2,3-双(4-羟苯基)丙腈的合成方法与流程
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法
技术领域
1.本发明涉及电解液添加剂技术领域,具体为2,3
‑
双(4
‑
羟苯基)丙腈的合成方法。
背景技术:
2.锂离子电池时目前最常见的电能存储装置,其相较于传统蓄电池有着更高的储电量和更稳定的性能,影响锂离子电池性能的关键因素在于有机电解液的各方面性能,在电池生产中在电解液中添加含有2,3
‑
双(4
‑
羟苯基)丙腈的电解液添加剂以增加电解液氢离子浓度从而降低电能转换过程中的损耗。
3.目前对2,3
‑
双(4
‑
羟苯基)丙腈的制造和提纯方法还不够完善,由制造过程的各个参数的不确定性导致产能较低。
技术实现要素:
4.本发明的目的在于提供2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,以解决上述背景技术中提出的问题。
5.为实现上述目的,本发明提供如下技术方案:
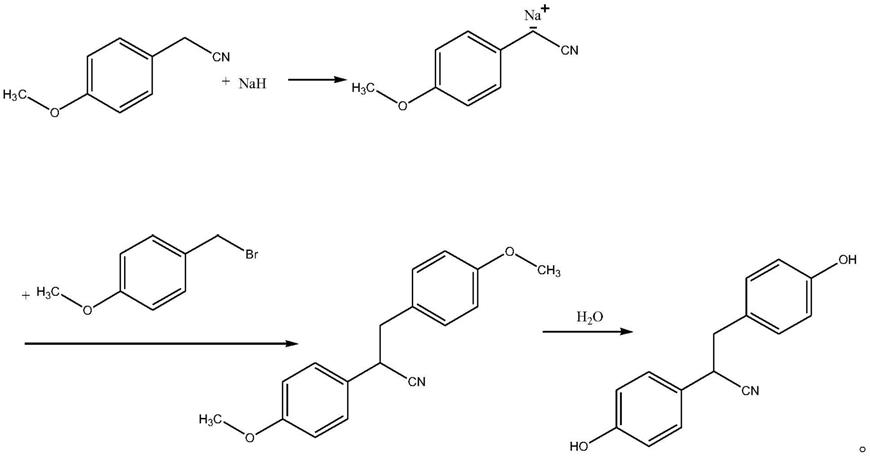
6.2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,是取对甲氧基苯乙腈溶于有机溶剂中,
‑
5~0℃加入氢化钠,维持
‑
5~0℃反应15~25min,再缓慢滴加4
‑
甲氧基溴苄,进行亲核取代反应,反应完成后加水淬灭,再经后处理,即得所述2,3
‑
双(4
‑
羟苯基)丙腈,具体化学反应式如下:
[0007][0008]
进一步地,所述亲核取代反应的温度为室温、时间为8~15h。
[0009]
进一步地,所述对甲氧基苯乙腈、氢化钠、4
‑
甲氧基溴苄及水四者之间的摩尔比为1:1.1~1.3:1.8~2.2:2.3~3。
[0010]
进一步地,所述对甲氧基苯乙腈与有机溶剂的重量体积比为1g:8~12ml。
[0011]
进一步地,所述的有机溶剂为四氢呋喃。
[0012]
进一步地,所述后处理是在加水淬灭后所得体系中,加入乙酸乙酯萃取,再经浓缩、柱层析纯化。
[0013]
进一步地,所述柱层析的洗脱剂是体积比为1~4:1的正己烷与乙酸乙酯的混合溶液。
[0014]
进一步地,所述萃取后所得乙酸乙酯相还需加入无水硫酸镁进行干燥。
[0015]
与现有技术相比,本发明的有益效果是:
[0016]
本发明以对甲氧基苯乙腈和4
‑
甲氧基溴苄为原料,以氢化钠拔除对甲氧基苯乙腈的氰基临位碳上的氢,形成碳负与钠离子结合,同时释放一分子氢气,然后再用碳负进攻溴的临位碳发生亲核取代反应,再水解得粗产品,粗产品经纯化得最终的2,3
‑
双(4
‑
羟苯基)丙腈,并在生产过程中详细的阐述产品原料的使用范围,各阶段反应条件的控制保证了该生产过程的产品收率和产品纯度的最大化。通过合理控制反应原料用量及反应过程,进一步提高产品的收率,使收率达到84.94%以上,纯度达99.51%以上。
附图说明
[0017]
图1为本发明实施例1中制备的2,3
‑
双(4
‑
羟苯基)丙腈产物的氢核磁谱图;
[0018]
图2为本发明实施例1中制备的2,3
‑
双(4
‑
羟苯基)丙腈产物的碳核磁谱图。
具体实施方式
[0019]
下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0020]
实施例一:
[0021]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0022]
取1.47g对甲氧基苯乙腈(10mmol)加至15ml四氢呋喃中搅拌溶解,降温至0℃,搅拌过程中加入0.44g含量为60wt%氢化钠的矿物油分散液(11mmol的nah),维持0℃反应20min,反应完成后,维持0℃缓慢滴加2.9ml的4
‑
甲氧基溴苄(19.9mmol),滴加时长为15min,滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应10h后加入44ml水淬灭,再加入乙酸乙酯萃取(50ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.31g粗产物;具体化学反应式为:
[0023][0024]
以体积比为2:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.08g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为87.03%,熔点为114~115℃,纯度为99.62%。
[0025]
2,3
‑
双(4
‑
