聚乙交酯的合成方法、聚乙交酯、聚乙交酯的应用以及合成装置与流程
1.本发明涉及聚合物合成技术领域,具体涉及一种聚乙交酯的合成方法、聚乙交酯、聚乙交酯的应用以及合成装置。
背景技术:
2.随着石油基塑料污染的严峻形势,限塑已成为全球共识。聚乳酸、聚乙交酯等环状聚酯作为典型的可降解聚酯,得到了市场的广泛关注。聚乙交酯因其优良的物理性能,在手术缝合线、人造皮肤及血管、骨骼固定及修复、药物控缓释、组织工程支架等医用领域得到了应用。另外,在页岩油、页岩气等非常规油气开采领域中,由聚乙交酯制备的可降解桥塞及暂堵剂具有良好的机械性能和快速降解的特点,可替代传统的镁合金材料,减少环境污染,在工业领域也得到了广泛的应用。
3.现有的聚乙交酯合成方法主要存在如下缺陷:
4.①
副反应多;
5.②
反应不可控。
6.有鉴于此,特提出本发明。
技术实现要素:
7.本发明的目的之一在于提供一种聚乙交酯的合成方法,以缓解现有聚乙交酯合成方法中的副反应多和反应不可控的技术问题。
8.本发明的目的之二在于提供一种聚乙交酯,具有产品分子量大和力学性能好的优点。
9.本发明的目的之三在于提供一种聚乙交酯在医学领域的应用。
10.本发明的目的之四在于提供一种聚乙交酯的合成装置,操作简单,过程可控,产品质量稳定,适用于大规模工业化生产。
11.为解决上述技术问题,本发明特采用如下技术方案:
12.本发明的第一方面提供了一种聚乙交酯的合成方法,将乙交酯、催化剂和引发剂混合后熔解得到的熔解液;再将得到的熔解液进行聚合得到所述的聚乙交酯。
13.所述熔解的温度为90
‑
150℃;
14.所述聚合的温度为150
‑
200℃。
15.可选地,所述熔解的温度为110
‑
130℃。
16.优选地,所述聚合的温度为150~165℃。
17.可选地,所述聚合的时间为5~96h。
18.优选地,所述聚合的时间为12~78h。
19.优选地,所述聚合的时间为48
‑
72h。
20.可选地,所述催化剂包括锡类催化剂。
21.优选地,所述锡类催化剂包括氯化亚锡、二水合氯化亚锡、氯化锡、五水合氯化锡、辛酸亚锡和乙酸亚锡中的至少一种。
22.优选地,所述催化剂的添加量为乙交酯质量的0.001
‑
0.1%。
23.优选地,所述催化剂的添加量为乙交酯质量的0.001
‑
0.05%。
24.可选地,所述引发剂包括一元醇。
25.优选地,所述一元醇包括正十二醇、正十八醇或丁醇中的至少一种。
26.优选地,所述引发剂的添加量为乙交酯质量的0.001
‑
0.1%。
27.优选地,所述引发剂的添加量为乙交酯质量的0.05
‑
0.1%。
28.本发明的第二方面提供了一种聚乙交酯,由第一方面所述的合成方法合成得到。
29.本发明的第三方面提供了第二方面所述的聚乙交酯在外科手术缝线、组织修复材料和药物控制释放体系中的应用。
30.本发明的第四方面提供了一种聚乙交酯的合成装置,包括相互连通的熔解瓶和聚合瓶;
31.其中,所述熔解容器还连接有抽真空装置。
32.可选地,所述熔解容器内部设置有搅拌桨。
33.本发明提供的聚乙交酯的合成方法,在熔解和聚合阶段采用不同的温度控制,在相对低温下使乙交酯充分熔解,液体状态下的乙交酯与引发剂、催化剂等接触机会均等,反应均一性好;之后在相对高温下聚合,有利于降低产物的分子量分布范围,使产物的分子量分布范围变窄。同时避免了在熔解过程中,因乙交酯的竞聚率大而发生的部分单体尚未熔解而部分单体开始聚合,从而引发的分子量分布问题。采用不同的温度控制可以降低分子内的酯交换和分子间的酯交换反应,还可以降低降解等副反应的发生,有利于反应的正向进行,使聚乙交酯的分子量加大,得到的聚乙交酯的分子量在30
‑
