一种聚酯/尼龙复合材料、其制备方法及用途
1.本发明涉及高分子复合材料领域,特别是涉及一种聚酯/尼龙复合材料、其制备方法及用途。
背景技术:
2.尼龙是分子主链上含有重复酰胺基团—[nhco]—的热塑性树脂总称,是应用广泛的聚合物材料,年产量在千万吨级别。脂肪族的尼龙作为工程塑料,具有比重低、抗拉强度高、抗冲性能优异、耐磨和自润滑性好、成型时不易热裂解的优势;作为纤维具有耐磨性、回弹性、色牢度、抗静电、抗起球性佳的优点,并且面料手感柔软;缺点是易吸水、收缩率比较大、尺寸稳定性差、耐光性不足、不耐酸碱、织物保形性差。例如:尼龙6(pa6)和尼龙66(pa66)。(半)芳香族尼龙具有优异的耐热性能和力学性能,但是熔点高、加工困难。例如:聚己二酰间苯二甲胺(mxd6)、聚间(对)苯二甲酰己二胺(pa6icot)。
[0003]
聚酯是由多元醇和多元酸缩聚而得,主链上含有重复酯键—[coo]—的热塑性树脂总称,是应用广泛的聚合物材料,年产量在千万吨的级别。聚酯以(半)芳香族高分子为主,作为工程塑料具有强度高、刚性大、抗蠕变性佳、吸湿性低、电绝缘性突出、热膨胀系数小、高温尺寸稳定性好、耐uv性能优异的优点;作为化纤耐热性较好,所制得的面料挺括、不易变形。缺点是:比重较高、加工时易降解、在水中的长期使用温度低、不耐酸及醇、纤维回弹不足、吸湿性差、织物透气性差、易产生静电积累、易起毛。如聚对苯二甲酸乙二酯(pet)和聚对苯二甲酸丁二酯(pbt),前者为用量最大的合成纤维,后者主要作为工程塑料使用。另外,脂肪族聚酯,是主要的可生物降解高分子材料,但热性能和力学性能较差,应用范围受到限制。例如:聚乳酸(pla)和聚丁二酸丁二酯(pbs)等。
[0004]
随着对高性能材料需求的不断增加,对单一组分的尼龙、聚酯进行改性日显重要。而在两相体系中,不同组分之间会发生一定程度的渗透,进而形成一个新的界面,此处界面张力可以驱动两相聚集的发生。界面张力越小,说明两相相容性越好,具体表现为界面变得不规则且分散相体积变小,分散相与基体相的界限变得模糊。
[0005]
聚酯和尼龙受结构和端基的影响,混合焓非常高,热力学不相容,在共混时呈现不相容的状态,导致宏观上的相分离,很大程度上影响了复合材料的热性能和力学性能(如:huang y q,liu y x,zhao c h.morphology and properties of pet/pa
‑
6/e
‑
44blends[j].1998,69(8):1505
‑
1515.)。如对比例1,尼龙含量较高,作为基体相时,直接将聚酯和尼龙进行反应得到的复合材料进行刻蚀后sem图像如图9(b)/图3(a)/图10(a)所示,聚酯颗粒呈现为球状颗粒团聚堆积的形式,说明两相间界面张力非常大,是相容性差的表现。如对比例2,聚酯含量较高,作为基体相时,直接将聚酯和尼龙进行反应得到的复合材料进行刻蚀后sem图像如图9(a)/图3(c)所示,被刻蚀的孔洞界面形状是较规整的圆形,说明两相间界面张力非常大,是相容性差的表现。因此通过提高两种聚合物的相容性,以改变共聚物微观链结构和聚集态结构,得到一种宏观相容、微观分离的复合材料,使其表现两种材料的优异性能,具有非常重要的意义。
exchange reaction between pbs and pa6icot[j],2012,51(2):751
‑
757.)在聚丁二酸丁二酯(pbs)和聚间(对)苯二甲酰胺己二胺(pa6icot)的体系中添加tsoh,并在卧式反应釜中进行酯交换反应0.5h~2h,以提高合成的可生物降解聚合物的热性能和机械性能。使用氯仿作为溶剂做溶解度实验,并对产物结构做了详细表征分析。结果表明,添加tsoh后,两相间界面张力明显降低,分散相尺寸变小,均匀性提升,相容性大幅度提升,如图2所示(文献中figure 4)。定量分析后,发现tsoh是酯酰胺交换反应的有效催化剂。当两种聚合物的比例接近1:1时,共聚物的无规度和反应程度达到最大值。samperi等人(samperi f,montaudo m,puglisi c,et al.essential role of chain ends in the ny6/pbt exchange.a combined nmr and maldi approach[j].macromolecules,2003,36(19):7143
‑
7154.)就着重利用这种方法对pa6/pbt共混体系的酯交换反应进行了定量分析,结果证明羧基端基在交换反应中起到了决定性的作用,只有pbt和pa6的羧基端基能够在pa6/pbt共混物中反应且共聚物的组成取决于交换反应的反应程度。tsoh既可以催化聚酯与单一尼龙的酯交换,也可以催化聚酯与多种尼龙之间的酯交换反应。evstatiev等人(evstatiev m,schultz j m,fakirov s,et al.in situ fibrillar reinforced pet/pa
‑
6/pa
‑
66blend[j].polymeng sci,2001,41(2):192
‑
204.)制备了pet/pa6/pa66三元共混物,并在其中添加了tsoh作为催化剂,改善了体系相容性的同时提升了共混物的力学性能。
[0010]
直接添加外增容剂使工艺步骤变得复杂且成本提高,反应过程中存在基团反应活性差异,反应难以控制,会发生交联等不理想的副反应,而且添加的第三组分外增容剂还可能引起某些性能的弱化(如kim s j,kim d k,cho w j,et al.morphology and properties of pbt/nylon6/eva
‑
g
‑
mah ternary blends prepared by reactive extrusion[j].polymeng sci,2003,43(6):1298
‑
1311.)。直接添加外催化剂促进酯
‑
酰胺交换反应一般需要较长的时间,在对比例3中,我们将聚酯和尼龙在催化剂存在的条件下反应3min得到的复合材料进行刻蚀后sem图像如图10(c)所示,聚酯颗粒呈现为球状颗粒团聚堆积的形式,说明两相间界面张力非常大,相容性差;且对反应釜要求较高,需要氮气持续吹扫,这样就增加了生产过程的成本,且延长了复合材料制备工艺的周期,同时长时间的高温可能会使聚合物降解,分子链的结构被破坏。
[0011]
目前,在不添加第三组分(外增容剂或外催化剂)增容剂的条件下,无法获得聚酯和尼龙界面相容的复合材料。如果添加外增容剂,会导致工艺步骤变得复杂且成本提高,反应过程中会发生交联等不理想的副反应,还会引起某些性能的弱化;如果添加外催化剂则需要较长的反应时间,不利于聚酯/尼龙复合材料的应用。
技术实现要素:
[0012]
针对现有技术的不足,本发明提供了一种聚酯/尼龙复合材料、其制备方法及用途,所述聚酯/尼龙复合材料在于将磺酸根化合物中的磺酸基团直接接枝到聚酯的大分子链上形成改性聚酯,所述磺酸基团与尼龙的酰胺基团发生化学反应得到内增容链段,解决聚酯/尼龙复合材料的相容性问题。
[0013]
不同于目前通过添加第三组分(外增容剂或外催化剂)提升聚酯/尼龙复合材料相容性的方法,令人意外的,本发明不添加第三种物质,而是利用含有磺酸根的改性聚酯与尼龙反应,可在短短的几分钟之内,通过简单的熔融反应工艺,得到聚酯/尼龙复合材料,改性
聚酯的磺酸根基团与尼龙的酰胺基团发生化学反应得到内增容链段,所述聚酯/尼龙复合材料中聚酯的质量含量为20%~80%;所述内增容链段包含—so2—nh—基团且具有聚酯/尼龙内增容界面,所述内增容链段在核磁碳谱中具有化学位移在118ppm~119ppm区间的特征肩膀峰,所述的内增容链段起到了相容剂的作用,在不添加第三组分(外增容剂或外催化剂)的条件下,解决了聚酯与尼龙之间的相容性问题,得到具有较好的力学性能的聚酯/尼龙复合材料。