羟苯基)丙腈的氢核磁谱图参见图1,氢核磁表征数据为:1hnmr(500mhz,cdcl3)δ7.15(d,j=8.8hz,2h),7.03(d,j=8.6hz,2h),6.87(d,j=8.8hz,2h),6.82(d,j=8.8hz,2h),3.91(dd,j=8.0,6.5hz,1h),3.81(s,3h),3.79(s,3h),3.15
‑
3.00(m,2h)。
[0026]
2,3
‑
双(4
‑
羟苯基)丙腈的碳核磁谱图参见图2,碳核磁表征数据为:
13
cnmr(126mhz,cdcl3)δ159.3,158.8,130.3,128.6,128.4,127.2,120.7,114.3,113.9,55.3,55.2,41.5,39.2。
[0027]
实施例二:
[0028]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0029]
取1.47g对甲氧基苯乙腈(10mmol)加至17.6ml四氢呋喃中搅拌溶解,降温至
‑
5℃,搅拌过程中加入0.52g含量为60wt%氢化钠的矿物油分散液(13mmol的nah),维持
‑
5℃反应25min,反应完成后,维持
‑
5℃缓慢滴加3.2ml的4
‑
甲氧基溴苄(22mmol),滴加时长为20min,滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应12h后加入54ml水淬灭,再加入乙酸乙酯萃取(70ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.27g粗产物;
[0030]
以体积比为4:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.03g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为84.94%,熔点为113~114℃,纯度为99.60%。
[0031]
实施例三:
[0032]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0033]
取1.47g对甲氧基苯乙腈(10mmol)加至11.8ml四氢呋喃中搅拌溶解,降温至
‑
3℃,搅拌过程中加入0.48g含量为60wt%氢化钠的矿物油分散液(12mmol的nah),维持
‑
3℃反应20min,反应完成后,维持
‑
3℃缓慢滴加2.6ml的4
‑
甲氧基溴苄(18mmol),滴加时长为10min,
滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应8h后加入41.4ml水淬灭,再加入乙酸乙酯萃取(50ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.27g粗产物;
[0034]
以体积比为1:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.05g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为85.77%,熔点为114~116℃,纯度为99.53%。
[0035]
实施例四:
[0036]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0037]
取1.47g对甲氧基苯乙腈(10mmol)加至14ml四氢呋喃中搅拌溶解,降温至
‑
2℃,搅拌过程中加入0.48g含量为60wt%氢化钠的矿物油分散液(12mmol的nah),维持
‑
2℃反应18min,反应完成后,维持
‑
2℃缓慢滴加3ml的4
‑
甲氧基溴苄(20.5mmol),滴加时长为15min,滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应15h后加入50ml水淬灭,再加入乙酸乙酯萃取(60ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.29g粗产物;
[0038]
以体积比为3:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.07g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为86.61%,熔点为113~116℃,纯度为99.51%。
[0039]
实施例五:
[0040]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0041]
取1.47g对甲氧基苯乙腈(10mmol)加至16ml四氢呋喃中搅拌溶解,降温至0℃,搅拌过程中加入0.44g含量为60wt%氢化钠的矿物油分散液(11mmol的nah),维持0℃反应23min,反应完成后,维持0℃缓慢滴加3.1ml的4
