钆和基于非对映异构体富集的PCTA的螯合配体的络合物及其制备和纯化方法与流程
钆和基于非对映异构体富集的pcta的螯合配体的络合物及其制备和纯化方法
1.相关申请
2.本发明是申请日为2020年1月17日、申请号为202080009625.8、发明名称为“钆和基于非对映异构体富集的pcta的螯合配体的络合物及其制备和纯化方法”专利申请的分案申请。
技术领域
3.本发明涉及一种用于制备和纯化钆和基于非对映异构体富集的pcta的螯合配体的络合物的新方法,该方法使得能够优选地获得所述络合物的立体异构体,这些立体异构体具有在医学成像领域,尤其是在磁共振成像领域中作为造影剂应用的特别有利的物理化学性质。本发明还涉及非对映异构体富集的络合物本身,涉及包含所述络合物的组合物,并且还涉及一种通过解络合所述络合物以制备相应的螯合配体的方法,以及该配体本身。
背景技术:
4.已知许多基于镧系元素(顺磁性金属),尤其是钆(gd)的螯合物的造影剂,例如在us 4647447中所述。这些产品通常在术语gbca(基于钆的造影剂)下归为一组。有几种产品上市,其中有大环螯合物,例如基于dota(1,4,7,10
‑
四氮杂环十二烷
‑
n,n',n",n'"
‑
四乙酸)的葡甲胺钆酸盐、基于do3a
‑
butrol的钆布醇、基于hpdo3a的钆特醇,以及线性螯合物,尤其是基于dtpa(二亚乙基三胺五乙酸)或dtpa
‑
bma(钆二酰胺配体)的线性螯合物。
5.其他产品,其中一些正在开发中,代表了新一代的gbca。它们基本上是大环螯合物的络合物,例如双环多氮杂大环羧酸(ep 0 438 206)或pcta衍生物(即,最少包含3,6,9,15
‑
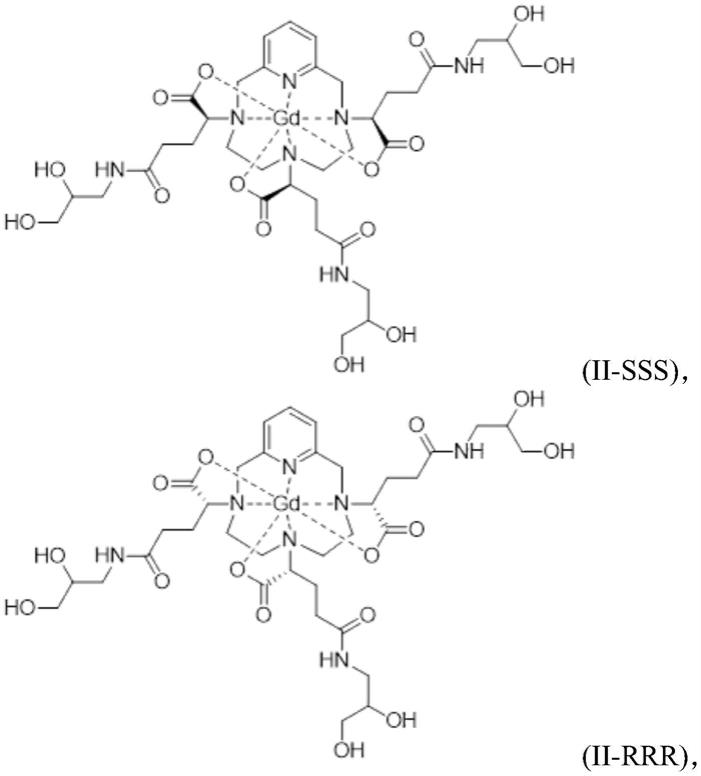
四氮杂双环[9,3,1]十五碳
‑
1(15),11,13
‑
三烯
‑
3,6,9
‑
三乙酸化学结构的衍生物),如ep 1 931 673中所述。
[0006]
ep 1 931 673中描述的基于pcta的螯合配体的络合物特别地具有以下优势:化学合成相对容易,并且还具有优于当前市场上的其他gbca的弛豫率的弛豫率(弛豫率r1在水中最高可达11
‑
12mm
‑1.s
‑1),这种弛豫率对应于这些产品的效率,并且因此对应于它们的对比能力。
[0007]
在生物体中,镧系元素(并且尤其是钆)的螯合物(或络合物)处于化学平衡状态(通过其热力学常数k
therm
表征),这可能导致所述镧系元素的不希望的释放(参见下面的反应式1):
[0008][0009]
螯合物或配体(ch)与镧系元素(l
n
)之间的络合化学平衡产生络合物ch
‑
l
n
。
[0010]
自2006年以来,一种被称为nsf(肾源性系统性纤维化或纤维源性皮肤病)的疾病至少部分与体内游离钆的释放有关。这种疾病已经导致卫生当局针对某些类别患者销售基于钆的造影剂发出警报。
[0011]
因此,已采取一些策略以完全安全的方式解决患者耐受性的复杂问题,并限制或甚至消除施用后镧系元素不希望的释放的风险。由于无论在诊断检查期间还是在剂量调整和监测治疗效果的过程中都经常重复施用造影剂,因此使该问题更加难以解决。
[0012]
此外,自2014年以来,人们曾提到在重复施用基于钆的产品(尤其是线性钆螯合物)后钆可能在大脑中沉积,特别是钆大环螯合物(例如)的这种沉积很少或根本没有被报导过。因此,鉴于稳定性不足,各国决定从市场上撤出大部分线性螯合剂,或大幅度限制其使用指示。
[0013]
因此,限制镧系元素释放到体内的风险的第一种策略是选择以尽可能高的热力学和/或动力学稳定性为特征的络合物。其原因是,络合物越稳定,随着时间推移镧系元素的释放量将越受限制。
[0014]
在现有技术中描述了用于改善镧系元素(尤其是钆)的螯合物的耐受性的其他方法。30多年前的us 5876695报告了,例如,除镧系元素螯合物外还包含额外络合剂的制剂,其旨在通过络合释放的镧系元素(gd