[0014]
本发明采用如下的技术方案:
[0015]
本发明提供了一种聚酯/尼龙复合材料,所述聚酯/尼龙复合材料包含酰胺基团—[nhco]—、酯基—[coo]—以及内增容链段;所述聚酯/尼龙复合材料中聚酯的质量含量为20%~80%;所述内增容链段包含—so2—nh—基团且具有聚酯/尼龙内增容界面,所述内增容链段在核磁碳谱中具有化学位移在118ppm~119ppm区间的特征肩膀峰;优选的,所述聚酯/尼龙复合材料中聚酯的质量含量为40%~60%;优选的,所述聚酯/尼龙复合材料中聚酯的质量含量的检测方法为:所述聚酯/尼龙复合材料经甲酸刻蚀2h以上,余下部分进行真空干燥24h,检测方法得到的聚酯/尼龙复合材料中聚酯的质量含量为其中,w表示经甲酸刻蚀前,所述聚酯/尼龙复合材料的质量,w1表示经真空干燥后,所述余下部分的质量;优选的,所述检测方法得到的聚酯/尼龙复合材料中聚酯的质量含量与实际的聚酯/尼龙复合材料中聚酯的质量含量之间允许存在20%的误差,所述实际的聚酯/尼龙复合材料中聚酯的质量含量的范围为其中,w表示经甲酸刻蚀前,所述聚酯/尼龙复合材料的质量,w1表示经真空干燥后,所述余下部分的质量。
[0016]
以下还提供了若干可选方式,但并不作为对上述总体方案的额外限定,仅仅是进一步的增补或优选,在没有技术或逻辑矛盾的前提下,各可选方式可单独针对上述总体方案进行组合,还可以是多个可选方式之间进行组合。
[0017]
优选的,所述聚酯/尼龙内增容界面的检测方法为:所述聚酯/尼龙复合材料经甲酸刻蚀2h以上,余下部分的形貌以放大倍数为1000倍~20000倍的sem图像观察,所述内增容界面表现为不规则结构或无定形结构;优选的,所述刻蚀时间为2h~6h;优选的,所述内增容界面不存在球状颗粒团聚堆积结构或规整孔洞结构,所述球状颗粒的粒径为0.1μm~3μm,所述规整孔洞的孔径为0.5μm~3μm。
[0018]
优选的,所述规整孔洞结构为规整的圆形结构。优选的,所述不规则结构选自不规则沟壑状结构、不规则蜂窝状结构。
[0019]
优选的,所述不规则沟壑状结构的长径比≥3。
[0020]
优选的,所述不规则沟壑状结构的表面带有球状颗粒;所述球状颗粒的粒径为10nm~500nm。
[0021]
优选的,所述不规则蜂窝状结构的表面带有微孔;所述微孔的孔径为50nm~800nm。
[0022]
优选的,所述无定形结构为平滑的无定形结构。
[0023]
优选的,所述平滑的无定形结构表面带有球状颗粒;所述球状颗粒的粒径为100nm~500nm。
[0024]
优选的,所述无定形结构由不规则颗粒相互粘结堆积而成,所述不规则颗粒的粒
径为1nm~50nm。
[0025]
优选的,所述不规则颗粒相互粘结堆积而成的无定形结构的表面带有微孔,所述微孔的孔径为10nm~100nm。
[0026]
本发明还提供一种聚酯/尼龙复合材料的制备方法,包括如下步骤:将改性聚酯和尼龙进行混合,在熔融状态下进行反应,并快速冷却到特定温度以下得到所述聚酯/尼龙复合材料;
[0027]
所述改性聚酯包含磺酸根;优选的,所述改性聚酯由磺酸根化合物将聚酯改性后得到;更优选的,所述磺酸根化合物选自2
‑
羟基
‑3‑
烯丙氧基丙磺酸钠、2
‑
羟基
‑3‑
甲基丙烯酰氧基丙磺酸钠、异戊二烯磺酸盐、磺酸钠、磺酸钾中的一种或多种;
[0028]
所述改性聚酯与所述尼龙的质量比为8∶2~2∶8;优选的,所述改性聚酯与所述尼龙的质量比为6∶4~4∶6;
[0029]
所述特定温度低于所述改性聚酯和所述尼龙两者中玻璃化转变温度较低者的温度;优选的,所述特定温度比所述玻璃化转变温度较低者低20℃及以上;
[0030]
所述快速冷却的时间≤2min。即在2min内冷却到所述改性聚酯和所述尼龙两者的玻璃化转变温度以下(温度低于所述改性聚酯和所述尼龙两者玻璃化转变温度较低者的温度)。
[0031]
优选的,所述聚酯选自单组份聚酯或修饰性聚酯,所述单组份聚酯选自聚对苯二甲酸乙二醇酯、聚对苯二甲酸丁二醇酯、聚乳酸、聚丁二酸丁二醇酯中的一种或多种,所述修饰性聚酯选自单组份聚酯的阳离子可染改性聚酯、抗静电改性聚酯、阻燃改性聚酯中的一种或多种;
[0032]
优选的,所述尼龙选自单组份聚酰胺或共聚酰胺,所述单组份聚酰胺选自尼龙6、尼龙66、尼龙610、尼龙612、尼龙6i、聚对苯二甲酰己二胺、聚间(对)苯二甲酰己二胺、聚己二酰间苯二甲胺、改性尼龙中的一种或多种;所述改性尼龙选自单组分聚酰胺的增强尼龙、阻燃尼龙、透明尼龙、耐磨尼龙、增韧尼龙中的一种或多种。
[0033]
优选的,所述磺酸根化合物接枝在所述聚酯的主链上。
[0034]
优选的,所述磺酸根化合物占所述改性聚酯的1wt%~8wt%。
[0035]
优选的,所述改性聚酯占所述聚酯/尼龙复合材料的20wt%~80wt%。
[0036]
优选的,所述尼龙占所述聚酯/尼龙复合材料的20wt%~80wt%。
[0037]
优选的,所述充分干燥是利用高温真空进行;所述高温的温度范围为:80℃~120℃;所述真空的压力范围为100pa~50000pa;所述充分干燥的时间为8h~24h。
[0038]
优选的,所述快速冷却的时间为≤1min;更优选的,所述快速冷却的时间≤0.5min。
[0039]
优选的,所述反应在双螺杆挤出机中进行;所述反应的温度为150℃~350℃;所述反应的时间为1min~10min。
[0040]
优选的,所述反应的温度为250℃~280℃;所述反应的时间为1min~5min。
[0041]
优选的,包括如下步骤:所述反应结束后,在惰性气体氛围中、温度为120℃~280℃条件下进行固相反应1h~20h,得到所述聚酯/尼龙复合材料;优选的,所述惰性气体为氮气。
[0042]
本发明还提供一种复合物,包含所述的聚酯/尼龙复合材料;优选的,所述复合物
包含添加剂,所述的添加剂是指提高复合物的性能或者降低成本的辅助材料;优选的所述添加剂选自填料、颜料、增塑剂、抗氧剂中的一种或多种,所述填料选自无机填料、阻燃剂、抗冲改性材料、导电填料、导热填料、增强纤维中的一种或多种。
[0043]
本发明还提供一种聚酯/尼龙复合材料及包含聚酯/尼龙复合材料的复合物在纤维领域的用途。
[0044]
现有技术的聚酯/尼龙复合材料克服聚酯和尼龙的相容性必须加入外增容剂或者外催化剂,本发明的聚酯/尼龙复合材料制备方法克服了现有技术的技术偏见,通过将磺酸根接枝在聚酯上得到改性聚酯,改性聚酯与尼龙进行反应会形成内增容链段,内增容链段起到增容剂的作用,解决了聚酯与尼龙之间的相容性问题,得到具有较好的力学性能的聚酯/尼龙复合材料,且至少具有以下技术效果之一:
[0045]
1、本发明磺酸根化合物接到聚酯大分子链上形成新的改性聚酯,与尼龙进行反应一步生成聚酯/尼龙复合,改性聚酯的磺酸根基团与尼龙的酰胺基团发生化学反应得到内增容链段,所述聚酯/尼龙复合材料中聚酯的质量含量为20%~80%;所述内增容链段包含—so2—nh—基团且具有聚酯/尼龙内增容界面,所述内增容链段在核磁碳谱中具有化学位移在118ppm~119ppm区间的特征肩膀峰,所述内增容链段大幅度提升聚酯和尼龙的相容性。
[0046]
2、与现有技术相比,本发明反应时间短,可在短短几分钟内生成嵌段共聚物,并实现聚酯和尼龙的微观混合,效果优于现有技术反应三小时的结果。
[0047]
