一种mer-Ir(ppy)3的制备方法与流程
一种mer
‑
ir(ppy)3的制备方法
技术领域
1.本发明涉及有机铱配合物发光材料领域,尤其是涉及一种mer
‑
ir(ppy)3的制备方法。
背景技术:
2.铱磷光配合物具有良好的热稳定性,相对短的激发态寿命,发光效率高以及发光颜色容易调节等方面的优势,成为当前数量最庞大、性能最为优异的有机发光分子材料。
3.铱(iii)为d7电子构型,容易与有机配体形成六配位八面体配合物,配合物中配体的种类和结构容易改变,发光颜色可以通过改变配体的结构在整个可见光范围内进行调节。截止目前,铱磷光配合物是发光性能最优的有机电致磷光材料,在oled、lec和光催化等领域有着广泛应用。然而,铱磷光配合物的成本高,阻碍了相关产业的发展。因此,发展低成本高效率的制备新方法,提高产率,对降低材料的成本仍具有重要的现实意义和显著的科学价值。
4.ir(c^n)3型铱磷光配合物是由3个相同的环金属配体(c^n)与铱配位形成的均配型中性铱磷光配合物,存在面式和经式两种异构体(如图1所示)。
5.在面式异构体中,所有的ir
‑
c键处于吡啶环的反位,ir
‑
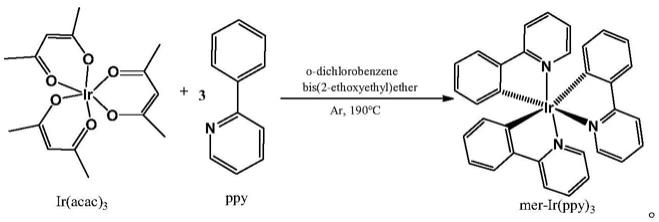
n键处于苯环的反位,所有ir
‑
c键长和ir
‑
n键长各自相等。而在经式异构体中,ir
‑
c键和ir
‑
n键的键长各不相同。面式和经式异构体的形成与温度有很大的关系。fac
‑
ir(c^n)3是热力学稳定产物,一般在高温(~200℃)条件下形成,mer
‑
ir(c^n)3是动力学稳定产物,一般在较低温度(~150℃)下形成,二者在一定条件下可以相互转化,mer
‑
ir(c^n)3在较高温度下可以转化为fac
‑
ir(c^n)3。如果温度控制不好,在合成中往往会生产两种异构体,二者之间分离较为困难,给后续的提纯工作带来很大的困难。
6.为此,发展了多种如下所述的fac
‑
ir(c^n)3的制备方法:
7.(a)
8.(b)
9.(c)
10.(d)
11.(e)
12.(f)
13.然而,mer
‑
ir(c^n)3的合成方法研究较少,通常是通过水合三氯化铱和环金属配体反应,先得到铱的氯桥二聚体,然后在较低温度和碱性条件下和环金属配体进一步反应
得到,并且通常夹杂fac
‑
ir(c^n)3的生成,导致了经式结构的研究较少。因此,通过研究mer
‑
ir(c^n)3的简单和高效制备方法,对促进其应用研究有着重要的意义。
技术实现要素:
14.鉴于上述的分析,本发明的目的在于提供一种制备mer
‑
ir(ppy)3的新方法,用于解决mer
‑
ir(ppy)3的高效合成问题。
15.本发明的目的主要是通过以下技术方案实现的:
16.一种mer
‑
ir(ppy)3的制备方法,以乙酰丙酮铱(ir(acac)3)为前驱体,以邻二氯甲苯或二乙二醇二乙醚为溶剂,在惰性气体气氛下加热回流,直接和环金属配体反应,并经过柱纯化,得到所述mer
‑
ir(ppy)3。
17.进一步地,本发明的制备方法包括以下步骤:
18.将适量乙酰丙酮铱(ir(acac)3)加入到三口瓶中,加入适量2
‑
苯基吡啶(ppy)和邻二氯甲苯;反复充放惰性气体三次后,惰性气体气氛下加热回流;混合物冷却至室温后,抽滤,得到滤饼;滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到所述mer
‑
ir(ppy)3固体产物。
19.进一步地,所述在惰性气体气氛下加热温度为160~250℃,回流时间6~10h。
20.进一步地,一种具体的反应路线和反应条件如下:
[0021][0022]
本发明的机理及有益效果
[0023]
(1)本发明通过简单的溶剂改变,实现了mer
‑
ir(ppy)3的高效合成。通常以乙酰丙酮铱为前驱体,甘油为反应溶剂,加热回流反应得到的是fac
‑
ir(ppy)3,然而甘油溶剂在低温下粘稠,不易处理,于是发明人试图采用邻二氯甲苯或二乙二醇二乙醚为溶剂制备fac
‑
ir(ppy)3,然而,发明人经过反复试验发现,在反应中并没有fac
‑
ir(ppy)3生成,生成的产物是mer
‑
ir(ppy)3。
[0024]
(2)mer
‑