3+
金属离子)来防止镧系元素在体内的不希望的释放。额外螯合剂可以其游离形式或以弱络合物(典型地为钙、钠、锌或镁)的形式引入制剂中。尽管它可能与活性络合物的组成配体不同,但重要的是它与释放的镧系元素形成的络合物不如活性络合物稳定,从而防止了活性络合物与额外螯合物之间的转位反应,这将特别地具有完全消耗所述额外配体的作用,然后不再捕获释放的镧系元素。当其以游离形式添加,而不是以例如钙络合物形式添加时,这种通过转位消耗额外螯合剂的风险更加明显。
[0015]
因此,在上述两种策略中,重要的是使活性络合物尽可能稳定。
[0016]
然而,包含ep 1 931 673中所述的pyclene型结构的基于pcta的螯合配体的络合物虽然具有良好的动力学稳定性,但其热力学常数通常低于其他基于环烯的大环的络合物的热力学常数。
[0017]
对于具有以下表示的式(ii)的络合物特别地是这种情况:
[0018][0019]
具体地,如在wo 2014/174120中特别地描述的,对应于用于形成具有式(ii)的络合物的反应的热力学平衡常数(也称为稳定常数)为10
14.9
(即log(k
therm
)=14.9)。出于比较目的,1,4,7,10
‑
四氮杂环十二烷
‑
n,n',n”,n”'
‑
四乙酸(dota
‑
gd)的钆络合物的稳定常数为10
25.6
(即log(k
therm
)=25.6)。
[0020]
然而,应当注意,具有式(ii)的络合物对应于几种立体异构体,特别地由于相对于侧链接枝到其上的大环的氮原子在该络合物的侧链上存在位于α位的三个不对称碳原子。这三个不对称碳在上面表示的式(ii)中标有星号(*)。
[0021]
因此,如ep 1 931 673中所述的具有式(ii)的络合物的合成导致立体异构体混合物的产生。
[0022]
具有式(ii)的络合物的侧链的氨基丙二醇基团还包含不对称碳。因此,具有式(ii)的络合物总共包含六个不对称碳,并且因此以64种构型立体异构体的形式存在。然而,在本说明书的其余部分中,为简单起见,针对给定侧链考虑的立体异构体的唯一来源将是对应于带有羧酸酯基团的不对称碳的来源,该不对称碳在上式(ii)中标有星号(*)。
[0023]
由于这三个不对称碳中的每个均可以具有r或s绝对构型,因此具有式(ii)的络合物以八个立体异构体家族的形式存在,以下称为ii
‑
rrr、ii
‑
sss、ii
‑
rrs、ii
‑
ssr、ii
‑
rss、ii
‑
srr、ii
‑
rsr和ii
‑
srs。更准确地,根据立体化学中通常的命名法,具有式(ii)的络合物以八个非对映异构体家族的形式存在。
[0024]
如前提及的,使用术语“家族”是合理的,因为这些家族中的每一种都将几种立体异构体组合在一起,特别地由于氨基丙二醇基团中不对称碳的存在。
[0025]
然而,由于在本说明书的其余部分中将不考虑与给定的氨基丙二醇基团的不对称碳相关的立体异构体,因此将不加区别地使用术语异构体、立体异构体或非对映异构体ii
‑
rrr、ii
‑
sss、ii
‑
rrs、ii
‑
ssr、ii
‑
rss、ii
‑
srr、ii
‑
rsr和ii
‑
srs,而无需说明每个对应于一个立体异构体家族。
[0026]
发明人已经成功地通过高效液相层析法(hplc)和超高效液相层析法(uhplc)分离和鉴定了根据现有技术的方法获得的具有式(ii)的络合物的四个未分辨的峰或异构体组,其对应于四个不同的通过它们在层析图上的保留时间表征的洗脱峰,在本说明书的其余部分中将被称为iso1、iso2、iso3和iso4。通过进行ep 1 931 673中描述的方法,所得混合物中iso1、iso2、iso3和iso4组的各自含量如下:20%、20%、40%和20%。
[0027]