50万,同时其力学强度提高,表现出优于传统塑料的力学性能。
34.本发明提供的聚乙交酯具有产品分子量大和力学性能好的优点,可广泛应用于工业、食品、医疗等领域。
35.本发明提供的聚乙交酯的合成装置,操作简单,过程可控,产品质量稳定,适用于大规模工业化生产。
附图说明
36.为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
37.图1为实施例1提供的聚乙交酯合成装置;
38.图2为实施例3合成的聚乙交酯的弯曲强度与其他塑料的性能对比图;
39.图3为实施例3合成的聚乙交酯的拉伸强度与其他塑料的性能对比图。
40.图标:1
‑
熔解瓶;11
‑
搅拌桨;12
‑
出料口;13
‑
导管;2
‑
聚合瓶;21
‑
进料口;22
‑
气球;3
‑
连接管。
具体实施方式
41.为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。本发明实施例的组件可以以各种不同的配置来布置和设计。
42.聚乙交酯作为生物可降解型的大分子聚合物,是目前最引人瞩目的绿色环保型高分子材料之一,在医疗、工业及食品领域有着广泛的应用。本发明对精制的乙交酯在无水、无氧及惰性气体氛围中,在催化剂、引发剂及一定的温度条件下进行开环聚合,制备出了分子量高、物理性能优异的乙交酯均聚物。
43.根据本发明的第一方面提供的一种聚乙交酯的合成方法,将乙交酯、催化剂和引发剂混合后熔解得到的熔解液;再将得到的熔解液进行聚合得到所述的聚乙交酯。
44.所述熔解的温度为90
‑
150℃;
45.所述聚合的温度为150
‑
200℃。
46.本发明提供的聚乙交酯的合成方法,在熔解和聚合阶段采用不同的温度控制,将熔解和聚合反应分步骤进行,避免乙交酯因高竞聚率导致的部分单体尚未熔解完全,部分单体已经开始低聚,生成的低聚物会包裹尚未熔解的单体,从而导致不能参与反应的单体偏多,影响产品品质。熔解和聚合分步骤进行可降低分子内的酯交换和分子间的酯交换反应,还可以降低降解等副反应的发生,有利于反应的正向进行,使聚乙交酯的分子量加大,其力学强度提高,表现出优于传统塑料的力学性能。
47.聚乙交酯,又名聚羟基乙酸或聚乙醇酸,英文简称pga,化学式(c4h4o4)
n
,是一种高结晶(结晶度为46%
‑
52%)、可生物降解的线性脂肪族聚酯。pga机械性能优异,具有良好的可加工性;因其热分解温度远高于其熔融温度,又表现出典型的非牛顿型假塑性流体特性,故能用通用的设备进行挤出、注塑、纺丝、吹塑等。pga的降解主要是通过水解,也可以在自然条件下通过微生物进行代谢,最终分解成水和二氧化碳。pga的水解是因为主链中酯键的存在,水解过程存在两个步骤:
48.(1)水先扩散到聚合物母体的非晶体区,使酯键裂解;
49.(2)在非晶区侵蚀后开始,聚合物的结晶区因水解而裂解,结晶区域的聚合物链瓦解崩溃。
50.pga的降解产物乙醇酸是无毒的,它能进入三羧酸循环,之后变成水和二氧化碳排出;降解的过程会存在一定程度上的自催化降解,当部分pga降解后,其降解产物的一端因含有羧基而呈酸性,此羧基官能团会继续攻击pga链段中的酯键,从而加速整个降解过程。
51.反应方程式如式(1)所示:
[0052][0053]
熔解是指乙交酯由固态转变为液态的相变过程。乙交酯的熔点是84℃,因为熔解过程中温度不变,从外界吸热。熔解的温度选择为乙交酯单体的熔解温度之上聚合温度之下,因此乙交酯熔解的温度选为90
‑
150℃,优选为110
‑
130℃。
[0054]
在本发明的一些实施方式中,乙交酯熔解的温度典型但非限制性的为90℃,100℃,110℃,115℃,120℃,125℃,130℃,140℃或150℃。