3、由于相容性的改善,本发明制备的复合产品性能突出,力学性能优于尼龙原料;同时由于产品中所含聚酯的成本优于尼龙,价廉质优,拥有广阔的应用场合。
[0048]
4、本反应时间短,工艺简单,副反应少,具有良好的工业化能力。
附图说明
[0049]
图1为pet/pa
‑
6/e
‑
44共混物断面sem图(huang y q,liu y x,zhao c h.morphology and properties of pet/pa
‑
6/e
‑
44blends[j].1998,69(8):1505
‑
1515.);
[0050]
图2为在pbs/pa6icot混合物中以pbs作为基质相的分散相的形态图[pbs/pa/催化剂(wt%),混合时间](yao z,sun j m,wang q,et al.study on ester
‑
amide exchange reaction between pbs and pa6icot[j],2012,51(2):751
‑
757.);
[0051]
图3为聚酯/尼龙体系相容性检测表征标准sem图(放大倍数为5.00k se);
[0052]
图4为原料c
‑
pet以及复合材料pet/pa6、c
‑
pet/pa6溶解度测试中可溶相的
13
c nmr谱图((a)c
‑
pet;(b)pa6;(c)pet/pa6复合材料可溶相;(d)&(e)c
‑
pet/pa6复合材料可溶相);
[0053]
图5为改性聚酯c
‑
pet和pa6的结构示意图;
[0054]
图6为苯环上碳的核磁信号峰图((a)&(c)c
‑
pet/pa6复合材料可溶相;(b)&(d)c
‑
pet);
[0055]
图7为原料c
‑
pet以及复合材料pet/pa6、c
‑
pet/pa6溶解度测试中不溶相的13c nmr谱图((a)c
‑
pet;(b)pet/pa6复合材料不溶相;(c)c
‑
pet/pa6复合材料不溶相);
[0056]
图8为苯环上碳的核磁信号峰图((a)&(d)c
‑
pet/pa6复合材料不溶相;(b)&(e)c
‑
pet;(c)&(f)pet);
[0057]
图9为不同比例pet/pa6复合材料sem图(放大倍数为5.00k se);
[0058]
图10为磺酸根化合物不同混合方法的pet/pa6复合材料sem图(放大倍数为5.00k se);
[0059]
图11为不同比例用磺酸根化合物改性的pet/pa6复合材料sem图(放大倍数为5.00kse)。
具体实施方式
[0060]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0061]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
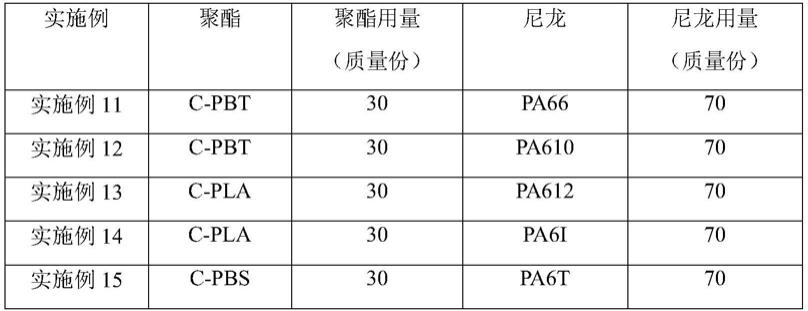
[0062]
以下还提供了若干可选方式,但并不作为对上述总体方案的额外限定,仅仅是进一步的增补或优选,在没有技术或逻辑矛盾的前提下,各可选方式可单独针对上述总体方案进行组合,还可以是多个可选方式之间进行组合。
[0063]
本发明提供了一种聚酯/尼龙复合材料,所述聚酯/尼龙复合材料包含酰胺基团—[nhco]—、酯基—[coo]—以及内增容链段;所述聚酯/尼龙复合材料中聚酯的质量含量为20%~80%;所述内增容链段包含—so2—nh—基团且具有聚酯/尼龙内增容界面,所述内增容链段在核磁碳谱中具有化学位移在118~119ppm区间的特征肩膀峰;优选的,所述内增容界面表现为不规则结构或无定形结构;优选的,所述聚酯/尼龙复合材料中聚酯的质量含量为40%~60%;优选的,优选的,所述聚酯/尼龙复合材料中聚酯的质量含量的检测方法为:所述聚酯/尼龙复合材料经甲酸刻蚀2h以上,余下部分进行真空干燥24h,检测方法得到的聚酯/尼龙复合材料中聚酯的质量含量为其中,w表示经甲酸刻蚀前,所述聚酯/尼龙复合材料的质量,w1表示经真空干燥后,所述余下部分的质量;优选的,所述检测方法得到的聚酯/尼龙复合材料中聚酯的质量含量与实际的聚酯/尼龙复合材料中聚酯的质量含量之间允许存在20%的误差,所述实际的聚酯/尼龙复合材料中聚酯的质量含量的范围为其中,w表示经甲酸刻蚀前,所述聚酯/尼龙复合材料的质量,w1表示经真空干燥后,所述余下部分的质量。
[0064]
其中,所述聚酯由多元醇和多元酸缩聚而得,主链上含有重复酯键—[coo]—的热塑性树脂总称。所述尼龙是分子主链上含有重复酰胺基团—[nhco]—的热塑性树脂总称。所述内增容链段包含—so2—nh—基团,通过核磁(碳谱一维谱图)检测,所述内增容链段的化学位移在118ppm~119ppm区间所对应的电子环境发生了变化,出现了新的特征肩膀峰;同时,所述内增容链段具有聚酯/尼龙内增容界面。
[0065]
所述聚酯/尼龙复合材料中聚酯的质量含量的检测方法中,所述聚酯/尼龙复合材料在经甲酸刻蚀前,称取质量为w;所述聚酯/尼龙复合材料经甲酸刻蚀2h以上,将余下部分
先进行离心倾析甲酸得到固体物,再将固体物进行真空干燥24h,得到的质量为w1;通过检测方法得到的聚酯/尼龙复合材料中聚酯的质量含量与实际的聚酯/尼龙复合材料中聚酯的质量含量之间允许存在20%的误差,即通过检测方法得到聚酯/尼龙复合材料中聚酯的质量含量为那么实际的聚酯/尼龙复合材料中聚酯的质量含量的范围为
[0066]
进一步的,所述聚酯/尼龙内增容界面的检测方法为:所述聚酯/尼龙复合材料经甲酸刻蚀2h以上,余下部分的形貌以放大倍数为1000倍~20000倍的sem图像观察,所述内增容界面表现为不规则结构或无定形结构;优选的,所述刻蚀时间为2h~6h;优选的,所述内增容界面不存在球状颗粒团聚堆积结构或规整孔洞结构;所述球状颗粒的粒径为0.1μm~3μm,所述规整孔洞的孔径为0.5μm~3μm;优选的,所述规整孔洞结构为规整的圆形结构。
[0067]
在两相体系中,不同组分之间会发生一定程度的渗透,进而形成一个新的界面(内增容界面),此处界面张力可以驱动两相聚集的发生。界面张力越小,说明两相相容性越好,具体表现为界面变得不规则且分散相体积变小,分散相与聚集相的界限变得模糊。
[0068]
所述聚酯/尼龙体系相容性的检测方法为:以sem图像表征相形态以评价相容性好坏。sem即扫描电子显微镜成像,其利用聚焦的很窄的高能电子束来扫描样品,通过光束与物质间的相互作用,来激发各种物理信息,对这些信息收集、放大、再成像以达到对物质微观相貌表征的目的。对于聚酯/尼龙体系来说,尼龙可溶于甲酸,而聚酯不溶于甲酸,因此,经甲酸刻蚀后尼龙部分被溶解掉,余下聚酯部分的形态可通过sem图像进行观察。
[0069]
基体相指材料中构成其基本组织的相,一般具有连续的空间分布。分散相:称被分散的物质为分散相。比如聚酯/尼龙复合材料的制备中,改性聚酯的含量大于尼龙的含量时,改性聚酯称为基体相,尼龙称为分散相。当聚酯含量较低,作为分散相时,若刻蚀后的样品为球状,如图3(a)/图9(b)/图10(a)所示,说明两相间界面张力非常大,是不相容的状态;若其形状变得不规则,不呈球形颗粒团聚堆积的形式,则说明两相间界面张力小,相容性好,某一种形态如图3(b)/图10(b)/图11(g)所示。当聚酯含量较高,作为基体相时,若被刻蚀的孔洞界面形状是圆形,如图3(c)/图9(a)所示,说明两相间界面张力非常大,是不相容的状态;若其是不规则的图形,被刻蚀的孔洞界面形状不呈规整的圆形,说明两相间界面张力小,相容性好,某一种形态甚至有沟壑状的形态出现,如图3(d)/图11(c)所示。其中,相容性是否被增强可以通过所述聚酯/尼龙内增容界面来明确,所述改性聚酯与所述尼龙的相容性得到增强,即在聚酯/尼龙复合材料内生成了内增容界面,其表现形式为:经过甲酸刻蚀后的改性聚酯部分界面不规则且分散相体积变小,分散相与聚集相的界面表现为为不规则结构或无定形结构,具体的,可以是不规则的沟壑状、不规则的蜂窝状等,而如果所述改性聚酯与所述尼龙的相容性差,则在聚酯/尼龙复合材料内没有生成内增容界面,所述复合材料的微观形貌表现为所述聚酯的颗粒呈现为球状颗粒团聚堆积的形式或者被刻蚀的孔洞界面形状是规整的圆形结构。