然后,他们发现这些不同的异构体组具有不同的物理化学性质,并确定了包含具有以下表示的式(ii
‑
rrr)和(ii
‑
sss)的异构体ii
‑
rrr和ii
‑
sss的混合物、被称为iso4的异构体组被证明是最有利的医学成像造影剂。
[0028]
[0029][0030]
因此,令人惊讶地,iso4的特征在于其热力学稳定性显著优于非对映异构体混合物的热力学稳定性,该非对映异构体混合物呈通过进行ep 1931673中描述的方法获得具有式(ii)的络合物的形式。具体地,其热力学平衡常数k
thermiso4
等于10
18.7
(即log(k
thermiso4
)=18.7),该值已经通过进行pierrard等人,contrast media mol.imaging,2008,3,243
‑
252和moreau等人,dalton trans.,2007,1611
‑
1620中的方法确定。
[0031]
而且,iso4是由发明人分离的四组中具有最佳动力学惯性(也称为动力学稳定性)的异构体组。具体地,发明人通过研究四组异构体在37℃下在酸性水溶液(ph=1.2)中的解络合动力学来评估其动力学惯性。在下表1中列出了针对各组异构体确定的半衰期时间值(t
1/2
),该半衰期时间对应于根据以下解络合反应(反应式2)初始存在的络合物量的50%解离的时间:
[0032][0033][0034][0035]
[表1]:异构体iso1至iso4的解络合动力学
[0036]
出于比较的目的,作为大环钆络合物的钆布醇或钆酸盐在相同条件下分别具有18小时和4天的动力学惯性,而线性钆络合物(例如钆二胺或钆喷酸盐)会瞬间解离。
[0037]
此外,特别地iso4在化学上比iso3更稳定。其原因是,具有式(ii)的络合物的酰胺官能团易于水解。酰胺官能团(反应式3)的水解反应导致形成双偶合杂质,伴随着3
‑
氨基
‑
1,2
‑
丙二醇的释放。发明人研究了具有式(ii)的络合物在ph为13的水溶液中的水解反应动力学,并观察到iso4的酰胺官能团相对于iso3的酰胺官能团在水解方面更稳定。
[0038][0039]
关于各组异构体的弛豫率(即它们作为造影剂的效率)所进行的测量表明,iso1、iso2和iso4组的对比能力相对相当,而iso3的效率降低(见表2)。
[0040][0041]
[表2]:异构体组iso1至iso4在37℃下的弛豫率
[0042]
本发明人已经成功地开发了一种用于制备和纯化具有式(ii)的络合物的新方法,其使得能够优选地获得具有特别有利的物理化学性质的所述络合物的非对映异构体ii
‑
rrr和ii
‑
sss。根据本发明的方法包括通过将最不稳定的立体异构体转化为最稳定的立体异构体以富集异构体的步骤,令人惊讶的是,当在六酸中间体络合物上而不是在最终络合物上进行时,其使得在绝大多数情况下获得具有式(ii)的络合物的最稳定的异构体成为可能。
[0043]
与制备立体异构体的混合物的替代方案相比,实施一种使得在大多数情况下获得目标非对映异构体成为可能的方法无疑是有利的,以便随后根据常规技术尝试分离非对映异构体,从而使用本领域熟知的任何分离技术分离目标异构体。具体而言,除了在工业规模上更容易进行没有非对映异构体分离步骤的方法之外,不进行分离一方面可以节省大量时间,另一方面使得能够通过尽可能地限制最终将被消除的不需要的非对映异构体的产生来提高方法的总产率。此外,常规的分离技术通常涉及大量使用溶剂,这超出了财务成本,出于环境原因,这是不可取的。此外,考虑到职业接触二氧化硅固有的健康风险,因此应特别
避免使用二氧化硅层析法,国际癌症研究机构将其归类为对人类致癌(第1组)。
[0044]
如前所述,由发明人开发的制备具有式(ii)的络合物的方法是基于具有以下表示的式(i)的中间体六酸钆络合物的异构体富集的步骤:
[0045][0046]
