一种甲磺酸奥希替尼的制备方法与流程
1.本发明属于化学合成技术领域,具体涉及一种甲磺酸奥希替尼的制备方法。
背景技术:
2.肺癌病人根据癌细胞形态分为“小细胞肺癌”和“非小细胞肺癌”。约85%肺癌病人都是“非小细胞肺癌”。表皮生长因子受体(egfr)突变是东亚患者包括中国非小细胞肺癌患者最常见的基因变异,约占50%~60%,大部分egfr突变的非小细胞肺癌患者使用表皮生长因子受体酪氨酸激酶抑制剂(egfr
‑
tki)可以取得比较好的治疗效果。目前上市的egfr
‑
tki靶向药物大致可分为三代,吉非替尼、厄洛替尼为代表的第一代egfr
‑
tki靶向药物,治疗效果好,患者有效率约为50%~70%,但是60%左右的患者在使用约10个月后出现egfr t790m作用位点耐药突变。阿法替尼、达克替尼为代表的第二代egfr
‑
tki靶向药物在临床实际运用过程中,其疗效并没有优于第一代的靶向药,而且相对而言第二代靶向药的副作用更大。更重要的是,第一代靶向药出现耐药后,第二代靶向药也不能克服耐药,所以现在第二代的靶向药临床运用并不广泛。使用现有egfr
‑
tki靶向药物治疗后产生t790m耐药突变的患者,后续药物治疗的选择非常有限。第三代egfr
‑
tki新药奥希替尼,可用于治疗t790m耐药突变、且一二代egfr
‑
tki药物治疗无效的转移性非小细胞肺癌患者。奥希替尼是目前唯一能有效治疗egfr t790m突变阳性转移性的非小细胞肺癌的药物,将使近30%的egfr突变患者生存时间延长。
3.甲磺酸奥希替尼(osimertinib mesilate,azd9291),中文化学名为:n
‑
(2
‑
((2
‑
(二甲基氨基)乙基)(甲基)氨基)
‑4‑
甲氧基
‑5‑
((4
‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑2‑
基)氨基)苯基)丙烯酰胺甲磺酸盐,由英国阿斯利康公司研发,并于2015年11月13日美国食品和药物管理局(fda)以加速批准的方式提前批准上市,商品名为:泰瑞沙(tagrisso)。
4.文献报道的甲磺酸奥希替尼的制备方法,主要有以下几种:
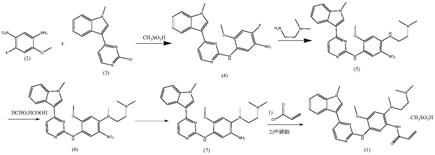
5.方法一:阿斯利康公司的专利cn103702990b中,以2
‑
甲氧基
‑4‑
氟苯胺、1
‑
甲基吲哚和2,4
‑
二氯嘧啶为原料,经过6步反应,最终得到产品奥希替尼,合成路线如下:
[0006][0007]
该路线中4
‑
氟
‑2‑
甲氧基苯胺(式(9))采用混酸硝化,氨基容易被硫酸氧化,造成收率偏低,且因4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺(式(2))与硫酸成盐,后处理需采用大量碱中和,产生远大于4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺重量的无机盐,三废难以处理。文献中制备式(4)化合物,采用对甲苯磺酸作催化剂,因对甲苯磺酸本身含水,在反应过程中极易造成3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚(式(3)化合物)与水反应,生成2
‑
羟基杂质,导致式(4)化合物纯度难以提高,且含有大量的对甲苯磺酸难以去除,而残留的对甲苯磺酸,在后继采用醇类溶剂的步骤中,极易产生具有潜在基因毒性的杂质,如对甲苯磺酸乙酯等,导致成品有潜在的致癌风险。
[0008]
制备式(6)化合物采用的n,n,n
’‑
三甲基乙二胺,市售的纯度均在98%左右,其主要杂质为n,n,n’,n
’‑
四甲基乙二胺和n,n
’‑
二甲基乙二胺,其中n,n
’‑
二甲基乙二胺参与反应,且生成多个副产物,导致式(6)化合物的纯度难以提高,进而影响成品甲磺酸奥希替尼的纯度和质量,且n,n,n