[0070]
进一步的,所述不规则沟壑状结构的长径比≥3。其中,以沟壑状结构中沟壑最长的方向为径向,与径向成90
°
方向的为纵向;所述长径比为径向长度/纵向长度。
[0071]
进一步的,所述不规则沟壑状结构的表面带有球状颗粒;所述球状颗粒的粒径为10nm~500nm。
[0072]
进一步的,所述不规则蜂窝状结构的表面带有微孔;所述微孔的孔径为50nm~800nm。
[0073]
进一步的,所述无定形结构为平滑的无定形结构。其中,所述无定形结构表现为没有固定形状的结构拼接或堆叠的形式;所述平滑的无定形结构表示无定形结构相对于沟壑状而言,呈现基本上连续平整界面相连的形式。
[0074]
进一步的,所述平滑的无定形结构表面带有球状颗粒;所述球状颗粒的粒径为100nm~500nm。
[0075]
进一步的,所述无定形结构由不规则颗粒相互粘结堆积而成,所述不规则颗粒的粒径为1nm~50nm。
[0076]
进一步的,所述无定形结构的表面带有微孔,所述微孔的孔径为10nm~100nm。
[0077]
其中,当聚酯含量较低,作为分散相时,本发明所述样品的聚酯部分相形态为由50nm~500nm不规则颗粒相互粘结堆积的无定形结构,可认定为所述聚酯和尼龙相容性好;所述聚酯部分形貌为由50nm~500nm不规则颗粒相互粘结堆积的无定形结构上有100nm~1000nm的微孔,也可认定为所述聚酯和尼龙相容性好;优选的,所述聚酯部分形貌为平滑的无定形结构,可认定为所述聚酯和尼龙相容性好;优选的,所述聚酯部分形貌为平滑的无定形结构上有100nm~500nm的球状颗粒,可认定为所述聚酯和尼龙相容性好。
[0078]
其中,当聚酯含量较高,作为基体相时,本发明所述样品的所述聚酯部分形貌为不规则蜂窝状结构,可认定为所述聚酯和尼龙相容性好;所述聚酯部分形貌为不规则蜂窝状结构上有直径为50nm~800nm的微孔,也可认定为所述聚酯和尼龙相容性好;优选的,所述聚酯部分形貌为不规则沟壑,可认定为所述聚酯和尼龙相容性好;优选的,所述聚酯部分形貌为长径比≥3的不规则沟壑状结构,可认定为所述聚酯和尼龙相容性好;优选的,所述聚酯部分形貌为不规则沟壑上有10nm~500nm的球状颗粒,可认定为所述聚酯和尼龙相容性好。
[0079]
本发明还提供一种聚酯/尼龙复合材料的制备方法,包括如下步骤:
[0080]
将改性聚酯和尼龙进行混合,在熔融状态下进行反应,并快速冷却到特定温度以下得到所述聚酯/尼龙复合材料;
[0081]
所述改性聚酯包含磺酸根;优选的,所述改性聚酯由磺酸根化合物将聚酯改性后得到;更优选的,所述磺酸根化合物选自2
‑
羟基
‑3‑
烯丙氧基丙磺酸钠、2
‑
羟基
‑3‑
甲基丙烯酰氧基丙磺酸钠、异戊二烯磺酸盐、磺酸钠、磺酸钾中的一种或多种;
[0082]
所述改性聚酯与所述尼龙的质量比为8∶2~2∶8;优选的,所述改性聚酯与所述尼龙的质量比为6∶4~4∶6;
[0083]
所述特定温度低于所述改性聚酯和所述尼龙两者中玻璃化转变温度较低者的温度;优选的,所述特定温度比玻璃化转变温度较低者低20℃及以上。
[0084]
所述快速冷却的时间≤2min。
[0085]
其中,所述改性聚酯可以通过将磺酸根化合物接枝在所述聚酯的大分子链上形成,所述改性聚酯与所述尼龙发生酯交换反应形成内增容链段,所述内增容链段起到相容剂的作用,增强所述改性聚酯与所述尼龙的相容性。所述熔融状态是指温度高于改性聚酯与尼龙两者熔点较高的熔点。
[0086]
通过对所述改性聚酯与所述尼龙的质量比,以及快速冷却条件的协同控制,使得
反应制得的聚酯/尼龙复合材料内部生成了特定功能的内增容链段,克服了现有技术的偏见,实现了本发明意想不到的技术效果。
[0087]
其中一实施例,称取充分干燥后的20~80质量份用磺酸根化合物改性的改性聚酯、20~80质量份尼龙,进行混合,在熔融状态下进行反应(温度高于聚酯和尼龙两者熔点较高者的熔点),并快速冷却到聚酯和尼龙两者的玻璃化转变温度以下(温度低于聚酯和尼龙两者玻璃化转变温度较低者的温度),得到所述聚酯/尼龙复合材料。
[0088]
以其中一实施例阐释聚酯/尼龙复合材料看的相容性大幅度提升的原因,将磺酸根化合物接到pet大分子链上形成新的改性聚酯(用c
‑
pet表示),c
‑
pet与pa6反应得到新的复合材料c
‑
pet/pa6,对pet/pa6和c
‑
pet/pa6进行核磁分析。将原料c
‑
pet与复合材料pet/pa6、c
‑
pet/pa6分别用甲酸刻蚀(温度为45℃,时间为4h)后,得到的可溶部分的核磁谱图以及各个特征峰部分放大的谱图如图4所示。图5是原料c
‑
pet和pa6的结构式。
[0089]
图4(ⅰ)是140ppm~180ppm范围内的核磁信号,对应的是pa6与pet中碳氧双键中的碳原子。通过对比几个谱图,可以发现以pet为原料的实验组可溶部分只有pa6,但是以c
‑
pet为原料的实验组可溶部分出现了157ppm聚酯的特征峰。
[0090]
图4(ⅱ)是124ppm~116ppm范围内的核磁信号,这一范围内包含的是聚酯苯环上的碳原子。通过对比几个谱图,可以发现以pet为原料的实验组可溶部分在此范围内没有聚酯的信号峰出现,以c
‑
pet为原料的实验组可溶部分出现了聚酯的特征峰(122ppm附近)。
[0091]
图4(ⅲ)是60ppm~0ppm范围内的核磁信号,这一范围包含了聚酯与pa6结构中的碳氢单键。同样地,以c
‑
pet为原料的实验组可溶部分出现了聚酯的特征峰(57ppm),但是以pet为原料的实验组可溶部分没有此特征峰。综上分析,经甲酸刻蚀后的可溶部分本应只有共混物中的pa6部分。通过对比各个部分的核磁谱图,可以发现pet/pa6实验组的核磁谱图与纯pa6的核磁谱图基本一致,但是c
‑
pet/pa6可溶物的核磁谱图中出现了聚酯的特征峰。由此说明,c
‑
pet/pa6实验组中的一部分聚酯链段与pa6结合在了一起,形成了内相容链段。
[0092]
如图6所示,进一步将聚酯苯环上的碳原子对应的信号峰放大后发现,复合材料c
‑
pet/pa6可溶部分中该信号峰与原料c
‑
pet是有区别的。复合材料c
‑
pet/pa6可溶物的苯环上与羰基相连的d’、f’碳原子所对应的特征峰变得更宽,苯环上其它碳原子(e’、g’、h’、i’)所对应的特征峰出现了新的肩膀。这说明c
‑
pet中苯环的电子环境发生了变化。
[0093]
对于不可溶的部分来说,如图7和图8所示,通过放大复合材料c
‑
pet/pa6不容部分聚酯的苯环上碳的特征峰,并与原料聚酯c
‑
pet比较来看,c
‑
pet/pa6实验组不可溶部分的聚酯苯环上的碳原子特征峰也是有所变宽,进一步表明c
‑
pet与pa6反应共混后有新的物质生成。
[0094]
上述结构分析中表明c
‑
pet与pa6相容性大幅度提升的原因是生成了新的物质。具体反应机理分析如下,c
‑
pet与pet结构是有区别的,pet的端基是羟基与羧基,c
‑