[0055]
聚合是指乙交酯单体通过相互连接成为链状大分子,得到分子量大的pga的过程。乙交酯聚合的温度选为150
‑
200℃,优选为150~165℃。
[0056]
当聚合温度大于200℃时,pga易碳化裂解使产物颜色偏深,影响pga分子量和反应的进行。
[0057]
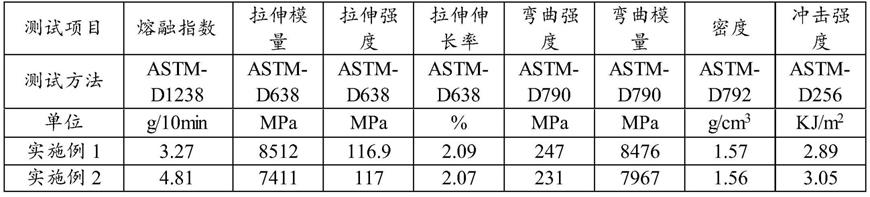
在本发明的一些实施方式中,乙交酯聚合的温度典型但非限制性的为150℃,155℃,160℃,165℃,170℃,180℃,190℃或200℃。
[0058]
pga合成过程中,体系中的水和氧会使链反应终止,因此反应在无水无氧及氮气氛围保护下进行。
[0059]
优选地,所述聚合的时间为5~96h,优选为12~78h,更优选为48
‑
72h。
[0060]
聚合时间对pga的影响是随着反应时间的延长,产率呈增加趋势,但pga的分子量呈先增加后减小的趋势,特别是当聚合时间大于96h时,pga分子在辛酸亚锡的催化下发生分子内酯交换反应,或分子链回咬生成环状低聚物,造成pga分子量的降低。
[0061]
在本发明的一些实施方式中,聚合的时间典型但非限制性的为5h,8h,12h,16h,20h,24h,28h,32h,36h,40h,44h,48h,52h,56h,60h,64h,68h,72h,76h,80h,84h,88h,92h或96h。
[0062]
可选地,所述催化剂包括锡类催化剂。
[0063]
优选地,所述锡类催化剂包括氯化亚锡、二水合氯化亚锡、氯化锡、五水合氯化锡、辛酸亚锡和乙酸亚锡中的至少一种。
[0064]
在本发明的一些实施方式中,锡类催化剂典型但非限制性的为氯化亚锡、二水合氯化亚锡、氯化锡、五水合氯化锡、辛酸亚锡或乙酸亚锡中的一种或几种。
[0065]
优选地,所述催化剂的添加量为乙交酯质量的0.001
‑
0.1%,优选为0.001
‑
0.05%。
[0066]
当催化剂添加量大于0.1%时,形成的活性位较多,使得pga的分子量较低,特性黏数较小。而且聚合反应本身为可逆反应,当催化剂添加量大于0.1%时,pga在后续热加工时会加速热降解。当催化剂添加量在0.001
‑
0.1%范围时,形成的活性位减少,因此分子量提高,特性黏数也增大。当催化剂添加量低于0.001%时,活性链数量进一步减少,长分子链易发生链转移反应,导致聚合产物的特性黏数减小。
[0067]
在本发明的一些实施方式中,催化剂的添加量典型但非限制性的为乙交酯质量的0.001%,0.005%,0.01%,0.05%或0.1%。
[0068]
引发剂可以引发乙交酯的链反应。可选地,所述引发剂包括一元醇。
[0069]
优选地,所述一元醇包括正十二醇、正十八醇或丁醇中的至少一种。
[0070]
在本发明的一些优选实施方式中,一元醇典型但非限制性的为正十二醇、正十八醇或丁醇。
[0071]
优选地,所述引发剂的添加量为乙交酯质量的0.001
‑
0.1%,优选为0.05
‑
0.1%。
[0072]
在本发明的一些实施方式中,引发剂的添加量典型但非限制性的为乙交酯质量的0.001%,0.005%,0.01%,0.05%或0.1%。
[0073]
根据本发明的第二方面提供的一种聚乙交酯由第一方面所述的合成方法合成得
到。
[0074]
本发明提供的pga具有产品分子量大和力学性能好的优点,可广泛应用于工业、食品、医疗等领域。
[0075]
根据本发明第三方面提供的聚乙交酯在外科手术缝线、组织修复材料或药物控制释放体系中的应用。
[0076]
根据本发明的第四方面提供的一种pga的合成装置,包括相互连通的熔解瓶和聚合瓶;