’‑
三甲基乙二胺高昂的市售价格,不利于奥希替尼产品的市场推广和使用。且式(6)的纯化需要通过柱层析纯化,不利于工业化生产。
[0009]
方法二:吉民、李元等人在专利cn104817541b中报道了4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺通过氨基保护,首先与n,n,n
’‑
三甲基乙二胺及丙烯酰氯反应,再与3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚反应制备得到奥希替尼的合成路线,合成路线如下:
[0010][0011]
该路线同样存在n,n,n
’‑
三甲基乙二胺的高昂价格问题及其引入的杂质难以去除问题,另外,其制备奥希替尼的步骤,采用对甲苯磺酸做催化剂,异丁醇做溶剂,在酸性环境下,极易产生具有遗传毒性的杂质对甲苯磺酸异丁醇酯,导致产品存在重大的安全风险。
[0012]
基于此,开发一种低成本、高收率、环境友好的甲磺酸奥希替尼的合成方法是当前需要解决的问题。
技术实现要素:
[0013]
本发明克服了上述现有技术的不足,提供一种甲磺酸奥希替尼的制备方法。本发明以4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺与3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚经缩合反应,再与n,n
‑
二甲基乙二胺发生亲核取代反应,通过eschweiler
‑
clarke胺还原烷基化得到高纯度的式(6)化合物,再以水为溶剂,醋酸等做助溶剂,经催化氢化,与丙烯酰氯发生酰胺化反应得到高纯度的奥希替尼,与甲磺酸成盐得到甲磺酸奥希替尼,相比专利cn103702990b,其操作简便、对环境污染少、收率高、成本低、产品质量好,更适合于工业化生产。
[0014]
本发明的技术方案是:一种甲磺酸奥希替尼的制备方法,其特征是,包括以下步骤:
[0015]
s1:4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺(式(2)化合物)与3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚(式(3)化合物)通过缩合反应得到n
‑
(4
‑
氟
‑2‑
甲氧基
‑
t
‑
硝基苯基)
‑4‑
(1h
‑3‑
吲哚基)嘧啶
‑2‑
胺(式(4)化合物)
[0016]
s2:将式(4)化合物中的氟替换为2
‑
(二甲基氨基)乙基)(甲基)氨基,得到式(6)化合物;
[0017]
s3:式(6)化合物的硝基还原成为氨基;
[0018]
s4:然后氨基上的氢再经丙烯酰基取代得到奥希替尼,最后与甲磺酸成盐得到甲磺酸奥希替尼;
[0019]
其特征是,
[0020]
所述步骤s1中溶剂采用有机溶剂,以无水液体酸(如甲磺酸)为催化剂;
[0021]
所述步骤s2具体为:式(4)化合物在醇类溶剂中,碱存在下,与n,n
‑
二甲基乙二胺反应,生成式(5)化合物,再通过eshweiler
‑
clarke胺烷基化反应,以甲酸为溶剂,与甲醛和甲酸反应得到式(6)化合物;
[0022]
所述步骤s3具体为:式(6)化合物在水中,用酸调ph值至固体溶解,通过催化剂催化加氢,得到式(7)化合物;
[0023]
所述步骤s4中,式(7)化合物与丙烯酰氯反应得到奥希替尼。
[0024]
其合成路线如下所示:
[0025][0026]
其中,
[0027]
所述步骤s1的无水液体酸为甲磺酸、三氟乙酸、三氯乙酸中的一种,优选甲磺酸。3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚的2位氯在酸性环境中,会与水(溶剂中的水以及催化剂酸引入的水)发生取代反应,生成2为羟基取代杂质,从而影响式(4)化合物的纯度,反应式如下。因此采用无水酸和非水溶剂,可以有效避免上述副反应的发生,提高产品纯度和收率。
