作为降糖药合成原料的5-溴-2-氯-苯甲酸的制备方法与流程
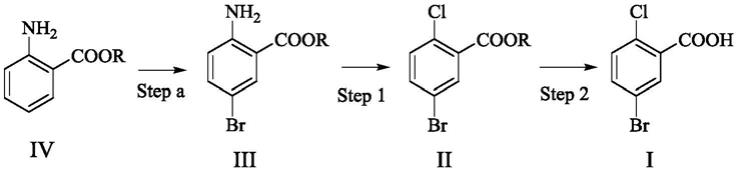
作为降糖药合成原料的5
‑
溴
‑2‑
氯
‑
苯甲酸的制备方法
技术领域
1.本发明涉及药物中间体的合成领域,特别是涉及作为降糖药合成原料的5
‑
溴
‑2‑
氯
‑
苯甲酸的制备方法及其应用。
背景技术:
2.式i化合物5
‑
溴
‑2‑
氯
‑
苯甲酸是制备抗糖尿病药物达格列净(dapagliflozin)和恩格列净(empagliflozin)的重要原料。
[0003][0004]
已报道的式i化合物的制备方法,主要有如下几种。
[0005]
方法一:中国专利cn105622382b报道了将2
‑
氯
‑
三氯甲基
‑
苯溴化,然后水解制备式i化合物。但反应原料2
‑
氯
‑
三氯甲基
‑
苯的成本相对较高,且较难水解完全,反应会产生较多的异构体杂质,需要进一步重结晶除去,从而影响收率和产品纯度。
[0006][0007]
方法二:中国专利cn107954852a、cn110105193a和cn110590541a都报道了用2
‑
氯苯甲酸为原料,采用例如溴化钠和高碘酸钠、溴、n
‑
溴代丁二酰亚胺、二溴海因等不同的溴化试剂进行溴化,但由于该反应的本质决定,溴化反应时都会产生约10%左右的异构体杂质,式i化合物产品收率和纯度受到影响,且会产生较多废酸,不适合工业化生产。
[0008][0009]
虽然中国专利cn110002989b报道了在2
‑
氯苯甲酸为原料,nbs/硫酸为反应体系下,加入抑制剂硫化钠、硫化钾或亚硫酸钠,可以高效制备式i化合物,抑制4
‑
溴
‑2‑
氯苯甲酸杂质的生成。但抑制剂在强酸体系是否存在,以及是否会产生4
‑
溴
‑2‑
氯苯甲酸杂质,有待商榷。
[0010]
方法三:中国专利cn108250060a报道了以水杨酸为原料制备式i化合物方法。虽然此方法可以有效减少异构体生成,但是氯化步骤使用120
‑
180℃较高的反应温度,对设备要求较高,且使用不环保的氯代试剂四氯化碳、三氯化硼、三氯化磷等,因此该方法并不理想。
[0011][0012]
方法四:中国专利cn111925289a报道了以2
‑
氯苯甲酸为原料,依次经过氯化、酰胺化、成环、溴化和水解制备式i化合物。该路线反应步骤较长,氯化反应时使用具有强腐蚀性的酰氯试剂,溴化反应时使用危险的丁基锂试剂,成本较高,生产周期长,不适合工业化生产。
[0013][0014]
方法五:中国专利cn112979448a报道了以2
‑
氯苯甲酸为原料、以二溴代氨基硅胶为溴化试剂来制备式i化合物的方法。该方法中氨基硅胶原料成本较高,制备二溴代氨基硅胶方法中使用了溴和2倍当量以上的碳酸钾作为缚酸剂,虽然氨基硅胶可以回收套用,但最终是作为固体废弃物处理。而且,溴代反应使用了1
‑
氯丁烷和1
‑
溴丁烷为反应溶剂,三氟甲磺酸铁为催化剂,60
‑
70℃反应,导致成本较高。
[0015][0016]
综上所述,目前已报道的式i化合物的制备方法存在反应步骤长、不够环保、异构体杂质较多、成本较高等问题,给工业化规模生产带来一定难度。因此,有必要进一步研究适合式i化合物的工业化生产的简单高效的合成方法。
技术实现要素:
[0017]
为了克服现有技术所存在的缺点与不足,本发明提供了一种新的合成式i化合物5
‑
溴
‑2‑
氯苯甲酸的方法。该方法具有操作便利,原辅料廉价易得,产物收率高,中间体和目标产物的纯度好等特点,且易于进行工业化生产。
[0018]
第一方面,本发明提供了一种式i化合物的制备方法,该方法包括:
[0019]
步骤(1):将式iii化合物经重氮化氯化制备式ii化合物,和
[0020]
步骤(2):将式ii化合物水解,得到式i化合物,
[0021][0022]
其中:
[0023]
r选自:c1‑6烷基、c2‑6烯基、c2‑6炔基、c3‑8环烷基、c3‑8环烷基
‑
c1‑6烷基、c6‑
10
芳基、c6‑
10
芳基
‑
c1‑6烷基、5
‑
10元杂芳基、3
‑
12元杂环烷基、5
‑
10元杂芳基
‑
c1‑6烷基、3
‑
12元杂环烷基
‑
c1‑6烷基、c1‑6烷氧基羰基、c1‑6烷氧基羰基c1‑6烷基、c6‑
10
芳基
‑
c1‑6烷氧基羰基、c6‑
10
芳基
‑
c1‑6烷氧基羰基c1‑6烷基,它们各自任选地被独立地选自以下的一个或多个基团取代:卤素、氨基、
‑
nh(c1‑6烷基)、
‑
n(c1‑6烷基)2、羟基、c1‑6烷基、c1‑6烷氧基、卤素取代的c1‑6烷基、卤素取代的c1‑6烷氧基、c3‑8环烷基、c6‑