[0077]
其中,所述熔解瓶还连接有抽真空装置。
[0078]
本发明提供的pga的合成装置,操作简单,过程可控,产品质量稳定,适用于大规模工业化生产。
[0079]
可选地,所述熔解容器内部设置有搅拌桨。
[0080]
下面结合实施例,对本发明的一些实施方式作详细说明。在不冲突的情况下,下述的实施例及实施例中的特征可以相互组合。
[0081]
本发明中实施例和对比例中所用的乙交酯规格为精制,可通过市售购买得到,其他原料均为常规原料,可通过市售购买得到。
[0082]
实施例1
[0083]
本实施例提供一种聚乙交酯的合成装置,如图1所示,本实施例提供的聚乙交酯合成装置包括熔解瓶1和聚合瓶2,所述熔解瓶1与聚合瓶2通过导管13连通。所述熔解瓶1出口设置有导管13,导管13连接有抽真空装置。熔解瓶1和聚合瓶2独立设置有加热装置。所述熔解瓶1内部设置有搅拌桨11。熔解瓶1和聚合瓶2设有进料口。所述熔解瓶1设有出料口12。熔解瓶的出料口12与聚合瓶的进料口21通过连接管3相连通。聚合瓶2设有正负压显示装置气球22。气球充满时,表示的是合成装置中为正压,气球瘪掉时表示合成装置中为负压。导管13外接数显真空计来显示内部的压力。
[0084]
实施例2
[0085]
本实施例提供一种聚乙交酯,使用实施例1提供的合成装置进行合成。取精制的乙交酯200g,催化剂辛酸亚锡0.1g,正十二醇0.2ml于熔解瓶中,将合成装置密封并抽真空,通入氮气置换3次,反应过程中全程氮气氛围保护。130℃熔融完全后将熔液通过连接管转移到聚合瓶中,聚合瓶中提前设置温度为170℃,反应48小时。反应结束后,用液氮将聚合瓶冻碎,取出成品,破碎机破碎,干燥,待检测。
[0086]
实施例3
[0087]
本实施例提供一种聚乙交酯,与实施例2不同的是,引发剂为正十八醇0.2g,熔融温度为140℃,聚合温度为160℃,反应60小时,其余步骤均与实施例2相同,在此不再赘述。
[0088]
实施例4
[0089]
本实施例提供一种聚乙交酯,与实施例2不同的是,催化剂为氯化亚锡0.05g,正十二醇的用量为0.1ml,熔融温度为120℃,聚合温度为150℃,反应72小时。其余步骤均与实施例2相同,在此不再赘述。
[0090]
实施例5
[0091]
本实施例提供一种聚乙交酯,与实施例3不同的是,熔融温度为90℃,聚合温度为150℃,反应96小时。其余步骤均与实施例3相同,在此不再赘述。
[0092]
实施例6
[0093]
本实施例提供一种聚乙交酯,与实施例3不同的是,熔融温度为110℃,聚合温度为165℃,反应72小时。其余步骤均与实施例3相同,在此不再赘述。
[0094]
实施例7
[0095]
本实施例提供一种聚乙交酯,与实施例3不同的是,催化剂辛酸亚锡的量为0.002g,其余步骤均与实施例3相同,在此不再赘述。
[0096]
实施例8
[0097]
本实施例提供一种聚乙交酯,与实施例3不同的是,引发剂的量为0.002g,其余步骤均与实施例3相同,在此不再赘述。
[0098]
实施例9
[0099]
本实施例提供一种聚乙交酯,与实施例3不同的是,催化剂辛酸亚锡的量为0.02g,其余步骤均与实施例3相同,在此不再赘述。
[0100]
实施例10
[0101]
本实施例提供一种聚乙交酯,与实施例3不同的是,引发剂的量为0.02g,其余步骤均与实施例3相同,在此不再赘述。
[0102]
对比例1
[0103]
本对比例提供一种聚乙交酯,取精制的乙交酯200g,催化剂辛酸亚锡0.1g和正十八醇0.2g于反应釜中进行反应,将反应釜密封并抽真空后通入氮气,置换三次。将反应釜升温至140℃反应60h后取出聚合物,干燥待检测。