pet结构中除了端基之外,在链的中间还会存在磺酸基团。苯磺酸基团进攻pa6中的酰胺键,与其氨基结合。此时pet与pa6通过苯磺酸基团相链接,形成内增容链段;同时,苯磺酸上的苯环电子环境发生了变化,导致了核磁谱图发生变化。该内增容链段起到了相容剂的作用,解决了聚酯与尼龙不相容的问题。
[0095]
进一步的,所述聚酯选自单组份聚酯或修饰性聚酯,所述单组份聚酯选自聚对苯二甲酸乙二醇酯(pet)、聚对苯二甲酸丁二醇酯(pbt)、聚乳酸(pla)、聚丁二酸丁二醇酯
(pbs)中的一种或多种,所述修饰性聚酯选自单组份聚酯的阳离子可染改性聚酯、抗静电改性聚酯、阻燃改性聚酯中的一种或多种;所述尼龙选自单组份聚酰胺或共聚酰胺,所述单组份聚酰胺选自尼龙6(pa6)、尼龙66(pa66)、尼龙610(pa610)、尼龙612(pa612)、尼龙6i(pa6i)、聚对苯二甲酰己二胺(pa6t)、聚间(对)苯二甲酰己二胺(pa6icot)、聚己二酰间苯二甲胺(mxd6)、改性尼龙中的一种或多种;所述改性尼龙选自单组分聚酰胺的增强尼龙、阻燃尼龙、透明尼龙、耐磨尼龙、增韧尼龙中的一种或多种。
[0096]
其中,所述尼龙可以选自单组份尼龙,如尼龙6和尼龙66等;也可以是共聚酰胺,如尼龙6和尼龙66的共聚酰胺等。由于共聚酰胺具有更好的收缩特性,更有选的,所述尼龙选自共聚酰胺。
[0097]
进一步的,所述磺酸根化合物接枝在所述聚酯的主链上。即所述磺酸根化合物接枝在聚合物大分子链上。
[0098]
进一步的,所述磺酸根化合物占所述改性聚酯的1wt%~8wt%。
[0099]
进一步的,所述改性聚酯占所述聚酯/尼龙复合材料的20wt%~80wt%。
[0100]
进一步的,所述尼龙占所述聚酯/尼龙复合材料的20wt%~80wt%。
[0101]
其中,改变改性聚酯的含量、尼龙的含量或者改性聚酯中磺酸根化合物的含量,可以使制备的聚酯/尼龙复合材料具有良好的力学性能。
[0102]
进一步的,所述充分干燥是利用高温真空进行;所述高温的温度范围为:80℃~120℃;所述真空的压力范围为100pa~50000pa;所述充分干燥的时间为8h~24h。
[0103]
进一步的,所述快速冷却的时间为≤1min。即在1min内冷却到所述改性聚酯和所述尼龙两者的玻璃化转变温度以下(温度低于所述改性聚酯和所述尼龙两者玻璃化转变温度较低者的温度低20℃及以上)。
[0104]
更进一步的,所述快速冷却的时间≤0.5min。即在0.5min内冷却到所述改性聚酯和所述尼龙两者的玻璃化转变温度以下(温度低于所述改性聚酯和所述尼龙两者玻璃化转变温度较低者的温度低20℃及以上)。
[0105]
进一步的,所述反应在双螺杆挤出机中进行;所述反应的温度为150℃~350℃;所述反应的时间为1min~10min。
[0106]
其中,所述双螺杆挤出机中反应的温度选择依据所述改性聚酯与所述尼龙两种聚合物的剪切粘度进行匹配,且样品尽可能不降解。具体的,选择同一温度时,所述改性聚酯与所述尼龙在同一剪切速率下,剪切粘度相近。
[0107]
进一步的,所述反应的温度为250℃~280℃;所述反应的时间为1min~5min。如用磺酸根化合物改性的pet和pa6/pa66在双螺杆挤出机中混合并反应,其中双螺杆挤出机的温度范围为250℃~280℃,停留时间在5min以内,挤出剪切速率在100r/min~500r/min。同时,在1min内冷却到50℃以下,得到所述聚酯/尼龙复合材料。
[0108]
进一步的,包括如下步骤:所述反应结束后,在惰性气体氛围中、温度为120℃~280℃条件下进行固相反应1h~20h,得到所述聚酯/尼龙复合材料。优选的,所述惰性气体为氮气。其中,所述反应结束后指的是在熔融状态下进行的反应结束后。
[0109]
本发明还提供一种复合物,包含所述的聚酯/尼龙复合材料;优选的,所述复合物包含添加剂,所述添加剂选自填料、颜料、增塑剂、抗氧剂中的一种或多种,所述填料选自无机填料、阻燃剂、抗冲改性材料、导电填料、导热填料、增强纤维中的一种或多种。
[0110]
本发明的聚酯/尼龙复合材料中可以添加无机填料、阻燃剂、抗冲改性材料、导电填料、导热填料、增强纤维等。无机填料可以是玻璃纤维、碳纤维、钛酸钾晶须、氧化锌晶须、硼酸铝晶须、芳族聚酰胺纤维、氧化铝纤维、碳化硅纤维、陶瓷纤维、石棉纤维、石膏纤维、金属纤维、硅灰石、沸石、绢云母、高岭土、云母、滑石、粘土、叶蜡石、膨润土、蒙脱石、锂蒙脱石、合成云母、石棉、铝硅酸盐、氧化铝、氧化硅、氧化镁、氧化锆、氧化钛、氧化铁、碳酸钙、碳酸镁、白云石、硫酸钙、硫酸钡、氢氧化镁、氢氧化钙、氢氧化铝、玻璃珠、陶瓷珠、氮化硼、碳化硅和二氧化硅等。这些材料可以是中空的,还可以使用两种以上的上述无机填充材料。此外,关于膨润土、蒙脱石、锂蒙脱石和合成云母等溶胀性层状硅酸盐,可以使用以有机铵盐将层间离子(interlayer ion)进行了阳离子交换的有机化蒙脱石。为了增强聚酯/尼龙复合材料的性能,在上述填充材料中,特别优选玻璃纤维、碳纤维。为了使复合物的表面外观优异,优选无机填充剂的平均粒径为0.001μm~10μm,更优选的平均粒径优选为0.01μm~5μm,进一步优选为0.05μm~3μm。另外,这些无机填充剂的平均粒径通过沉降法测定。为了增强复合物并且具有良好的表面外观,作为无机填充材料,优选使用滑石、高岭土、硅灰石、溶胀性层状硅酸盐。
[0111]
此外,为了获得更优异的机械强度,无机填充材料优选使用异氰酸酯系化合物、有机硅烷系化合物、有机钛酸酯系化合物、有机硼烷系化合物、环氧化合物等偶联剂进行预处理再使用。特别优选的是有机硅烷系化合物,作为其具体例,可列举
‑
环氧丙氧丙基三甲氧基硅烷、γ
‑
环氧丙氧丙基三乙氧基硅烷、γ
‑
(3,4
‑
环氧环己基)乙基三甲氧基硅烷等含有环氧基的烷氧基硅烷化合物、γ
‑
巯基丙基二甲氧基硅烷、γ
‑
巯基丙基二乙氧基硅烷等含有巯基的烷氧基硅烷化合物、γ
‑
脲基丙基二乙氧基硅烷、γ
‑
脲基丙基二甲氧基硅烷、γ
‑
(2
‑
脲基乙基)氨基丙基二甲氧基硅烷等含有脲基的烷氧基硅烷化合物、γ
‑
异氰酸酯基丙基三乙氧基硅烷、γ
‑
异氰酸酯基丙基三甲氧基硅烷、γ
‑
异氰酸酯基丙基甲基二甲氧基硅烷、γ
‑
异氰酸酯基丙基甲基二乙氧基硅烷、γ
‑
异氰酸酯基丙基乙基二甲氧基硅烷、γ
‑
异氰酸酯基丙基乙基二乙氧基硅烷、γ
‑
异氰酸酯基丙基三氯硅烷等含有异氰酸酯基的烷氧基硅烷化合物、γ
‑
(2
‑
氨基乙基)氨基丙基甲基二甲氧基硅烷、γ
‑
(2
‑
氨基乙基)氨基丙基二甲氧基硅烷、γ
‑
氨基丙基二甲氧基硅烷等含有氨基的烷氧基基硅烷化合物、γ
‑
羟基丙基二甲氧基硅烷、γ
‑
羟基丙基二乙氧基硅烷等含有羟基的烷氧基硅烷化合物、γ
‑
甲基丙烯酰氧基丙基二甲氧基硅烷、乙烯基二甲氧基硅烷、n
‑
n
’‑
(n
‑
乙烯基苄基氨基乙基)
‑
氨基丙基二甲氧基硅烷盐酸盐等含有碳碳不饱和基的烷氧基硅烷化合物、3
‑
二甲氧基甲硅烷基丙基琥珀酸酐等含有酸酐基的烷氧基硅烷化合物。特别优选使用γ
‑
甲基丙烯酰氧基丙基三甲氧基硅烷、γ
‑
(2
‑
氨基乙基)氨基丙基甲基二甲氧基硅烷、γ
‑
(2
‑
氨基乙基)氨基丙基三甲氧基硅烷、γ
‑
氨基丙基三甲氧基硅烷、3
‑
三甲氧基甲硅烷基丙基琥珀酸酐。阻燃剂可以是氢氧化镁、氢氧化铝、聚磷酸铵(app)、八溴醚、磷酸三苯酯、六溴环十二烷、三聚氰胺、三异氰酸三聚氰胺盐(mca)、聚磷酸铵三聚氰胺盐(mpp)、硼酸锌、十溴二苯乙烷、包覆红磷、对叔丁基邻苯二酚(tbc)、十溴二苯醚、磷氮系阻燃剂、四溴双酚a、溴代聚苯乙烯、六溴环十二烷及氯化石蜡、得克隆、氯化聚乙烯(cpe)。