10
芳基、5
‑
10元杂芳基或3
‑
12元杂环烷基;
[0024]
或者,当r是h时,进行如上所述的步骤(1),直接得到式i化合物。
[0025]
在一个优选的实施方案中,r选自:c1‑6烷基、c2‑6烯基、c3‑8环烷基、c3‑8环烷基
‑
c1‑6烷基、c6‑
10
芳基或c6‑
10
芳基
‑
c1‑6烷基,它们各自任选地被独立地选自以下的一个或多个基团取代:卤素、氨基、
‑
nh(c1‑6烷基)、
‑
n(c1‑6烷基)2、羟基、c1‑6烷基、c1‑6烷氧基、卤素取代的c1‑6烷基或卤素取代的c1‑6烷氧基。
[0026]
在另一个优选的实施方案中,r选自:甲基、乙基、丙基、异丙基、烯丙基、环丙基、正丁基、异丁基、叔丁基、环丁基、正戊基、正己基、环己基、苯基或苄基。
[0027]
在步骤(1)中,所述的式iii化合物经重氮化氯化反应制备式ii化合物是指:式iii化合物先经重氮化反应,然后氯化反应两步制备式ii化合物;或者式iii化合物经重氮化氯化反应一锅法制备式ii化合物。
[0028]
所述的两步反应中,重氮化反应是指式iii化合物在酸性体系中,控制温度低于20℃,例如
‑
10到15℃,加入重氮化试剂进行反应。
[0029]
所述的重氮化试剂可选自亚硝酸或其盐或c1‑6烷基酯,例如亚硝酸、亚硝酸钠、亚硝酸钾、亚硝酸甲酯、亚硝酸乙酯、亚硝酸异戊酯或亚硝酸叔丁酯。
[0030]
所述的氯化反应是在铜催化剂存在下,在含有氯离子的溶液中将重氮化基团进行氯代的反应。例如,所述的氯化反应可在
‑
10到40℃,在含有氯离子的溶液中,将上述重氮化基团进行氯代。所述含有氯离子的溶液可选自盐酸、氯化钠、氯化钾、氯化铜、氯化铁或氯化亚铁溶液。
[0031]
所述的铜催化剂选自金属铜、氯化亚铜、氯化铜、溴化亚铜或碘化亚铜。
[0032]
在一个特别优选的实施方案中,步骤(1)包括:将式iii化合物加入酸性溶剂中,控
制反应温度为
‑
10到15℃,滴加重氮化试剂,得到重氮化反应溶液。然后控制温度为
‑
10到40℃,将重氮化反应溶液,滴加至包含铜催化剂的含有氯离子的溶液中,析出式ii化合物,过滤,水洗,得到式ii化合物。该产物无需干燥,直接进行下步反应。
[0033]
在另一个优选的实施方案中,在步骤(1)中,所述式iii化合物通过一锅法制备式ii化合物,其包括:将式iii化合物加入至含有铜催化剂和氯离子的酸性反应液中,搅拌均匀,控制反应温度
‑
10到70℃,滴加重氮化反应试剂进行反应,获得式ii化合物。产物经过滤,水洗后,直接进行下步反应。
[0034]
在所述铜催化剂是金属铜单质的情况下,产物经过滤后,使用选自甲苯、己烷、正庚烷、甲醇、乙醇或异丙醇或其与水的混合溶液的溶剂将式ii化合物溶解,过滤,得到金属铜催化剂,该催化剂回收使用。然后将式ii化合物溶液蒸馏至干,直接进行下步反应,或结晶析出,离心后直接进行下步反应。
[0035]
在步骤(2)中,将式ii化合物在碱金属氢氧化物水溶液中水解得到式i化合物。
[0036]
在一个优选的实施方案中,将式ii化合物在碱金属氢氧化物水溶液中水解,通过有机溶剂提取除去非水溶性杂质,然后酸化,离心,干燥,得到式i化合物。
[0037]
所述的碱金属氢氧化物水溶液可选自氢氧化锂、氢氧化钠或氢氧化钾水溶液、或它们的混合物。除去杂质所用的有机溶剂可选自非极性或极性非质子有机溶剂,例如己烷、正庚烷、乙酸异丙酯、甲苯、二甲苯或氯苯、或它们的两种或多种的混合物。
[0038]
所述的酸化所使用的酸选自盐酸、氢溴酸、氢碘酸、硫酸、磷酸、硝酸、甲酸或乙酸、或它们的两种或多种的混合物。
[0039]
第二方面,本发明提供一种式iii化合物的制备方法,
[0040][0041]
该方法包括:
[0042]
步骤(a):将式iv化合物与溴化试剂反应得到式iii化合物,其中r如上文第一方面所定义。
[0043]
在一个优选的实施方案中,该方法包括:将式iv化合物溶解在极性溶剂中,控制反应温度为例如5
‑
50℃,滴加溴化试剂或其溶液进行反应而得到式iii化合物。该反应完毕后,将反应液加入自来水中,析出式iii化合物,离心后,可直接用于式ii化合物的制备。
[0044]
所述的极性溶剂选自:乙腈、四氢呋喃、甲基四氢呋喃、n,n
‑
二甲基甲酰胺、n,n
‑
二乙基甲酰胺、n
‑
甲基吡咯烷酮、二甲基亚砜、甲酸、乙酸或水、或它们的两种或多种的混合物。
[0045]
所述的溴化试剂选自:溴、氢溴酸、溴化锂、溴化钠、溴化钾、溴酸钠、二溴海因、n
‑
溴代乙酰胺、n
‑