ir(ppy)3在本发明所选取的溶剂邻二氯甲苯或二乙二醇二乙醚中的溶解度较小,生成的产物,很容易通过简单的过滤实现分离,产物在溶剂的溶解度较小,进一步分离得到了mer
‑
ir(ppy)3,从而提高了mer
‑
ir(ppy)3的收率,收率高达97%。
[0025]
由此可见,本发明提供的mer
‑
ir(ppy)3的合成方法简单、高效、成本低,容易实现批量制备,可为相关领域的研究及相关产业提供高品质的mer
‑
ir(ppy)3。
附图说明
[0026]
图1:面式和经式配合物[ir(c^n)3]的立体结构。
[0027]
图2:本发明的一种反应路线和反应条件示意图。
[0028]
图3:本发明制备的mer
‑
ir(ppy)3的氢谱。
[0029]
图4:本发明制备的mer
‑
ir(ppy)3的碳谱。
[0030]
图5:本发明制备的mer
‑
ir(ppy)3的质谱。
[0031]
图6:本发明制备的mer
‑
ir(ppy)3的光致光谱。
具体实施方式
[0032]
实施例1:mer
‑
ir(ppy)3的合成
[0033]
将5.00g(10.21mmol)乙酰丙酮铱(ir(acac)3)加入到250ml三口瓶中,加入7.92g(51.03mmol)2
‑
苯基吡啶(ppy)和50ml邻二氯甲苯,反复充放氩气三次后,氩气气氛下225℃加热回流8h,混合物冷却至室温后,抽滤,滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到橘黄色固体产物6.49g,产率约为97.0%。
[0034]
实施例2:mer
‑
ir(ppy)3的合成
[0035]
将5.00g(10.21mmol)乙酰丙酮铱(ir(acac)3)加入到250ml三口瓶中,加入7.92g(51.03mmol)2
‑
苯基吡啶(ppy)和50ml二乙二醇二乙醚,反复充放氩气三次后,氩气气氛下185℃加热回流8h,混合物冷却至室温后,抽滤,滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到橘黄色固体产物6.56g,产率约为98.1%。
[0036]
实施例3:mer
‑
ir(ppy)3的合成
[0037]
将5.00g(10.21mmol)乙酰丙酮铱(ir(acac)3)加入到250ml三口瓶中,加入7.92g(51.03mmol)2
‑
苯基吡啶(ppy)和50ml邻二氯甲苯,反复充放氩气三次后,氩气气氛下210℃加热反应8h,混合物冷却至室温后,抽滤,滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到橘黄色固体产物6.55g,产率约为97.9%。
[0038]
实施例4:mer
‑
ir(ppy)3的合成
[0039]
将5.00g(10.21mmol)乙酰丙酮铱(ir(acac)3)加入到250ml三口瓶中,加入7.92g(51.03mmol)2
‑
苯基吡啶(ppy)和50ml二乙二醇二乙醚,反复充放氩气三次后,氩气气氛下175℃加热反应8h,混合物冷却至室温后,抽滤,滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到橘黄色固体产物6.50g,产率约为97.2%。
[0040]
对比例1:fac
‑
ir(ppy)3的合成
[0041]
将5.00g(10.21mmol)乙酰丙酮铱(ir(acac)3)加入到250ml三口瓶中,加入7.92g(51.03mmol)2
‑
苯基吡啶(ppy)和50ml甘油,反复充放氩气三次后,氩气气氛下290℃加热回流8h,混合物冷却至室温后加入大量酸性水溶液,抽滤,滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到黄色固体产物4.12g,产率约为61.6%。
[0042]
对比例2:fac
‑
ir(ppy)3的合成
[0043]
将5.00g(10.21mmol)乙酰丙酮铱(ir(acac)3)加入到250ml三口瓶中,加入7.92g(51.03mmol)2
‑
