基做反应物,第一步的杂环和糖环的加成,第二步氰化反应,第三步的脱苄基反应均高于文献报道中直接使用裸露氨基的收率。第四步上丙酮叉保护,利用丙酮作为保护源,成本低廉且反应收率高,操作简单。第五步氨解使用醋酸铵和氨甲醇溶液做氨源,反应后处理简单方便,反应转化率高。所以此路线整体是一条非常高效简洁的路线,非常适合工业化生产。
具体实施方式
[0037]
为了使本领域技术人员可以更好地理解本发明,以下通过具体实施例对本发明技术方案进行进一步说明。需要理解的是,下属实施例只为更好地说明本发明而给出,并不是对本发明内容的限制。
[0038]
实施例1:
[0039]
化合物v的合成
[0040][0041]
n2保护下,加入化合物vi(21g,127mmol),500mlthf搅拌溶解,冷却至约-70℃,加入2m lda thf溶液(95ml,190mmol),搅匀溶解,略升温,继续降温至-70℃,缓慢滴入化合物vii(2,3,5-三苄氧基-d-核糖酸-1,4-内酯)/thf(54g,129mmol/420ml)溶液,滴毕,保温3小时,tlc检测,加入氯化铵水溶液,调节ph为8,搅拌30分钟,分层,下层水层用250ml乙酸乙酯萃取两次,合并有机层,加入无水硫酸钠干燥,过滤,滤液旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),浓缩得70.3g黄色油状物化合物v,hplc纯度98%,收率95%。
[0042]
esi-hrms理论值:c
33h33
n3o5s[m+h]
+
584.2141,实测值584.2143。
[0043]
实施例2:
[0044]
化合物v的合成
[0045][0046]
n2保护下,加入化合物vi(40g,242mmol),600mlthf搅拌溶解,冷却至约-78℃,加
入1m双(三甲基硅基)氨基锂的thf溶液(266ml,266mmol),搅匀溶解,略升温,继续降温至-78℃,缓慢滴入化合物vii(2,3,5-三苄氧基-d-核糖酸-1,4-内酯)/thf(92g,220mmol/500ml)溶液,滴毕,保温2.5小时,tlc检测,加入氯化铵水溶液,调节ph为7,搅拌1小时,分层,下层水层用300ml乙酸乙酯萃取两次,合并有机层,加入无水硫酸钠干燥,过滤,滤液旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),浓缩得119g黄色油状物化合物v,hplc纯度97%,收率93%。
[0047]
esi-hrms理论值:c
33h33
n3o5s[m+h]
+
584.2141,实测值584.2143。
[0048]
实施例3:
[0049]
化合物v的合成
[0050][0051]
n2保护下,加入化合物vi(35g,212mmol),100mlthf搅拌溶解,冷却至约-78℃,加入1m双(三甲基硅基)氨基钠的thf溶液(266ml,233mmol),搅匀溶解,略升温,继续降温至-78℃,缓慢滴入化合物vii(2,3,5-三苄氧基-d-核糖酸-1,4-内酯)/thf(83.6g,200mmol/200ml)溶液,滴毕,保温2小时,tlc检测,加入氯化铵水溶液,调节ph为7,搅拌1小时,分层,下层水层用220ml乙酸乙酯萃取两次,合并有机层,加入无水硫酸钠干燥,过滤,滤液旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),浓缩得106g黄色油状物化合物v,hplc纯度97%,收率91%。
[0052]
esi-hrms理论值:c
33h33
n3o5s[m+h]
+
584.2141,实测值584.2143。
[0053]
实施例4:
[0054]
化合物iv的合成
[0055][0056]
将化合物v(40g,68.5mmol),300mldcm搅拌溶解,冷却至约-20℃,,缓慢滴入三氟化硼乙醚(1.95g,13.7mmol,0.2equiv),保持温度-20℃,滴毕,保温30分钟,缓慢滴入tmscn(8.1g,82.2mmol,1.2equiv),滴毕保温1h。加入三乙胺(20ml),缓慢升温至室温,加入碳酸氢钠饱和水溶液(150ml)淬灭分液,150ml水洗,搅拌30分钟,过滤分层,水相用200ml二氯甲烷萃取两次,合并有机相,加入无水硫酸钠干燥,过滤,滤液旋干,经过硅胶柱层析(乙酸乙
酯/石油醚洗脱),浓缩得39.4克,hplc纯度99%,收率97%。
[0057]
esi-hrms理论值:c
34h32
n4o4s[m+h]
+
593.2144,实测值593.2142。1h-nmr(400mhz,cdcl3)δ8.18(s,1h),7.43-7.21(m,15h),6.80(d,j=4.7hz,1h),6.68(d,j=4.7hz,1h),4.71(s,2h),4.65-4.37(m,5h),4.35-4.13(m,2h),3.88(dd,j=10.5,2.9hz,1h),3.74(dd,j=10.5,3.5hz,1h),2.65(s,3h)。
[0058]
实施例5:
[0059]
化合物iv的合成
[0060][0061]