[0112]
抗冲改性剂可以是氯化聚乙烯(cpe)、醋酸乙烯酯共聚物(eva)、丙烯酸酯橡胶(acr)、abs、sbs、乙丙橡胶(epr)、三元乙丙橡胶(epdm)、天然橡胶(nbr)、丁苯橡胶、丁二烯橡胶等。
[0113]
导电填料可以是炭黑、碳纤维、石墨、碳纳米管、铝粉、铜粉、镍粉、铁粉、银粉、金粉、黄铜纤维、不锈钢纤维、铁纤维、镀银玻纤、玻璃微球镀银粉等。
[0114]
导热填料可以是氧化铝、氧化镁、氧化锌、氮化铝、氮化硼、碳化硅、纤维状碳粉、鳞片状碳粉、二氧化硅等。
[0115]
增强纤维可以是玻璃纤维、碳纤维、硼纤维、晶须、石棉纤维、金属纤维、芳纶纤维、奥纶纤维、聚酯纤维、尼龙纤维、维尼纶纤维、聚丙烯纤维、聚酰亚胺纤维等。
[0116]
增塑剂可以是苯磺酰胺(bsa)、n
‑
甲基对甲苯磺酰胺(mtsa)、n
‑
乙基对甲苯磺酰胺(neptsa)、n
‑
丁基苯磺酰胺(bbsa)、n
‑
(2
‑
羟丙基)苯磺酰胺(hpbsa)、n
‑
环已基对甲苯磺酰胺(ctsa)、n,n
‑
双(2
‑
羟乙基)对甲苯磺酰胺、甲苯磺酰胺甲醛树脂(mh/ms)等。
[0117]
抗氧剂选自常用商业化抗氧剂,如抗氧剂牌号1010、1076、168、bht、t501、246、tpp等。
[0118]
本发明还提供一种聚酯/尼龙复合材料及包含聚酯/尼龙复合材料的复合物在材料领域的用途。例如:纺织纤维领域、工程塑料领域。
[0119]
综上所述:(1)本发明用简单的方法得到了一种相容性好,力学性能好的聚酯/尼龙复合材料,所述聚酯/尼龙复合材料包含用磺酸根化合物改性的聚酯和尼龙,所述聚酯和尼龙发生反应,所述聚酯/尼龙复合材料中聚酯的质量含量为20%~80%;;所述内增容链段包含—so2—nh—基团且具有聚酯/尼龙内增容界面,所述内增容链段在核磁碳谱中具有化学位移在118ppm~119ppm区间的特征肩膀峰,所生成内增容链段起到相容剂作用,增强两者相容性。
[0120]
(2)与现有技术相比,本发明通过控制改性聚酯与尼龙的反应条件,可在短短几分钟内生成内增容链段,并实现聚酯和尼龙的微观混合,效果优于现有技术反应三小时的结果。
[0121]
(3)由于相容性的改善,本发明制备的复合产品性能突出,力学性能优于尼龙原料;同时由于产品中所含聚酯的成本优于尼龙,价廉质优,拥有广阔的应用场合。
[0122]
(4)本发明的反应时间短,工艺简单,副反应少,具有良好的工业化能力。
[0123]
(5)本发明的聚酯/尼龙复合材料成本低、稳定性强、性能的重复性高,可应用于纺织纤维、工程塑料等领域。
[0124]
以下结合具体实施例及对比例对本发明作进一步的说明。
[0125]
本发明中涉及的性能测试标准如下:
[0126]
拉伸模量:《gb/t 1040.1
‑
2018塑料拉伸性能的测定》;
[0127]
拉伸强度:《gb/t 1040.1
‑
2018塑料拉伸性能的测定》;
[0128]
断裂伸长率:《gb/t 1040.1
‑
2018塑料拉伸性能的测定》;
[0129]
冲击强度:《gb/t 1843
‑
2008塑料悬臂梁冲击强度的测定》。
[0130]
实施例1
[0131]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在100℃下真空干燥12h;其中,磺酸根化合物与pet的摩尔比为4:96;所述磺酸根化合物选自2
‑
羟基
‑3‑
烯丙氧基丙磺酸钠、2
‑
羟基
‑3‑
甲基丙烯酰氧基丙磺酸钠、异戊二烯磺酸盐、磺酸钠、磺酸钾中的一种或多种(以下实施例和对比例中的磺酸根化合物与本实施例中的磺酸根化合物选择相同)。
[0132]
制备过程:将20质量份改性pet粒料、80质量份pa6粒料进行混合(例如:5min),将
混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料c20/a80。
[0133]
性能测试:将复合粒料c20/a80在110℃下真空干燥10h,由注塑机制成样条,设定注塑温度为240℃~270℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0134]
sem测试:称取1.5g复合材料c20/a80置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果如图10(b)/图11(g)/图3(b)所示,聚酯部分作为基体相,刻蚀掉的尼龙作为分散相,聚酯部分形貌为由粒径为1nm~50nm的不规则颗粒相互粘结堆积的无定形结构;同时,无定形结构表面带有孔径为10nm~100nm的不规则微孔。
[0135]
复合材料c20/a80中聚酯含量的测定:称取1.50g复合材料c20/a80置于150ml甲酸,经甲酸刻蚀2h以上,将余下部分先进行离心倾析甲酸得到固体物,再将固体物进行真空干燥24h,称得固体物的质量为0.0935g,检测的复合材料c20/a80中聚酯质量含量为6.23%,则实际的复合材料c20/a80中聚酯质量含量为0~26.23%。
[0136]
对比例1
[0137]
预处理:将pet粒料和pa6粒料在100℃下真空干燥12h;
[0138]
制备过程:将20质量份pet粒料、80质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料p20/a80。
[0139]
性能测试:性能测试:因样品过于脆,即冲击强度过小,无法制成样条。
[0140]
sem测试:称取1.5g复合材料p20/a80置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察剩余的聚酯部分的形态,结果如图3(a)/图9(b)/图10(a)所示,聚酯部分作为分散相,刻蚀掉的尼龙作为基体相,聚酯以粒径为0.1μm~3μm的小球颗粒团聚在一起,颗粒尺寸较均匀。
[0141]
实施例2
[0142]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在110℃下真空干燥10h;其中,磺酸根化合物与pet的摩尔比为4:96;
[0143]
制备过程:将80质量份改性pet粒料、20质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料c80/a20。
[0144]
性能测试:因样品过于脆,即冲击强度过小,无法制成样条。
[0145]
sem测试:称取1.5g复合材料c80/a20置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果如图11(a)所示,聚酯部分作为基体相,刻蚀掉的尼龙作为分散相,聚酯部分形貌呈现不规则沟壑,同时,不规则沟壑表面上带有粒径为1nm~50nm的球状颗粒。
[0146]
复合材料c80/a20中聚酯含量的测定:称取1.50g复合材料c80/a20置于150ml甲
酸,经甲酸刻蚀2h以上,将余下部分先进行离心倾析甲酸得到固体物,再将固体物进行真空干燥24h,称得固体物的质量为1.145g,检测的复合材料c80/a20中聚酯质量含量为76.35%,则实际的复合材料c80/a20中聚酯质量含量为56.35%~96.35%。
[0147]
对比例2
[0148]
预处理:将pet粒料和pa6粒料在100℃下真空干燥12h;
[0149]