溴代丁二酰亚胺、苯基三甲基三溴化铵、溴化(溴甲基)三苯基鏻、或它们的两种或多种的混合物。
[0046]
第三方面,本发明提供了一种式i化合物的制备方法,该方法包括如上所述的步骤(a)、(1)和(2):
[0047][0048]
其中r如上文所定义。
[0049]
步骤(a)、(1)和(2)的反应条件和优选实施方案如上文所定义。
[0050]
第四方面,本发明提供了一种制备式vi化合物的方法,其包括以下步骤:
[0051][0052]
步骤(3):式i化合物通过傅克酰基化反应与式vii化合物反应制备式v化合物,
[0053][0054]
其中r1选自:h,c1‑6烷基,例如甲基或乙基,3
‑
四氢呋喃基,3
‑
s
‑
四氢呋喃基或3
‑
r
‑
四氢呋喃基;和
[0055]
步骤(4):式v化合物经还原得到式vi化合物。
[0056]
在一个优选的实施方案中,本发明提供了一种制备式vi化合物的方法,包括步骤(1)、(2)、(3)和(4):
[0057][0058]
其中r1选自:h,c1‑6烷基,例如甲基或乙基,3
‑
四氢呋喃基,3
‑
s
‑
四氢呋喃基或3
‑
r
‑
四氢呋喃基,且r如第一方面所定义,
[0059]
其中步骤(1)、(2)、(3)和(4)如上文所定义。
[0060]
在一个优选的实施方案中,该方法包括:(1)将式iii化合物经重氮化氯化反应制备式ii化合物,(2)式ii化合物水解得到式i化合物,(3)式i化合物通过傅克酰基化反应与式vii化合物反应得到式v化合物,以及(4)将式v化合物还原得到式vi化合物。
[0061]
步骤(1)和(2)的反应条件和优选实施方案如上文所定义。
[0062]
在该方法中,优选地,步骤(3)包括:式i化合物在酰化试剂作用下在弱极性溶剂中获得酰氯,然后在路易斯酸作用下,在弱极性溶剂中与式vii化合物反应得到式v化合物。
[0063]
在一个更优选的实施方案中,步骤(3)包括:式i化合物在酰化试剂作用下,在弱极性溶剂中获得酰氯,然后在路易斯酸作用下,在弱极性溶剂中与式vii化合物反应,控制反应温度在
‑
10到20℃反应直至完毕,反应物经浓缩结晶得到式v化合物。反应物的具体后处理可包括:加入水中淬灭,有机相水洗,浓缩,结晶,离心,烘干得到式v化合物。
[0064]
所述的酰化试剂选自草酰氯、氯化亚砜、三氯化磷、五氯化磷或三氯氧磷。
[0065]
所述路易斯酸选自氯化铝、氯化铁、氯化铜或溴化铝。
[0066]
所述弱极性溶剂选自四氢呋喃、甲基四氢呋喃、二氯甲烷、三氯甲烷、异丙醚或甲基叔丁基醚,或它们的两种或多种的混合物。
[0067]
结晶所用的溶剂选自甲苯、己烷、正庚烷、甲醇、乙醇、异丙醇、二氯甲烷、异丙醚、甲基叔丁基醚或水,或它们的两种或多种的混合物。
[0068]
在该方法中,步骤(4)包括:将式v化合物溶于有机溶剂中,在还原剂和助剂的作用下还原得到式vi化合物。
[0069]
在一个更优选的实施方案中,步骤(4)包括:将式v化合物溶于有机溶剂中,在还原剂和助剂的作用下,控制温度在
‑
10到80℃,还原,结晶,得到式vi化合物。
[0070]
所述的有机溶剂选自甲醇、乙醇、异丙醇、乙腈、异丙醚、甲基叔丁基醚、四氢呋喃、甲基四氢呋喃、甲苯、氯苯、戊烷、己烷、环己烷、正庚烷或水,或它们的两种或多种的混合物。
[0071]
所述的还原剂选自硼氢化锂、硼氢化钠、硼氢化钾、硼烷、三乙基硅烷、三甲基硅烷、四甲基二硅氧烷或氢化锂铝。
[0072]
所述的助剂选自氯化铝、氯化铁、氯化铜、溴化铝、三氟乙酸、三氟甲磺酸或三氟化硼乙醚。
[0073]
结晶所用溶剂选自甲醇、乙醇、异丙醇、乙腈、异丙醚、甲基叔丁基醚、四氢呋喃、甲基四氢呋喃、甲苯、氯苯、戊烷、己烷、环己烷、正庚烷或水,或它们的两种或多种的混合物。
[0074]
在另一个优选的实施方案中,本发明提供了制备式vi化合物的方法,其包括步骤(a)、(1)、(2)、(3)和(4):
[0075][0076]
其中步骤(a)、(1)、(2)、(3)和(4)及优选实施方案如上文所定义,r和r1如上文所定义。
[0077]
第五方面,本发明提供了式iii化合物在制备降糖药达格列净或恩格列净中的应用。
[0078]
具体而言,本发明提供了制备达格列净或恩格列净的方法,该方法包括如上文第一、二、三、四方面所述的任何一个或多个步骤,即,该方法包括上文所述的步骤(a)、(1)、(2)、(3)和(4)中的任何一个或多个步骤,其中r和r1如上文所定义。
[0079]
例如,本发明提供了制备达格列净的方法,该方法包括从式iii化合物制备式vi化合物的步骤,
[0080][0081]