‑
甲氧基溴苄(21.3mmol),滴加时长为20min,滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应11h后加入48ml水淬灭,再加入乙酸乙酯萃取(55ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.37g粗产物;
[0042]
以体积比为3:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.04g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为85.36%,熔点为114~116℃,纯度为99.54%。
[0043]
实施例六:
[0044]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0045]
取1.47g对甲氧基苯乙腈(10mmol)加至17ml四氢呋喃中搅拌溶解,降温至
‑
4℃,搅拌过程中加入0.44g含量为60wt%氢化钠的矿物油分散液(11mmol的nah),维持
‑
4℃反应23min,反应完成后,维持
‑
4℃缓慢滴加3ml的4
‑
甲氧基溴苄(20.5mmol),滴加时长为15min,滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应9h后加入45ml水淬灭,再加入乙酸乙酯萃取(60ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.29g粗产物;
[0046]
以体积比为2:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.06g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为86.19%,熔点为114~117℃,纯度为99.52%。
[0047]
实施例七:
[0048]
2,3
‑
双(4
‑
羟苯基)丙腈的合成方法,其步骤如下:
[0049]
取1.47g对甲氧基苯乙腈(10mmol)加至15ml四氢呋喃中搅拌溶解,降温至
‑
1℃,搅拌过程中加入0.44g含量为60wt%氢化钠的矿物油分散液(11mmol的nah),维持
‑
1℃反应23min,反应完成后,维持
‑
1℃缓慢滴加2.8ml的4
‑
甲氧基溴苄(19.2mmol),滴加时长为15min,滴加完成后,自然升温至室温,搅拌进行亲核取代反应,反应13h后加入43ml水淬灭,再加入乙酸乙酯萃取(50ml
×
2),合并乙酸乙酯相,加入无水硫酸镁干燥,减压浓缩,得2.33g粗产物;
[0050]
以体积比为4:1的正己烷和乙酸乙酯的混合溶液为洗脱剂,对所得粗产物进行柱层析纯化,收集、浓缩洗脱液,即得2.07g的2,3
‑
双(4
‑
羟苯基)丙腈,收率为86.61%,熔点为114~115℃,纯度为99.59%。
[0051]
对比例一:
[0052]
购买市场上常见的2,3
‑
双(4
‑
羟苯基)丙腈成品,通过气相色谱仪检测其纯度为98.01%。
[0053]
将实施例一至实施例七的成品产物的纯度与对比例一的产品收率和纯度进行比对,其比对结果如表1所示:
[0054]
表1
[0055][0056][0057]
应用实验:
[0058]
以三元材料ncm(622)锂为正极材料,负极采用中间相碳微球,正负极集流体分布为铝箔和铜箔,隔膜采用陶瓷隔膜组成软包电池,注入电解液后,在手套箱中组装成软包电池,静置8小时后进行测试。在室温25℃恒温下分别以1/10c 3.0v到4.2v以上进行充放电对电池进行活化,得到待测试电池。所测试的电解液包括基础电解液e1和电解液e2,其成分如下所示:
[0059]
1、基础电解液e1
[0060]
ec:solution
‑
1:dec=3:3:4(v:v:v),lipf6:1.0m,0.5%lifsi,1%vc
[0061]
2、电解液e2
[0062]
ec:solution
‑
1:dec=3:3:4(v:v:v),lipf6:1.0m,0.5%lifsi,1%vc,1%2,3
‑
双(4
‑
羟苯基)丙腈添加剂
[0063]
测试结果:
[0064]
1、60℃循环后测试结果如下:
[0065]
表2
[0066][0067]
2、将电池搁置在低温箱中,分别控制温度为
‑
30℃或
‑
40℃,搁置时间240min,随后测量电池的容量保持率。
[0068]
表3
[0069]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1