具有式(i)的络合物对应于几种立体异构体,这是由于相对于侧链接枝到其上的大环的氮原子在该络合物的侧链上存在位于α位的三个不对称碳原子。这三个不对称碳在上面表示的式(i)中标有星号(*)。
[0047]
由于带有羧酸酯官能团的三个不对称碳中的每个均可以具有r或s绝对构型,因此具有式(i)的络合物以八种立体异构体的形式存在,以下称为i
‑
rrr、i
‑
sss、i
‑
rrs、i
‑
ssr、i
‑
rss、i
‑
srr、i
‑
rsr和i
‑
srs。更准确地,根据立体化学中通常的命名法,具有式(i)的络合物以四对作为相互非对映异构体的对映体(enantiomer)的形式存在。
[0048]
发明人已经成功地通过高效液相层析法(hplc)和超高效液相层析法(uhplc)分离和鉴定了根据ep 1 931 673中所述的方法获得的具有式(i)的络合物的四个未分辨的峰或异构体组,其对应于四个不同的通过它们在层析图上的保留时间表征的洗脱峰,在本说明书的其余部分中将被称为isoa、isob、isoc和isod。
[0049]
isod在水中结晶。x射线衍射分析使发明人能够确定这组异构体的晶体结构,并因此发现其包含具有式(i)的络合物的具有以下表示的式(i
‑
rrr)和(i
‑
sss)的非对映异构体i
‑
rrr和i
‑
sss。
[0050][0051]
应当注意,具有式(i)的络合物的非对映异构体i
‑
rrr和i
‑
sss是彼此的对映体。
[0052]
本发明方法的异构体富集步骤涉及在isod中富集具有式(i)的中间体六酸钆络合物。
[0053]
具有式(ii)的络合物的合成特别地涉及将具有式(i)的中间体六酸络合物的羧酸官能团转化为酰胺官能团。此酰胺化反应不会改变具有式(i)的络合物的三个不对称碳原子的绝对构型。
[0054]
因此,当对先前获得的isod中富集的具有式(i)的六酸络合物进行酰胺化反应时,能够获得iso4中富集的具有式(ii)的络合物。
[0055]
此外,由发明人开发的纯化方法使得在按照具有上述式(ii)的络合物的制备方法时,能够获得具有优化的异构体特征的具有式(ii)的络合物,而且显著改善了杂质分布。
[0056]
因此,可以用游离的大环配体(例如游离dota)代替dota的钙络合物来配制这种具有改善的稳定性的非对映异构体富集和纯化的络合物,如wo 2014/174120中所推荐。从工业的角度来看,使用游离dota特别地具有优势,从某种意义上说它使得可能消除如wo 2014/174120中所述的用于合成制剂的方法步骤,即添加cacl2。
技术实现要素:
[0057]
具有式(ii)的络合物
[0058]
因此,本发明首先涉及一种具有式(ii)的络合物:
[0059][0060]
由至少80%的非对映异构体过量构成,其包含具有下式的异构体ii
‑
rrr和ii
‑
sss的混合物:
[0061][0062][0063]
在本发明的上下文中,术语“非对映异构体过量”旨在代表关于具有式(ii)的络合物的以下事实,即所述络合物主要以选自以下非对映异构体的异构体或异构体组的形式存
在:ii
‑
rrr、ii
‑
sss、ii
‑
rrs、ii
‑
ssr、ii
‑
rss、ii
‑
srr、ii
‑
rsr和ii
‑
srs。所述非对映异构体过量以百分比表示,并对应于由主要异构体或异构体组表示的量相对于具有式(ii)的络合物总量。应当理解,该百分比可以基于摩尔或基于质量,因为根据定义,异构体具有相同的摩尔质量。
[0064]
在一个具体实施方式中,根据本发明的具有式(ii)的络合物具有至少85%、特别地至少90%、具体地至少92%、优选地至少94%、有利地至少97%、更有利地至少99%的包含异构体ii
‑
rrr和ii
‑
sss的混合物的非对映异构体过量。
[0065]
优选地,所述非对映异构体过量由异构体ii