其中步骤(1)、(2)、(3)和(4)如上文所定义,r和r1如上文所定义。
[0082]
以式vi化合物为原料,可参考专利申请wo03099836a1和wo2005092877a1的方法分别制备达格列净和恩格列净。专利申请wo03099836a1和wo2005092877a1整体引入本文作为参考。
[0083]
在一个具体的实施方案中,本发明提供了制备达格列净的方法,该方法包括以下步骤:
[0084][0085]
其中步骤(1)、(2)、(3)和(4)如上文所定义,r如上文所定义,r1为乙基,且步骤(5)、(6)、(7)和(8)如wo03099836a1的实施例中所述的方法。
[0086]
本发明相对于现有技术,具有以下优势:
[0087]
1.本发明制备的式i化合物收率高,纯度好,异构体杂质少;
[0088]
2.本发明制备式i化合物的方法步骤少,工艺简单,所用的溶剂、催化剂和酸可以回收并重复使用,从而显著地降低成本和适合工业化生产;
[0089]
3.使用本发明式i化合物进一步制备的降糖药关键中间体式vi化合物的收率高,纯度达到99.9%,从而为制备最终药物提供可靠的质量保证。
[0090]
定义:
[0091]
为了解释本说明书,将使用以下定义,并且只要适当,以单数形式使用的术语也可以包括复数,并且反之亦然。要理解,本文所用的术语仅是为了描述具体的实施方案,并且不意欲是限制性的。
[0092]
本文所用的术语“卤素”或“卤代”意指f、cl、br或i。此外,术语“被卤素取代的”基团旨在包括单卤代或多卤代基团,其中一个或多个相同或不同的卤素取代基团中的一个或多个氢。
[0093]
本文所用的术语“烷基”指由碳原子和氢原子组成的直链或支链的饱和烃基团。具体地,烷基具有1
‑
10个,例如1至6个、1至5个、1至4个、1至3个或1至2个碳原子。例如,如本文中所使用,术语“c1‑
c6烷基”指具有1至6个碳原子的直链或支链的饱和烃基团,其实例例如甲基、乙基、丙基(包括正丙基和异丙基)、丁基(包括正丁基、异丁基、仲丁基或叔丁基)、戊基(包括正戊基、异戊基、新戊基)、正己基、2
‑
甲基戊基等。
[0094]
本文所用的术语“烯基”指由碳原子和氢原子组成的包含至少一个双键的直链或支链的不饱和烃基团。具体地,烯基具有2
‑
8个,例如2至6个、2至5个、2至4个或2至3个碳原子。例如,如本文中所使用,术语“c2‑
c6烯基”指具有2至6个碳原子的直链或支链的烯基,例如乙烯基、丙烯基、烯丙基、丁烯基、戊烯基等。
[0095]
本文所用的术语“炔基”指由碳原子和氢原子组成的包含至少一个叁键的直链或支链的不饱和烃基团。具体地,炔基具有2
‑
8个,例如2至6个、2至5个、2至4个或2至3个碳原子。例如,如本文中所使用,术语“c2‑
c6炔基”指具有2至6个碳原子的直链或支链的炔基,例如乙炔基、丙炔基、炔丙基、丁炔基等。
[0096]
本文所用的术语“烷氧基”意指基团
‑
o
‑
烷基,其中烷基具有本文所述的含义。具体地,该术语包括基团
‑
o
‑
c1‑6烷基,更具体的
‑
o
‑
c1‑3烷基。烷氧基的代表性实例包括但不限于甲氧基、乙氧基、丙氧基(包括正丙氧基、异丙氧基)、丁氧基(包括正丁氧基、异丁氧基、叔丁氧基)、戊氧基(包括正戊氧基、异戊氧基、新戊氧基)、己氧基(包括正己氧基、异己氧基)等。
[0097]
如本文中所使用的术语“卤素取代的c1‑
c6烷基”指上文所述的c1‑
c6烷基,其中一个或多个(例如1、2、3、4或5个)氢原子被卤素代替。本领域技术人员应当理解,当卤素取代基多于一个时,卤素可以相同也可以不同,并且可以位于相同或不同的c原子上。“卤素取代的c1‑
c6烷基”的实例包括例如
‑
ch2f、
‑
chf2、
‑
cf3、
‑
ccl3、
‑
c2f5、
‑
c2cl5、
‑
ch2cf3、
‑
ch2cl、
‑
ch2ch2cf3或
‑
cf(cf3)2等。
[0098]
如本文中所使用的术语“环烷基”指具有指定环原子数的单环、稠合多环、桥接多环或螺环非芳族饱和单价烃环结构。环烷基可具有3至12个碳原子(即c3‑
c
12
环烷基),例如3至10个,3至8个,3至7个,3至6个,5至6个碳原子。适合的环烷基的实例包括但不限于单环结构,如环丙基、环丁基、环戊基、环己基、环庚基或环辛基;或多环(例如双环)结构,包括螺环、稠合或桥连系统,诸如双环[1.1.1]戊基、双环[2.2.1]庚基、螺[3.4]辛烷基、双环[3.1.1]己烷基、双环[3.1.1]庚基或双环[3.2.1]辛基等。
[0099]