制备过程:将80质量份pet粒料、20质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料p80/a20。
[0150]
性能测试:性能测试:因样品过于脆,即冲击强度过小,无法制成样条。
[0151]
sem测试:称取1.5g复合材料p80/a20置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察剩余的聚酯部分的形态,结果如图9(a)/图3(c)所示,聚酯部分作为基体相,刻蚀掉的尼龙作为分散相,平滑连续的基体相上分散着孔径为0.5μm~3μm的规整孔洞结构。
[0152]
图9(a)/图3(c)所示的p80/a20材料和图9(b)/图3(a)/图10(a)所示的p20/a80材料,两种情况下都可以清晰地看到分散相为球状颗粒团聚堆积结构,说明两相间界面张力非常大,是不相容的状态。
[0153]
实施例3
[0154]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在80℃下真空干燥24h;其中,磺酸根化合物与pet的摩尔比为4:96;
[0155]
制备过程:将70质量份改性pet粒料、30质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为10min,经水冷在0.5min内降到30℃,得到复合材料c70/a30。
[0156]
性能测试:因样品过于脆,即冲击强度过小,无法制成样条。
[0157]
sem测试:称取1.5g复合材料c70/a30置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果如图11(b)所示,聚酯部分作为基体相,刻蚀掉的尼龙作为分散相,聚酯部分形貌为不规则蜂窝状结构;同时,不规则蜂窝状结构表面带有孔径为50nm~800nm的微孔。
[0158]
实施例4
[0159]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在120℃下真空干燥8h;其中,磺酸根化合物与pet的摩尔比为4:96;
[0160]
制备过程:将60质量份改性pet粒料、40质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为1min,经水冷在2min内降到30℃,得到复合材料c60/a40。
[0161]
性能测试:因样品过于脆,即冲击强度过小,无法制成样条。
[0162]
sem测试:称取1.5g复合材料c60/a40置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果如
图3(d)/图11(c)所示,聚酯部分形貌为不规则蜂窝状结构;同时,不规则蜂窝状结构表面带有孔径为100nm~800nm的微孔。
[0163]
实施例5
[0164]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在110℃下真空干燥10h;其中,磺酸根化合物与pet的摩尔比为4:96;
[0165]
制备过程:将50质量份改性pet粒料、50质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为5min,经水冷在1min内降到30℃,得到复合材料c50/a50。
[0166]
性能测试:将复合粒料c50/a50在110℃下真空干燥10h,由注塑机制成样条,设定注塑温度为240℃~270℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0167]
sem测试:称取1.5g复合材料c50/a50置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果图11(d)所示,聚酯部分形貌为连续、长径比≥3的不规则沟壑;同时,不规则沟壑表面带有粒径为10nm~50nm的颗粒。
[0168]
复合材料c50/a50中聚酯含量的测定:称取1.50g复合材料c50/a50置于150ml甲酸,经甲酸刻蚀2h以上,将余下部分先进行离心倾析甲酸得到固体物,再将固体物进行真空干燥24h,称得固体物的质量为0.6344g,检测的复合材料c50/a50中聚酯质量含量为42.29%,则实际的复合材料c80/a20中聚酯质量含量为22.29%~62.29%。
[0169]
其中,实施例1、实施例2及实施例5制得的复合材料中聚酯含量测定结果如下表,表中的不溶物比例即为检测的复合材料中聚酯质量含量。
[0170]
实施例样品名称不溶物比例(%)可溶物比例(%)实施例1c20/a806.2393.77实施例2c80/a2076.3523.65实施例5c50/a5042.2957.71
[0171]
实施例6
[0172]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在110℃下真空干燥10h;其中,磺酸根化合物与pet的摩尔比为4:96;
[0173]
制备过程:将40质量份改性pet粒料、60质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料c40/a60。
[0174]
性能测试:将复合粒料c40/a60在110℃下真空干燥10h,由注塑机制成样条,分别设定注塑温度为240℃~270℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0175]
sem测试:称取1.5g复合材料c40/a60置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果图11(e)所示,聚酯部分形貌为连续的不规则沟壑;同时,不规则沟壑表面带有粒径为10nm~
500nm的颗粒。
[0176]
实施例7
[0177]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在110℃下真空干燥10h;其中,磺酸根化合物与pet的摩尔比为4:96;
[0178]
制备过程:将30质量份改性pet粒料、70质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料c30/a70。
[0179]
性能测试:将复合粒料c30/a70在110℃下真空干燥10h,由注塑机制成样条,分别设定注塑温度为240℃~270℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0180]
sem测试:称取1.5g复合材料c30/a70置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果图11(f)所示,聚酯部分作为分散相,刻蚀掉的尼龙作为基体相,聚酯部分形貌为平滑的无定形结构;同时,平滑的无定形结构表面带有粒径为1nm~50nm的球状颗粒。
[0181]
从图11中聚酯部分的形貌,可以发现将磺酸根化合物接枝到pet大分子链上,聚酯部分形状变得不规则且体积大幅度减小,甚至有沟壑状的形态出现。说明含磺酸基团的pet与pa6熔融挤出后界面张力很小,两相互相渗透程度提高,相容性大幅度提升。同时,对于原料含量不同的实施例来说,用磺酸根化合物改性的pet与pa6添加量比例相同的时候,复合材料的相容性是最好的。
[0182]
实施例8