[0028][0029]
所述步骤s1的有机溶剂为正丁醇、二氧六环、正戊醇、2
‑
戊醇、正丙醇中的一种或两种混合,优选二氧六环。
[0030]
所述步骤s2中的醇类溶剂为乙醇、异丙醇、正丙醇、正丁醇、正戊醇、异戊醇中的一种或两种混合,优选正丁醇。从正丁醇的易回收性、价格低廉、后处理简洁性方面考虑,其他溶剂的反应性均与正丁醇类似。
[0031]
所述步骤s2中碱为二异丙基乙胺、甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾等,优选二异丙基乙胺。
[0032]
所述步骤s3中的酸为醋酸、甲酸、磷酸、三氟乙酸、三氯乙酸中的一种,优选醋酸。
[0033]
所述步骤s3中的催化剂为钯碳、铂碳、雷尼镍中的一种,优选铂碳。
[0034]
所述步骤s4中的有机溶剂为二氯甲烷、三氯甲烷、四氢呋喃、二氧六环中的一种或
两种混合,优选二氯甲烷。成盐溶剂为醋酸异丙酯。
[0035]
所述步骤s1的反应温度为70~90℃,所述步骤s2的制备式(5)化合物的反应温度为100~120℃,制备式(6)化合物反应温度为80~100℃;所述步骤s4制备奥希替尼的反应温度为
‑
5~10℃。
[0036]
优选的,本发明具体包括以下步骤:
[0037]
s1:将4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺和3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚加入到溶剂1,4
‑
二氧六环中,再加入甲磺酸,70~90℃保温反应3~8h,降温析晶,打浆,烘干得式(4)化合物;
[0038]
s2:式(4)化合物、醇类溶剂、碱及n,n
‑
二甲基乙二胺在100~120℃保温反应18~22h,反应毕,过滤,洗涤,烘干,得到式(5)化合物;将式(5)化合物、甲酸及甲醛水溶液在70~90℃下保温1.5~3.5h,减压蒸除溶剂,经后处理得式(6)化合物;
[0039]
s3:将式(6)化合物加入水中,用醋酸调ph值至固体溶解,以铂碳为催化剂,采用氢气还原反应;经后处理得到式(7)化合物;
[0040]
s4:将式(7)化合物溶解在有机溶剂中,冰浴下,将丙烯酰氯滴加到上述混合液中,0~5℃下搅拌反应,反应毕,加入水,并用碱调ph值至10以上,静置分层,有机层减压蒸干,剩余物加入醋酸异丙酯升温至回流,然后加入甲磺酸成盐,得甲磺酸奥西替尼。
[0041]
其中,步骤s2的后处理为:减压蒸除溶剂后的剩余物加入水和活性炭脱色,用氢氧化钠溶液调ph值至10,然后加入醋酸异丙酯,搅拌过夜,析出灰白色固体,过滤,水洗涤、醋酸异丙酯洗得式(6)化合物。
[0042]
其中,步骤s3的后处理为:滤除铂碳,水洗,铂碳套用,滤液用氢氧化钠溶液调ph值至10,析出固体,过滤,水洗,烘干,得到式(7)化合物。
[0043]
所述步骤s1中的无水液体酸与3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚的摩尔比为0.2:1~2:1,优选0.3:1~1:1,更进一步的优选0.5:1。
[0044]
所述步骤s2中式(4)化合物与n,n
‑
二甲基乙二胺的摩尔比为1:1.2~1:2.0,优选1:1.5;式(5)化合物与甲醛的摩尔比为1:1.5~1:3,优选为1:1.5~1:2。
[0045]
所述步骤s4中式(7)化合物与丙烯酰氯的摩尔比为1:1.0~1.2,式(7)化合物与甲磺酸的摩尔比为1:1.0~1.2。
[0046]
本发明的有益效果是:
[0047]
1、本发明式(4)化合物的制备过程中,以二氧六环等溶剂为溶剂,以甲磺酸等无水酸为催化剂,反应条件温和,避免了难去除的3
‑
(2
‑
羟基