本文中所使用的术语“环烷基”还包括“环烯基”。“环烯基”意指具有指定环原子数的单环、稠合多环、桥接多环或螺环非芳族不饱和烃环结构,包含至少一个(例如1、2、或3个)碳碳双键。环烯基可具有3至12个碳原子(即c3‑
c
12
环烯基),例如3至10个,3至8个,3至7个,3至6个,5至6个碳原子。适合的环烯基的实例包括但不限于单环结构,如环丙烯基、环丁烯基、环戊烯基、环戊二烯基、环己烯基、环己二烯基、环庚烯基、环庚二烯基、环庚三烯基或环辛烯基。
[0100]
本文所用的术语“杂环烷基”意指包括一或多个(例如1、2、3或4个)独立地选自o、n及s的杂原子及指定环原子数的单环、稠合多环、螺环或桥接多环非芳族饱和环结构,或其
n
‑
氧化物,或其s
‑
氧化物或s
‑
二氧化物。杂环烷基可具有3至12个环成员(可称为3
‑
12元杂环烷基),例如3至10个环成员,3至8个环成员,3至7个环成员,4至7个环成员、4至6个环成员、5至6个环成员。杂环烷基通常含有至多4个(例如1个、2个、3个或4个)杂原子。适合的杂环烷基的实例包括但不限于氮杂环丁烷基、氧杂环丁烷基、硫杂环丁基、吡咯烷基(例如1
‑
吡咯烷基、2
‑
吡咯烷基及3
‑
吡咯烷基)、四氢呋喃基(例如1
‑
四氢呋喃基、2
‑
四氢呋喃基及3
‑
四氢呋喃基)、四氢噻吩基(例如1
‑
四氢噻吩基、2
‑
四氢噻吩基及3
‑
四氢噻吩基)、哌啶基(例如1
‑
哌啶基、2
‑
哌啶基、3
‑
哌啶基及4
‑
哌啶基)、四氢吡喃基(例如4
‑
四氢吡喃基)、四氢噻喃基(例如4
‑
四氢噻喃基)、吗啉基(例如吗啉代)、硫吗啉基、二噁烷基、哌嗪基或氮杂环庚烷基、二氮杂环庚烷基例如1,4
‑
二氮杂环庚基、3,6
‑
二氮杂
‑
双环[3.1.1]庚基或3
‑
氮杂
‑
双环[3.2.1]辛基。杂环烷基中与化合物其余部分连接的原子可以是碳原子,也可以是杂原子,只要化学上可行即可。
[0101]
本文所用的术语“杂环烷基”还包括“杂环烯基”,是指包含至少一个(例如1、2或3个)双键的本文所定义的“杂环烷基”,例如吡咯啉基(例如1
‑
吡咯啉基、2
‑
吡咯烷基、3
‑
吡咯啉基、4
‑
吡咯啉基或5
‑
吡咯啉基)、二氢呋喃基(例如1
‑
二氢呋喃基、2
‑
二氢呋喃基、3
‑
二氢呋喃基、4
‑
二氢呋喃基或5
‑
二氢呋喃基)、二氢噻吩基(例如1
‑
二氢噻吩基、2
‑
二氢噻吩基、3
‑
二氢噻吩基或4
‑
二氢噻吩基)、四氢吡啶基(例如1
‑
、2
‑
、3
‑
、4
‑
、5
‑
或6
‑
四氢吡啶基)、四氢吡喃基(例如4
‑
四氢吡喃基)或四氢噻喃基(例如4
‑
四氢噻喃基)。
[0102]
本文所用的术语“芳基”意指通过从芳族环系统中的单个碳原子上移除一个氢原子而衍生的单价芳族烃基。具体地,芳基是指具有指定环原子数的单环或稠合多环芳族环结构。具体地,该术语包括包含6至14个、例如6至10个、优选6个环原子的基团。特定的芳基包括苯基及萘基,最具体的芳基为苯基。
[0103]
本文所用的术语“杂芳基”意指包括一或多个(例如1、2、3或4个)独立地选自o、n及s的杂原子及指定环原子数的单环或稠合多环芳族环结构,或其n
‑
氧化物,或其s
‑
氧化物或s
‑
二氧化物。具体地,该芳族环结构可具有5至10个环成员。杂芳基可为例如5
‑
6元单环、或由稠合的两个6元环、稠合的两个5元环、稠合的6元环和5元环、或稠合的5元环和4元环形成的稠合双环结构。杂芳基环通常将含有至多4个杂原子、更通常至多3个杂原子、更通常至多2个、例如单个独立地选自o、n及s的杂原子,其中n和s可以是氧化状态如n氧化物、s=o或s(o)2。在一个实施方案中,杂芳基环含有至少一个环氮原子、至少一个环硫原子或至少一个环氧原子。例如,杂芳基可以是包含1、2、3或4个独立地选自n、o或s的杂原子的稠合环,例如苯并呋喃、苯并噻吩、吲哚、苯并咪唑、吲唑、苯并三唑、吡咯并[2,3
‑
b]吡啶、吡咯并[2,3
‑
c]吡啶、吡咯并[3,2
‑
c]吡啶、吡咯并[3,2
‑
b]吡啶、咪唑并[4,5
‑
b]吡啶、咪唑并[4,5
‑
c]吡啶、吡唑并[4,3
‑
d]吡啶、吡唑并[4,3
‑
c]吡啶、吡唑并[3,4
‑
c]吡啶、吡唑并[3,4
‑
b]吡啶、异吲哚、嘌呤、中氮茚、咪唑并[1,2
‑
a]吡啶、咪唑并[1,5
‑
a]吡啶、吡唑并[1,5
‑
a]哒嗪、吡咯并[1,2
‑
b]嘧啶、咪唑并[1,2
‑
c]嘧啶、5h
‑
吡咯并[3,2
‑
b]吡嗪、1h
‑
吡唑并[4,3
‑
b]吡嗪、1h
‑
吡唑并[3,4
‑
d]嘧啶、7h
‑
吡咯并[2,3
‑