‑
rrr和ii
‑
sss的混合物的至少70%、特别地至少80%、有利地至少90%、优选地至少95%构成。
[0066]
有利地,所述非对映异构体过量由异构体ii
‑
rrr和ii
‑
sss的混合物组成。
[0067]
经过引申,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”还涵盖了仅存在一种异构体的情况,无论存在的是ii
‑
rrr还是ii
‑
sss。然而,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”优选地代表异构体ii
‑
rrr和ii
‑
sss中的每个以可变但非零的量存在的所有情况。
[0068]
在一个优选的实施方式中,异构体ii
‑
rrr和ii
‑
sss以65/35与35/65之间、特别地60/40与40/60之间、具体地55/45与45/55之间的比率存在于所述混合物中。有利地,异构体ii
‑
rrr和ii
‑
sss以50/50的比率存在于混合物中。
[0069]
更具体地,如前所定义的非对映异构体过量对应于uhplc图中的峰4(即,按洗脱顺序排列异构体的第四个未分辨的峰,并对应于iso4),其通过保留时间为6.0与6.6分钟之间,典型地为约6.3分钟的保留时间来表征,所述图是使用下述uhplc方法获得的。
[0070]
在本发明的含义内,术语“uhplc图”是指对于给定的组成和给定的洗脱液流速,在固定相上化合物(在这种情况下为化合物的异构体)的混合物通过和分离后,检测器测得的浓度随时间变化的曲线。uhplc图由所分析化合物或化合物混合物的各种峰或未分辨的峰特征构成。
具体实施方式
[0071]
uhplc方法:
[0072]
watersuplc t3 150x2.1mm
‑
1.6μm柱。
[0073]
这是一种具有球形颗粒的反相uplc柱,这些颗粒由优选地非常坚硬的核心构成,核心由二氧化硅制成,并被三官能c18(十八烷基)接枝的多孔二氧化硅包围,并且硅烷醇已用封端剂(端部封端)处理。它的特征还在于150mm的长度、2.1mm的内径、1.6μm的粒径、的孔隙率和4.7%的碳含量。
[0074]
优选地,所使用的固定相应当与水性流动相兼容。
[0075]
‑
分析条件:
[0076][0077]
‑
流动相梯度(%v/v):
[0078][0079]
包含具有式(ii)的络合物的组合物
[0080]
其次,本发明涉及一种组合物,其包含:
[0081]
‑
具有式(ii)的络合物,其由至少80%的非对映异构体过量构成,所述非对映异构体过量包含异构体ii
‑
rrr和ii
‑
sss的混合物,以及
[0082]
‑
游离的大环配体。
[0083]
在本说明书中,可以不加区别地使用术语“大环配体”或“大环螯合物”。
[0084]
在本发明的上下文中,术语“大环”代表典型地包含至少九个原子的环,无论它们是碳原子还是杂原子,并且术语“大环配体”或“大环螯合物”是多齿的,至少是双齿的配体。
[0085]
在本发明的含义中,术语“游离的大环配体”是指游离形式的大环配体,即,不与以下,特别是金属(包括镧系元素和锕系元素)或碱土金属阳离子(例如钙或镁)络合。具体地,如us 5876695中所述,游离的大环配体不是呈与钆的络合物形式,并且不以弱络合物(典型地是钙、钠、锌或镁)的形式引入组合物中,然而,不排除组合物和因此相应络合物中所述阳离子的痕量存在。
[0086]
如先前所讨论的,具有游离大环配体的具有式(ii)的络合物而不是ep 1 931 673中推荐的所述大环配体的弱络合物的制剂,是通过改善根据本发明的非对映异构体富集的具有式(ii)的络合物的稳定性而使得成为可能。
[0087]