[0104]
试验例1
[0105]
将实施例1
‑
10和对比例1得到的聚乙交酯制成条状pga(10*30*2mm)的制品,置于23℃以上在六氟异丙醇中7天后测得的质量损失,结果如表1所示。
[0106]
表1聚乙交酯质量损失表
[0107]
项目质量损失实施例1a实施例2a实施例3a实施例4a实施例5a实施例6a实施例7a实施例8a实施例9a实施例10a对比例1b
[0108]
备注:a:质量损失≤1%;b:质量损失>1%。
[0109]
实施例1
‑
10得到的聚乙交酯在溶剂内浸泡7天后基本无质量损失,且无溶胀现象。而对比例1得到的聚乙交酯,质量损失为>1%,证明实施例1
‑
10得到的聚乙交酯分子量均大于对比例1得到的聚乙交酯分子量,基本不溶于六氟异丙醇。
[0110]
将对比例1得到的聚乙交酯进行分子量测试:取约10mgpga加入二甲基亚砜0.5ml,在150℃下加热溶解,冷却至室温。使用凝胶色谱gpc(检测器ri,标准物质聚苯乙烯)对该溶液进行测试,使用含有5mmol/l三氟乙酸钠的六氟异丙醇,进行分子量测试,测得对比例1得到的聚乙交酯分子量为121600。
[0111]
试验例2
[0112]
将实施例1
‑
10和对比例1得到的聚乙交酯进行熔融指数的测定。
[0113]
根据astm
‑
d1238或者gb/t 3682.1
‑
2018《塑料
‑
热塑性塑料熔体质量流动速率(mfr)和熔体体积流动速率(mvr)的测定中的相关规定,采用质量法测定聚乙交酯的熔融指数,其中测试温度设定为250℃,负载为2.16kg(在测定熔融指数之前,物料在70℃的真空干燥箱中干燥24小时及以上,充分除去水分,并在熔指测试之前测试水分残留需≤100ppm,减少水分对熔指结果的影响),得到的数据如表2所示。
[0114]
试验例3
[0115]
将实施例1
‑
10和对比例1得到的聚乙交酯进行力学性能的测定。
[0116]
根据astm
‑
d638塑料拉伸性能的试验方法、astm
‑
d790增强和未增强塑料弯曲试验标准方法、astm
‑
d256塑料冲击试验方法进行标准样品的制作及检测,得到的数据如表2所示。
[0117]
表2聚乙交酯性能数据表
[0118][0119][0120]
试验例4
[0121]
将实施例3得到的pga和传统的塑料如pa6、pps和peek进行力学性能的测定。
[0122]
pa6是指尼龙6,购买的厂家为美国杜邦,型号为7304nc010。
[0123]
pps是指聚苯硫醚,购买的厂家为日本东丽,型号为ar10m。
[0124]
peek是指聚醚醚酮,购买的厂家为德毅新材料,型号为peek100。
[0125]
根据astm
‑
d638塑料拉伸性能的试验方法、astm
‑
d790增强和未增强塑料弯曲试验标准方法、astm
‑
d256塑料冲击试验方法进行标准样品的制作及检测,得到的结果如图2和图3所示。
[0126]
通过表1和表2可以看出,本发明实施例1
‑
10提供的聚乙交酯因分子量大,不溶于
六氟异丙醇,因此无法用传统的测量pga分子量的方法进行测量。本发明制得的聚乙交酯的熔融指数只有3.27g/10min,根据熔融指数计算出本发明实施例1
‑
10提供的聚乙交酯的分子量在30
‑
50万之间。可以看出本发明提供的聚乙交酯合成方法得到的聚乙交酯分子量更大,反应过程中副反应和降解反应相对较少,小分子量的聚乙交酯能聚合成更高分子量的聚乙交酯,使其力学强度提高,如图2和图3所示,优于传统塑料的力学性能。
[0127]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1