d]嘧啶、喹啉、异喹啉、噌啉、喹唑啉、喹喔啉、酞嗪、1,6
‑
萘啶、1,7
‑
萘啶、1,8
‑
萘啶、1,5
‑
萘啶、2,6
‑
萘啶、2,7
‑
萘啶、吡啶并[3,2
‑
d]嘧啶、吡啶并[4,3
‑
d]嘧啶、吡啶并[3,4
‑
d]嘧啶、吡啶并[2,3
‑
d]嘧啶、吡啶并[2,3
‑
b]吡嗪、吡啶并[3,4
‑
b]吡嗪、嘧啶并[5,4
‑
d]嘧啶、吡嗪并[2,3
‑
b]吡嗪和嘧啶并[4,5
‑
d]嘧啶。例如,杂芳基可以是包含1或2个独立地选自n、o或s的杂原子的5
‑
6元杂芳基。适合的5元单环杂芳基的实例包括
但不限于吡咯基、呋喃基、噻吩基、咪唑基、呋咱基、噁唑基、噁二唑基、噁三唑基、异噁唑基、噻唑基、异噻唑基、吡唑基、三唑基及四唑基;适合的6元单环杂芳基的实例包括但不限于吡啶基、吡嗪基、哒嗪基、嘧啶基及三嗪基。杂芳基中与化合物其余部分连接的原子可以是碳原子,也可以是杂原子,只要化学上可行即可。
[0104]
取代基被描述为“任选取代的”意指基团可以是未取代的或被一个或多个(例如0、1、2、3、4或5或更多个,或其中可衍生的任何范围)对该基团所列的取代基取代,其中所述取代基可以相同或不同。在一个实施方案中,任选取代的基团被1个取代基取代。在另一个实施方案中,任选取代的基团被2个取代基取代。在另一个实施方案中,任选取代的基团被3个取代基取代。在另一个实施方案中,任选取代的基团被4个取代基取代。
[0105]
如本文所用,术语“包含”或“包括”是指包括所述的要素、整数或步骤,但是不排除任意其他要素、整数或步骤。在本文中,当使用术语“包含”或“包括”时,除非另有指明,否则也涵盖由所述及的要素、整数或步骤组合的情形。
具体实施方式
[0106]
通过以下实施例对本发明的方法进行进一步的说明。应当理解,提供以下实施例的目的仅仅是为了能够更好的理解本发明,而不是以任何方式限定本发明的范围。
[0107]
除非另有说明,以下各种溶剂和试剂在市场上均有销售。所用的起始原料来自市场销售,或者经过本领域熟知的常规反应进一步加工制备。
[0108]
式iii化合物的制备
[0109]
实施例1:5
‑
溴
‑2‑
氨基
‑
苯甲酸乙酯的制备
[0110][0111]
将41.5kg 2
‑
氨基苯甲酸乙酯加入175kg四氢呋喃中,搅拌溶清,控制反应釜温度10
‑
35℃,分批投入n
‑
溴代丁二酰亚胺44.5kg,加入完毕后,搅拌反应30
‑
60分钟。加入饱和亚硫酸氢钠水溶液2.5kg,然后减压蒸馏回收四氢呋喃,其可重复使用。向浓缩釜中加入150kg自来水打浆,离心,30kg自来水洗涤,得式iii化合物5
‑
溴
‑2‑
氨基苯甲酸乙酯湿品73.2kg,无需干燥,直接用于下步反应,收率按100%计,纯度99.1%。
[0112]
实施例2:5
‑
溴
‑2‑
氨基
‑
苯甲酸苄酯
[0113][0114]
将227g 2
‑
氨基苯甲酸烯丙酯加入900g dmf中,搅拌溶清,控制反应釜温度10
‑
35℃,分批加入163g溴,加入完毕后,搅拌反应40
‑
60分钟。将上述反应液加入至含有35kg亚硫酸钠和350g自来水溶液中,析出固体,离心,50g自来水洗涤,得5
‑
溴
‑2‑
氨基
‑
苯甲酸苄酯湿品346g,无需干燥,直接进行下步反应,收率按100%计,纯度99.1%。
[0115]
实施例3:5
‑
溴
‑2‑
氨基
‑
苯甲酸烯丙酯的制备
[0116][0117]
将176.2g 2
‑
氨基苯甲酸烯丙酯加入1000g dmf中,搅拌溶清,控制反应釜温度10
‑
35℃,分批加入205g二溴海因,加入完毕后,搅拌反应40
‑
60分钟。将上述反应液加入至含有30g亚硫酸钠和300g自来水溶液中,析出固体,离心,20g自来水洗涤,得5
‑
溴
‑2‑
氨基
‑
苯甲酸烯丙酯湿品289.3g,无需干燥,直接进行下步反应,收率按100%计,纯度99.2%。
[0118]
式i化合物的制备
[0119]
实施例4:5
‑
溴
‑2‑
氯
‑
苯甲酸的制备
[0120][0121]
方法一:
[0122]
将73.2kg实施例1制备的5
‑
溴
‑2‑
氨基苯甲酸乙酯湿品加入300kg 20%的盐酸溶液,然后加入8.0kg铜粉,搅拌均匀后,控制反应温度0
‑
20℃,滴加40kg亚硝酸钠水溶液(含亚硝酸钠17.6kg),滴加完毕后,继续搅拌30分钟。然后向反应釜中加入80kg甲苯,搅拌过滤,回收铜催化剂,其可重复使用。分层,酸性水相留待下批重复使用。有机相用20kg自来水洗涤一次,有机相浓缩至干回收甲苯,其可重复使用。加入80kg己烷,加热至50
‑