在一个优选的实施方式中,存在于本发明的组合物中的具有式(ii)的络合物具有至少85%、特别地至少90%、具体地至少92%、更具体地至少94%、优选地至少97%、有利地至少99%的包含异构体ii
‑
rrr和ii
‑
sss的混合物的非对映异构体过量。
[0088]
优选地,所述非对映异构体过量由至少70%、特别地至少80%、有利地至少90%、优选地至少95%的异构体ii
‑
rrr和ii
‑
sss的混合物构成。
[0089]
有利地,所述非对映异构体过量由异构体ii
‑
rrr和ii
‑
sss的混合物组成。
[0090]
经过引申,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”还涵盖了仅存在一种异构体的情况,无论存在的是ii
‑
rrr还是ii
‑
sss。然而,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”优选地代表异构体ii
‑
rrr和ii
‑
sss中的每个以可变但非零的量存在的所有情况。
[0091]
在一个优选的实施方式中,异构体ii
‑
rrr和ii
‑
sss以65/35与35/65之间、特别地60/40与40/60之间、具体地55/45与45/55之间的比率存在于所述混合物中。有利地,异构体ii
‑
rrr和ii
‑
sss以50/50的比率存在于混合物中。
[0092]
在一个有利的实施方式中,根据本发明的组合物具有小于1ppm(m/v),优选地小于0.5ppm(m/v)的游离钆浓度。
[0093]
在本说明书中,除非另有说明,否则不加区别地使用术语“gd”、“钆”和“gd
3+”来代表gd
3+
离子。经过引申,它也可能是游离钆的来源,例如氯化钆(gdcl3)或氧化钆(gd2o3)。
[0094]
在本发明中,术语“游离gd”代表钆的非络合形式,其优选地可用于络合。典型地是溶解在水中的gd
3+
离子。经过引申,它也可能是游离钆的来源,例如氯化钆(gdcl3)或氧化钆(gd2o3)。
[0095]
游离形式的钆典型地通过比色法测定,通常是二甲酚橙或偶氮胂(iii)来测量。在没有金属离子(例如钆)的情况下,这些指示剂具有特定的颜色:在酸性ph下,二甲酚橙为黄色,而偶氮胂为粉红色。在钆存在的情况下,它们的颜色变为紫色。
[0096]
目测确定溶液颜色的变化使得能够验证溶液中钆的存在与否。
[0097]
此外,有可能通过返滴定法定量测量溶液中的游离钆,例如使用edta作为“弱”钆螯合物。在这种测定中,添加有色指示剂,直至出现紫色。然后将edta(钆配体)滴加到混合物中。由于edta是比有色指示剂更强的络合剂,因此钆会改变配体并使有色指示剂优选地与edta络合。因此,有色指示剂逐渐恢复其非络合形式。
[0098]
当添加的edta量等于游离gd的初始量时,有色指示剂完全呈游离形式,溶液“变成”黄色。由于添加的edta的量是已知的,因此能够知道待测溶液中游离gd的初始量。
[0099]
这些方法是本领域技术人员熟知的,并且特别地在barge等人(contrast media and molecular imaging 1,2006,184
‑
188)中所述。
[0100]
因此,这些比色法通常在ph为4与8之间的溶液中进行。其原因是在这些ph范围之外,测量的准确性可能由于颜色变化的改变(或甚至抑制)而受到影响。
[0101]
因此,如果需要,将待测样品的ph调节为4与8之间。尤其是,如果样品的ph是酸性的,并且具体地小于4,则通过添加碱来有利地调节ph,然后在调节后的ph下对样品进行游离gd的测量。
[0102]
因此,根据本发明的组合物随时间推移具有稳定性,即在至少3年,优选地至少4年或更优选地至少5年的时间内,其组成保持符合有关游离钆的浓度的规定(具体地其游离gd浓度保持小于1ppm(m/v)),特别地就游离顺磁性金属的含量而言。根据ich指南,观察在40℃下6个月的稳定性被认为是25℃下3年稳定性的良好指示。