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚,反应收率由55%提高到92%以上,纯度提高到99.5%以上。
[0048]
2、本发明采用醇类溶剂,n,n
‑
二甲基乙二胺反应,再通过eschweiler
‑
clarke胺还原烷基化反应,制备得到式(6)化合物,虽然延长了反应步骤,但避免了难以去除的杂质,同时,工艺过程不需要使用柱层析纯化步骤,更有利于工业化。
[0049]
3、本发明创造性的以水为溶剂,将式(6)化合物通过醋酸等调ph的方式,钯碳、铂碳、雷尼镍等催化加氢还原,为环保部推荐的绿色工艺,三废少,溶剂可回收套用。
[0050]
4、本发明直接采用丙烯酰氯制备奥希替尼,简化了工艺,提高了反应收率。再通过醋酸异丙酯中与甲磺酸成盐,因醋酸异丙酯的难降解性,避免了基因毒性杂质甲磺酸乙酯,甲磺酸异丙酯的生成,提高了产物甲磺酸奥希替尼的安全性,避免了因潜在的毒性杂质引
起的病人的潜在基因突变。
[0051]
综合以上,相比中国专利cn103702990b,本发明的方法操作简便、对环境污染少、收率高(总收率70%左右)、成本低、产品质量好,更适合于工业化生产。
具体实施方式
[0052]
下面结合具体实施例对本发明作更进一步的说明,以便本领域的技术人员更了解本发明,但并不因此限制本发明。
[0053]
实施例1:n
‑
(4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯基)
‑4‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑2‑
胺(式(4)化合物)的制备
[0054]
将61.4g(0.33mol)4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺,73.1g(0.3mol)3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚加入到装有1200ml的1,4
‑
二氧六环的2000ml反应瓶中,再加入14.4g(0.15mol)甲磺酸,开加热,升温至80℃,保温80℃反应5h,降温至0~10℃析晶,过滤,70%乙醇水打浆洗涤,烘干得113.2g黄色固体,收率96.0%,hplc纯度99.5%。
[0055]
实施例2:n
‑
(4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯基)
‑4‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑2‑
胺(式(4)化合物)的制备
[0056]
将61.4g(0.33mol)4
‑
氟
‑2‑
甲氧基
‑5‑
硝基苯胺,73.1g(0.3mol)3
‑
(2
‑
氯
‑4‑
嘧啶基)
‑1‑
甲基
‑
1h
‑
吲哚加入到装有1200ml的1,4
‑
二氧六环的2000ml反应瓶中,再加入17.1g(0.15mol)三氟乙酸,开加热,升温至80℃,保温80℃反应5h,降温至0~10℃析晶,过滤,70%乙醇水打浆洗涤,烘干得111.4g黄色固体,收率94.5%,hplc纯度99.7%。
[0057]
实施例3:n
’‑
(2
‑
(二甲胺基)乙基)
‑5‑
甲氧基
‑
n
’‑
(4
‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑3‑
基)硝基苯
‑
1,4
‑
二胺(式(5)化合物)的制备
[0058]
将78.6g(0.2mol)的式(4)化合物,800ml正丁醇,38.7g(0.3mol)二异丙基乙胺及26g(0.3mol)n,n
‑
二甲基乙二胺,升温至110℃,保持110℃反应20h,反应毕,降温至室温,析出黄色固体,过滤,正丁醇洗涤,烘干得84.8g黄色固体,收率92.0%,hplc纯度99.7%。