55℃,搅拌溶清后,降温析晶,0
‑
10℃搅拌30分钟,离心,得到5
‑
溴
‑2‑
氯
‑
苯甲酸乙酯湿品72.5kg,纯度99.6%。
[0123]
将72.5kg上述式ii化合物加入反应釜中,然后加入150kg自来水,控制温度40
‑
55℃,滴加30%naoh溶液38.0kg,搅拌溶清后,滴加31.0kg浓盐酸,析出固体,离心,50
‑
70℃干燥6
‑
8h,得到式i化合物53.7kg,三步总收率91.2%,纯度99.7%。
[0124]
方法二:
[0125]
将73.2kg实施例1制备的5
‑
溴
‑2‑
氨基苯甲酸乙酯湿品加入至120kg 20%的盐酸溶液,搅拌均匀后,控制反应温度
‑
5至6℃,滴加40kg亚硝酸钠水溶液(含亚硝酸钠17.6kg),滴加完毕后,继续搅拌30分钟,得到5
‑
溴
‑2‑
氨基苯甲酸乙酯重氮化物溶液,待用。
[0126]
在另一个反应釜中,加入浓盐酸100kg,氯化亚铜12.4kg,搅拌溶清后,控温20
‑
30℃,滴加上述重氮化溶液,析出大量固体。过滤,滤液重复使用两次后按废液处理。滤饼,水洗,离心,得到5
‑
溴
‑2‑
氯苯甲酸乙酯湿品,71.8kg,纯度99.5%。
[0127]
将71.8kg上述5
‑
溴
‑2‑
氯苯甲酸乙酯加入反应釜中,然后加入160kg自来水,控制温度40
‑
55℃,滴加30%naoh溶液38.0kg,搅拌溶清后,滴加31.0kg浓盐酸,析出固体,离心,50
‑
70℃干燥8h,得到式i化合物5
‑
溴
‑2‑
氯苯甲酸52.9kg,三步总收率89.8%,纯度99.6%。
[0128]
方法三:
[0129]
将346g实施例2制备的5
‑
溴
‑2‑
氨基
‑
苯甲酸苄酯湿品加入至1050g 20%的盐酸溶液,搅拌均匀后,控制反应温度
‑
5至10℃,滴加212g亚硝酸钠水溶液(含亚硝酸钠70.4g),滴
加完毕后,继续搅拌30分钟,得到5
‑
溴
‑2‑
氨基苯甲酸乙酯重氮化物溶液,待用。
[0130]
在另一个反应瓶中,加入浓盐酸150g,氯化亚铜50g,搅拌溶清后,加入甲苯260g。控温20
‑
40℃,滴加上述重氮化溶液。滴毕,搅拌60分钟,分液,水相待处理。有机相用100g
×
2自来水洗涤。然后加入1200g自来水,加热至45
‑
55℃,滴加30%naoh溶液190g,搅拌溶清后,滴加200g浓盐酸,析出固体,离心,50
‑
70℃干燥6
‑
9h,得到式i化合物5
‑
溴
‑2‑
氯苯甲酸198g,三步总收率87.2%,纯度99.4%。
[0131]
按照方法三,以实施例3制备的5
‑
溴
‑2‑
氨基
‑
苯甲酸烯丙酯湿品为原料,制备式i化合物5
‑
溴
‑2‑
氯苯甲酸207.7g,收率88.2%,纯度99.7%。
[0132]
式vi化合物的制备
[0133]
实施例5:5
‑
溴
‑2‑
氯
‑
4'
‑
乙氧基二苯甲烷的制备
[0134][0135]
将5
‑
溴
‑2‑
氯苯甲酸235.5kg加入600kg二氯甲烷中,然后加入2kg dmf,控制反应温度5
‑
30℃,滴加130kg草酰氯,滴加完毕后搅拌溶清。常压蒸馏,回收二氯甲烷用于下批此步骤使用。蒸毕,加入200kg二氯甲烷,得酰氯溶液,搅拌溶清后待用。
[0136]
将1000kg二氯甲烷、133kg无水三氯化铝和123kg苯乙醚加入反应釜中,控制反应温度在
‑
10至10℃,滴加上述制备的酰氯的二氯甲烷溶液,滴毕,搅拌1h。加入10kg饮用水淬灭反应,析出三氯化铝水合物,过滤,回收。滤液加入150kg
×
2饮用水洗涤两次,常压回收二氯甲烷,脱水后可重复使用。然后在浓缩釜中,加入600kg 95%乙醇加热溶清,然后降温结晶,5
‑
10℃离心,得湿品,45
‑
55℃干燥,得到式v化合物5
‑
溴
‑2‑
氯
‑
4'
‑
乙氧基二苯甲酮279.2kg,收率82.4%,纯度99.8%。
[0137]
将279.2kg式v化合物5
‑
溴
‑2‑
氯
‑
4'
‑
乙氧基二苯甲酮加入600kg甲醇中,分批加入30kg硼氢化钠,加毕,控温10
‑
30℃搅拌1h,然后加入10kg三氯化铝,加热至55
‑
64℃反应6
‑
8h,反应完毕后,减压回收甲醇,其可在本步骤重复使用。蒸毕,加入450kg乙酸乙酯,自来水120kg
×
2洗涤,分液,有机相浓缩至干,溶剂可在本步骤重复使用。加入375kg甲醇,45
‑
55℃加热溶清,然后降温析晶,离心,27
‑
32℃减压干燥至干,得到249.0kg式vi化合物5
‑
溴
‑2‑
氯
‑
4'
‑
乙氧基二苯甲烷,收率93.0%,纯度99.9%。