[0183]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在100℃下真空干燥10h;其中,磺酸根化合物与pet的摩尔比为2:98;
[0184]
制备过程:将50质量份改性pet粒料、50质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区230℃、ii区240℃、iii区250℃、iv区250℃、v区250℃、vi区250℃、vii区250℃、viii区250℃,机头250℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料ec50/a50。
[0185]
性能测试:将复合粒料ec50/a50在100℃下真空干燥10h,由注塑机制成样条,设定注塑温度为240℃~255℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0186]
实施例9
[0187]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在100℃下真空干燥12h;其中,磺酸根化合物与pet的摩尔比为2:98;
[0188]
制备过程:将30质量份改性pet粒料、70质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区230℃、ii区240℃、iii区250℃、iv区250℃、v区250℃、vi区250℃、vii区250℃、viii区250℃,机头250℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料ec30/a70。
[0189]
性能测试:将复合粒料ec30/a70在100℃下真空干燥10h,由注塑机制成样条,分别设定注塑温度为240℃~255℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性
能测试结果见表1。
[0190]
实施例10
[0191]
预处理:将用磺酸根化合物改性的pet粒料和pa6粒料在100℃下真空干燥12h;其中,磺酸根化合物与pet的摩尔比为2:98;
[0192]
制备过程:将30质量份改性pet粒料、70质量份pa6粒料进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区230℃、ii区240℃、iii区250℃、iv区250℃、v区250℃、vi区250℃、vii区250℃、viii区250℃,机头250℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料ec30/a70,将复合材料ec30/a70在200℃,氮气流速为10l/min的条件下进行固相缩聚反应8h,得到复合材料ec30/a70
‑
8。
[0193]
性能测试:将复合粒料ec30/a70
‑
8在100℃下真空干燥12h,由注塑机制成样条,分别设定注塑温度为240℃~255℃,在23℃~27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0194]
实施例11~15
[0195]
预处理及制备方法均采用如实施例9的方法,仅改变原料(聚酯和尼龙),具体见下表格。
[0196][0197]
其中:c
‑
pbt表示用磺酸根化合物改性pbt得到的改性聚酯;
[0198]
c
‑
pla表示用磺酸根化合物改性pla得到的改性聚酯;
[0199]
c
‑
pbs表示用磺酸根化合物改性pbs得到的改性聚酯。
[0200]
实施例11~15均能得到性能良好的聚酯/尼龙复合材料。
[0201]
对比例3
[0202]
预处理:将pet粒料和pa6粒料在100℃下真空干燥12h;
[0203]
制备过程:将19质量份改性pet粒料、80质量份pa6粒料、1份对甲苯磺酸钠直接进行混合(例如:5min),将混合的物料加入双螺杆挤出机,双螺杆挤出机反应温度设置为:i区250℃、ii区260℃、iii区265℃、iv区265℃、v区265℃、vi区265℃、vii区265℃、viii区265℃,机头265℃;反应时间为3min,经水冷在1min内降到30℃,得到复合材料p20/a80
‑
s。
[0204]
性能测试:因样品过于脆,即冲击强度过小,无法制成样条。
[0205]
sem测试:称取1.5g复合材料p20/a80
‑
s置于150ml甲酸,在45℃磁搅拌4h。静置24h后,离心分离固相,真空烘箱干燥12h。通过sem图像观察未溶解的聚酯部分的形态,结果图10(c)所示,聚酯部分作为分散相,刻蚀掉的尼龙作为基体相,聚酯部分以粒径为0.1~3μm的球状颗粒团聚堆积在一起,且颗粒尺寸差异大。
[0206]
通过图10(b)/图11(g)/图3(b)可以看出,将磺酸根化合物接枝到pet大分子链上,聚酯部分形状变得不规则且体积大幅度减小,说明含磺酸基团的pet与pa6熔融挤出后界面张力很小,相容性大幅度提升。通过对比图10(a)/图3(a)/图9(b)和图10(c)可以看出p20/a80
‑
s中,pet作为分散相仍以小球的形状存在,二者界面张力较大,相容性较差,与p20/a80相差无几。在酯交换反应催化剂磺酸根化合物——对甲苯磺酸钠存在的情况下,pet和pa6在较短时间内反应程度极低,而将磺酸根化合物接枝到pet大分子链上,可在短短几分钟内生成嵌段共聚物,并实现聚酯和尼龙的微观相容混合。
[0207]
对比例4
[0208]
将pa6粒料在100℃下真空干燥10h,由注塑机制成样条,分别设定注塑温度为200℃
‑
240℃,在23℃
‑
27℃、50%湿度的环境中放置48h后进行测试,性能测试结果见表1。
[0209]
表1各实施例与对比例的性能测试结果
[0210][0211]
由实施例1、对比例1及对比例3的测试结果对比可知,直接将聚酯和尼龙进行熔融反应、或者直接将磺酸根化合物、聚酯和尼龙进行熔融反应,得到的复合材料的相容性太差,无法注塑制成样条;而采用磺酸根化合物改性的聚酯与尼龙进行熔融反应得到的复合材料,通过甲酸的刻蚀,sem观察到的图像表明具有良好的形容性,且拉伸模量能达到1.33gpa,拉伸强度能达到70.4mpa,断裂伸长率能达到401.9%,冲击强度能达到1.9kj/m2。
[0212]
将用磺酸根化合物改性得到的改性聚酯和尼龙进行熔融挤出,改性聚酯质量含量高的复合材料也无法注塑制成样条;改性聚酯质量含量低,磺酸根化合物含量较高的复合材料拉伸性能较好,部分性能指标甚至优于原料;改性聚酯质量含量低,磺酸根化合物含量
较低的复合材料拉伸性能较好,与原料尼龙相差无几,冲击强度相较于原料尼龙有大幅度提升。
[0213]
将用磺酸根化合物改性得到的改性聚酯和尼龙进行熔融挤出后进行固相缩聚反应,得到的复合材料拉伸模量与原料尼龙相差无几,拉伸强度、断裂伸长率、冲击强度均大幅度上升,优于原料尼龙。
[0214]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。不同实施例中的技术特征体现在同一附图中时,可视为该附图也同时披露了所涉及的各个实施例的组合例。
[0215]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对申请专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1