[0059]
实施例4:n
’‑
(2
‑
(二甲胺基)乙基)
‑5‑
甲氧基
‑
n
’‑
(4
‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑3‑
基)硝基苯
‑
1,4
‑
二胺(式(5)化合物)的制备
[0060]
将78.6g(0.2mol)的式(4)化合物,800ml正丁醇,28.8g(0.3mol)叔丁醇钠及26g(0.3mol)n,n
‑
二甲基乙二胺,升温至110℃,保持110℃反应20h,反应毕,降温至室温,析出黄色固体,缓慢加入200ml水,过滤,正丁醇洗涤,水洗涤,烘干得83.9g黄色固体,收率91.0%,hplc纯度99.8%。
[0061]
实施例5:n
’‑
(2
‑
(二甲胺基)乙基)
‑5‑
甲氧基
‑
n
’‑
(4
‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑3‑
基)硝基苯
‑
1,4
‑
二胺(式(5)化合物)的制备
[0062]
将78.6g(0.2mol)的式(4)化合物,800ml正丁醇,33.6g(0.3mol)叔丁醇钾及26g(0.3mol)n,n
‑
二甲基乙二胺,升温至110℃,保持110℃反应20h,反应毕,降温至室温,析出黄色固体,缓慢加入200ml水,过滤,正丁醇洗涤,水洗涤,烘干的82.7g黄色固体,收率89.7%,hplc纯度99.9%。
[0063]
实施例6:n
’‑
(2
‑
(二甲胺基)乙基)
‑5‑
甲氧基
‑
n
’‑
甲基
‑
n
’‑
(4
‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑3‑
基)硝基苯
‑
1,4
‑
二胺(式(6)化合物)的制备
[0064]
将75g(0.162mol)的式(5)化合物,250ml甲酸及19.5g(37%,0.24mol)甲醛水溶液
加入2000ml反应瓶中,缓慢升温至90℃,保温2h,减压蒸除溶剂,剩余物加入1000ml水,加入5g活性炭,继续搅拌1h,过滤除去不溶物,用氢氧化钠溶液调ph值至10,加入500ml醋酸异丙酯,搅拌过夜,析出灰白色固体,过滤,水洗涤、醋酸异丙酯洗得71.44g,收率92.8%,hplc纯度99.7%。
[0065]
实施例7:n
’‑
(2
‑
(二甲胺基)乙基)
‑5‑
甲氧基
‑
n
’‑
甲基
‑
n
’‑
(4
‑
(1
‑
甲基
‑
1h
‑
吲哚
‑3‑
基)嘧啶
‑3‑
基)苯基
‑
1,4
‑
二胺(式(7)化合物)的制备
[0066]
将70g(0.147mol)式(6)化合物,1000ml水加入2000ml反应瓶中,用醋酸调ph值至固体溶解,将物料转移至2000ml高压釜中,氮气保护下加入3.5g铂碳(以干品计,5%含量),氮气置换3次,通氢气反应3h。滤除铂碳,水洗,铂碳套用,滤液用氢氧化钠溶液调ph值至10,析出固体,过滤,水洗,烘干得类白色固体63g,收率96.3%,hplc纯度99.8%。
[0067]
实施例8:制备甲磺酸奥西替尼
[0068]
将63g(0.14mol)的式(7)化合物溶解在800ml二氯甲烷中,冰浴下,缓慢将15.2g(0.168mol)丙烯酰氯滴加到上述混合液中,将该混合物于0~5℃下搅拌反应2h,反应毕,加入500ml水,用氢氧化钠溶液调ph值至10以上,弃去水层,有机层再用500ml水洗涤一次,有机层减压蒸干,剩余物加入800ml醋酸异丙酯,升温至回流,缓慢加入14.8g(0.154mol)甲磺酸,得澄清液体,通过0.45um滤膜过滤,滤液降温0~5℃,析出白色固体,过滤,冷醋酸异丙酯洗涤得75g,收率90.0%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1