[0138]
实施例6:(3s)
‑3‑
[4
‑
[(5
‑
溴
‑2‑
氯苯基)甲基]苯氧基]四氢呋喃的制备
[0139][0140]
将5
‑
溴
‑2‑
氯苯甲酸141.3kg加入300kg二氯甲烷中,控制反应温度
‑
5至25℃,滴加78kg氯化亚砜,滴加完毕搅拌溶清。常压蒸馏,回收二氯甲烷用于下批此步骤使用。蒸毕,加入150kg二氯甲烷,得酰氯溶液,搅拌溶清后待用。
[0141]
将600kg二氯甲烷、80kg无水三氯化铝和98.0kg(3s)
‑
苯氧基四氢呋喃加入反应釜中,控制反应温度
‑
10
‑
10℃,滴加上述制备的酰氯的二氯甲烷溶液,滴毕,搅拌1
‑
2h。加入
6.0kg饮用水淬灭反应,析出三氯化铝水合物,过滤,回收。滤液加入900kg
×
2饮用水洗涤两次,常压回收二氯甲烷,脱水后可重复使用。然后在浓缩釜中,加入360kg甲醇加热溶清,然后降温结晶,0
‑
10℃离心,得湿品,40
‑
50℃干燥,得到式v化合物(r)
‑3‑
(4
‑
(2
‑
氯
‑5‑
碘苯甲酰基)苯氧基)四氢呋喃185.2kg,收率81.0%,纯度99.7%。
[0142]
将185.2kg式v化合物(r)
‑3‑
(4
‑
(2
‑
氯
‑5‑
碘苯甲酰基)苯氧基)四氢呋喃加入420kg甲醇中,分批加入18.0kg硼氢化钠,加毕,控温10
‑
30℃搅拌1.5h,然后加入15kg三氟化硼乙醚,加热至50
‑
64℃反应5
‑
7h,反应完毕后,减压回收甲醇,其可在本步骤重复使用。蒸毕,加入300kg甲苯,自来水70kg
×
2洗涤,分液,有机相浓缩至干,溶剂可在本步骤重复使用。加入220kg甲醇和10kg纯化水,45
‑
64℃加热溶清,然后降温析晶,离心,35
‑
40℃减压干燥至干,得到162.7kg式vi化合物(3s)
‑3‑
[4
‑
[(5
‑
溴
‑2‑
氯苯基)甲基]苯氧基]四氢呋喃,收率91.2%,纯度99.8%。
[0143]
实施例7:dapagliflozin(达格列净)的制备
[0144]
使用上面所获得的5
‑
溴
‑2‑
氯
‑
4'
‑
乙氧基二苯甲烷,参考专利申请wo03099836a1的实施例的方法,制备达格列净,步骤如下:
[0145][0146]
式viii化合物的合成:
[0147]
反应瓶中加入式vi化合物120g,330g四氢呋喃,600g甲苯,开始搅拌,降温至
‑
70℃,控制内温≤
‑
70℃滴加150ml正丁基锂,滴毕,保温30
‑
60分钟,缓慢转入至装有185g式xi化合物(参考wo03099836a1的实施例的步骤c的方法制备)和640g甲苯的反应瓶中,控制内温≤
‑
70℃,继续保温搅拌3
‑
4小时,反应完毕,加入55g甲磺酸和600g甲醇溶液淬灭,搅拌过夜。加入碳酸钠水溶液调节ph=6~7,分层,有机层水洗,浓缩至干得130.5g式viii化合物,收率80.6%,纯度95.1%。
[0148]
式ix化合物的合成:
[0149]
反应瓶中加入114.4g式viii化合物,1200g甲苯,139g三乙胺,开始搅拌,降温至0℃。加入217.5g乙酸酐,保温30分钟,关闭冷却,搅拌过夜。反应完毕,加入270g 85%h3po4和800g自来水配制的h3po4溶液稀释,搅拌30分钟,分层,水层130g
×
2甲苯提取两次,合并有机层,150g自来水洗洗涤,分层,有机层浓缩至干,得到150.6g式ix化合物,收率95.2%,纯度97.5%。
[0150]
式x化合物的合成:
[0151]
反应瓶中加入140g式ix化合物,800g乙腈,开始搅拌,降温至0℃。加入60.8g三乙基硅烷和48g三氟化硼乙醚,保温1小时,升温至室温,加入36g三乙基硅烷和20g三氟化硼乙醚,搅拌过夜。反应完毕,加入800g乙酸乙酯提取物料,依次用饱和碳酸氢钠溶液洗涤、自来水洗涤,过滤,有机相减压浓缩至干。加入560g乙醇,升温至65~75℃搅拌溶清,关闭加热,搅拌过夜,过滤,烘干,得109.8g产物,收率82.5%,纯度99.4%。
[0152]
dapagliflozin的合成:
[0153]
反应瓶中加入式x化合物80g,200g四氢呋喃,400g甲醇,150g自来水,开始搅拌,加入7.2g一水合氢氧化锂,室温下搅拌8h。反应完毕,减压浓缩蒸除有机溶剂,加入700g乙酸乙酯溶解,200g水洗,分液,有机相减压浓缩至干,得到54.2g达格列净,收率95.6%,纯度99.8%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1