将化合物v(40g,68.5mmol),300mldcm搅拌溶解,冷却至约-78℃,,缓慢滴入tmsotf(31.3g,140.8mmol),保持温度-78℃,滴毕,保温30分钟,缓慢滴入tmscn(29.3g,294.7mmol),滴毕保温2h。加入三乙胺(36ml),缓慢升温至室温,加入碳酸氢钠(55g),150ml水,搅拌30分钟,过滤分层,水相用250ml二氯甲烷萃取两次,合并有机相,加入无水硫酸钠干燥,过滤,滤液旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),浓缩得36.9克,hplc纯度98%,收率91%。
[0062]
esi-hrms理论值:c
34h32
n4o4s[m+h]
+
593.2144,实测值593.2142。1h-nmr(400mhz,cdcl3)δ8.18(s,1h),7.43-7.21(m,15h),6.80(d,j=4.7hz,1h),6.68(d,j=4.7hz,1h),4.71(s,2h),4.65-4.37(m,5h),4.35-4.13(m,2h),3.88(dd,j=10.5,2.9hz,1h),3.74(dd,j=10.5,3.5hz,1h),2.65(s,3h)。
[0063]
实施例6:
[0064]
化合物iii的合成
[0065][0066]
化合物iv(31g,52.3mmol)在甲醇(100ml)搅拌溶解,加入3.1g钯碳(5%)和3.1g氢氧化钯碳(20%)在0.4mpa氢气搅拌1.5小时,tlc显示原料反应完,过滤浓缩得36.4克,hplc纯度96%,收率99%。
[0067]
esi-hrms理论值:c
13h14
n4o4s[m+h]
+
323.0736,实测值323.0740。
[0068]
实施例7:
ii,hplc纯度95%,收率94%。
[0082]
esi-hrms理论值:c
16h18
n4o4s[m+h]
+
363.1049,实测值362.1046。
[0083]
实施例10:
[0084]
化合物i的合成
[0085][0086]
化合物ii(10g,27.6mmol),加入100ml甲醇溶解,加入醋酸铵(21.3g,276mmol)至高压釜,升温至120℃,搅拌过夜,旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),旋干得9.04g i,hplc纯度98%,收率99%。
[0087]
esi-hrms理论值:c
15h17
n5o4[m+h]
+
332.1281,实测值332.1284。1h-nmr(400mhz,cd3od):δ7.94(s,1h),7.09(d,j=4.6hz,1h),6.65(d,j=4.6hz,1h),5.80(s,2h),5.43(d,j=6.6hz,1h),5.24(dd,j=6.6,2.4hz,1h),4.69
–
4.65(m,1h),4.53(s,1h),3.99(dd,j=12.5,1.8hz,1h),3.85(d,j=12.5hz,1h),1.81(s,3h),1.40(s,3h)。
[0088]
实施例11:
[0089]
化合物i的合成
[0090][0091]
化合物ii(10g,27.6mmol),加入100ml甲醇溶解,加入醋酸铵(21.3g,276mmol)至高压釜,升温至120℃,搅拌过夜,旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),旋干得8.68g i,hplc纯度96%,收率95%。
[0092]
esi-hrms理论值:c
15h17
n5o4[m+h]
+
332.1281,实测值332.1284。1h-nmr(400mhz,cd3od):δ7.94(s,1h),7.09(d,j=4.6hz,1h),6.65(d,j=4.6hz,1h),5.80(s,2h),5.43(d,j=6.6hz,1h),5.24(dd,j=6.6,2.4hz,1h),4.69
–
4.65(m,1h),4.53(s,1h),3.99(dd,j=12.5,1.8hz,1h),3.85(d,j=12.5hz,1h),1.81(s,3h),1.40(s,3h)。
[0093]
实施例12:
[0094]
化合物i的合成
[0095][0096]
化合物ii(10g,27.6mmol),加入100ml甲醇溶解,加入7m氨的甲醇溶液(16ml,112mmol),至高压釜,升温至110℃,搅拌过夜,旋干,经过硅胶柱层析(乙酸乙酯/石油醚洗脱),旋干得8.40g i,hplc纯度96%,收率92%。
[0097]
esi-hrms理论值:c
15h17
n5o4[m+h]
+
332.1281,实测值332.1284。1h-nmr(400mhz,cd3od):δ7.94(s,1h),7.09(d,j=4.6hz,1h),6.65(d,j=4.6hz,1h),5.80(s,2h),5.43(d,j=6.6hz,1h),5.24(dd,j=6.6,2.4hz,1h),4.69
–
4.65(m,1h),4.53(s,1h),3.99(dd,j=12.5,1.8hz,1h),3.85(d,j=12.5hz,1h),1.81(s,3h),1.40(s,3h)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1