苯基吡啶(ppy)和50ml甘油,反复充放氩气三次后,氩气气氛下175℃反应8h,混合物冷却至室温后,抽滤,滤饼烘干后用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到橘黄色固体产物4.32g(通过柱层析硅胶分离,3.82g为fac
‑
ir(ppy)3,0.50g为mer
‑
ir(ppy)3),经式和面式混合物产率约为64.6%)。
[0044]
对比例3:mer
‑
ir(ppy)3的合成
[0045]
在圆底烧瓶中加入7.07g(44.7mmol)2
‑
苯基吡啶,用90ml乙二醇单乙醚溶解后,加入7.16g(20.3mmol)三水合三氯化铱和30ml去离子水,反复充放氩气三次后,氩气保护下加
热120℃回流反应24h。冷却到室温,抽滤,滤饼依次用丙酮、去离子水、丙酮洗涤,干燥,得到二聚体(ppy)2ir2cl2(ppy)2固体8.50g,产率为78.9%(以水合三氯化铱铱含量54%算)。
[0046]
将5.00g(4.67mmol)二聚体(ppy)2ir2cl2(ppy)2加入到250ml三口瓶中,加入1.81g(11.68mmol)2
‑
苯基吡啶、6.45g(46.70mmol)无水碳酸钾和50ml甘油,反复充放氩气三次后,氩气气氛下145℃反应24h,混合物冷却至室温后,加入蒸馏水,过滤,滤饼在用蒸馏水洗涤2次,烘干后,用二氯甲烷溶解,快速过柱,除去溶剂,过滤,得到橘黄色固体产物4.56g(通过柱层析硅胶分离,4.12g为mer
‑
ir(ppy)3),0.44g为fac
‑
ir(ppy)3,经式和面式混合物产率约为74.5%)。
[0047]
本发明的mer
‑
ir(ppy)3的结构解析:
[0048]
(1)元素分析,理论值(%):c 60.53,n 6.42,h 3.69;实测值(%):c 60.52,n 6.43,h 3.68。实测值和理论值相吻合。
[0049]
(2)氢谱(1h nmr,500mhz,cdcl3,如图3所示),化学位移(ppm):6.43(d,1h),6.62
‑
6.98(s,11h),7.44
‑
7.47(s,9h),7.50
‑
7.60(s,2h),7.62(d,1h)。
[0050]
(3)碳谱(
13
c nmr,500mhz,cdcl3,如图4所示),化学位移(ppm):118.37,118.40,118.56,118.80,118.99,120.85,121.20,121.32,121.97,122.23,123.93,124.12,124.19,129.57,130.57,132.77,133.99,135.47,136.45,138.01,142.30,144.80,145.34,147.86,151.24,153.27,159.43,167.94,168.55,170.60,175.37,177.81。
[0051]
(4)质谱(esi
‑
ms,如图5所示):出现了678分子离子峰,是[mer
‑
ir(ppy)3+na]
+
的分子量,501分子离子峰是[mer
‑
ir(ppy)3‑
ppy]
+
的分子量,说明测试结果与目标产物一致。
[0052]
(5)mer
‑
ir(ppy)3的光致发光光谱(如图6所示),在常温下样品在ch2cl2溶液中显示出强的黄绿光发射,它的最大发射波长为561nm。而文献报道了fac
‑
ir(ppy)3的最大发射波长为525nm,显示绿光发射(yang c h,fang k h,chen c h,su i w.chem.commun.,2004,2232
‑
2233.)。
[0053]
元素分析、氢谱、碳谱和质谱的结果表明,样品与mer
‑
ir(ppy)3目标化合物相吻合。光致光谱测试发现显示黄绿光发射。
[0054]
综上所述,本发明实施案例提供了一种简单、高效和低成本的mer
‑
ir(ppy)3的合成新方法。
[0055]
以上所述,仅为本发明较佳的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,都应涵盖在本发明的保护范围内。
相关技术
网友询问留言
已有1条留言
-
 0访客 来自[中国] 2023年09月18日 11:15我司有6公斤铱13676075190
0访客 来自[中国] 2023年09月18日 11:15我司有6公斤铱13676075190
1