[0103]
在一个具体实施方式中,根据本发明的组合物具有0.01与1.5mol.l
‑1之间、优选地0.2与0.7mol.l
‑1之间、更优选地0.3与0.6mol.l
‑1之间的上述具有式(ii)的络合物浓度。
[0104]
通过本领域技术人员已知的方法测定具有式(ii)的络合物。特别地,可以通过原子发射光谱法(也称为icp
‑
aes或icp原子发射光谱法)在矿化和测定组合物中存在的钆的
总量的后进行测定。
[0105]
具有式(ii)的络合物的含量使该组合物具有最佳的对比能力,同时具有令人满意的粘度。具体地,低于0.01mol.l
‑1的上述具有式(ii)的络合物,其作为对比产品的性能质量不能令人满意,并且当浓度高于1.5mol.l
‑1时,该组合物的粘度变得太大而变得难以处理。
[0106]
在一个具体实施方式中,根据本发明的组合物包含相对于具有式(ii)的络合物的0.002与0.4mol/mol%之间、特别地0.01与0.3mol/mol%之间、优选地0.02与0.2mol/mol%之间并且更优选地0.05与0.15mol/mol%之间的游离的大环配体。
[0107]
有利地,大环配体选自下组,该组由以下各项构成:dota、nota、do3a、bt
‑
do3a、hp
‑
do3a、pcta、dota
‑
ga及其衍生物。
[0108]
优选地,其为dota(1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7,10
‑
四乙酸)。
[0109]
组合物中游离dota的浓度典型地通过用铜进行返滴定来测量,例如使用硫酸铜作为铜离子源。
[0110]
在本领域技术人员熟知的该方法中,优选地使用含有已知的硫酸铜初始浓度q0的溶液,该浓度大于溶液中游离配体的量。将含有待确定的q1量的游离dota的待测溶液添加到该硫酸铜溶液中。dota是非常好的铜络合剂:因此观察到了dota
‑
铜络合物的形成。
[0111]
然后有利地通过电位差计进行溶液中保持游离的铜的返滴定。为此,例如将edta滴加到混合物中。edta使溶液中的游离铜络合,但不会使dota
‑
铜解络合,因为dota是比edta更强的络合剂。当添加的edta量q2等于溶液中游离铜的量时,观察到溶液电位突然下降。
[0112]
已知铜的初始量q0和添加的edta量q2,将这两个值相减q0‑
q2得到待测溶液中的游离dota量q1。
[0113]
可替代地,可以使用hplc方法,特别地hilic lc
‑
uv方法。
[0114]
这些测量方法(具体地电位测量方法)是在ph有利地介于4与8之间的溶液中进行的。因此,如果需要,将待测样品的ph调节为4与8之间。尤其是,如果样品的ph是酸性的,并且具体地小于4,则通过添加碱(例如葡甲胺)来有利地调节ph,然后在调节后的ph下对样品进行游离dota的测量。
[0115]
优选地,本发明中特别是以上所规定的比例是在组合物灭菌之前的比例。
[0116]
有利地,组合物的ph在4.5与8.5之间、优选地在5与8之间、有利地在6与8之间、特别地在6.5与8之间。这些ph范围特别地使得能够限制某些杂质的出现并促进顺磁性金属离子m的络合。
[0117]
具体地,根据本发明的组合物可以是缓冲的,即,其也可以包含选自针对5至8的ph范围建立的常见缓冲液的缓冲液,优选地乳酸盐、酒石酸盐、苹果酸盐、马来酸盐、琥珀酸盐、抗坏血酸盐、碳酸盐、tris(三(羟甲基)氨基甲烷)、hepes(2
‑
[4
‑
(2
‑
羟乙基)
‑1‑
哌嗪]乙磺酸)和mes(2
‑
吗啉乙磺酸)缓冲液及其混合物,并且优选地选自tris、乳酸盐、酒石酸盐、碳酸盐和mes缓冲液及其混合物的缓冲液。有利地,根据本发明的组合物包含tris缓冲液。
[0118]
作为本发明主题的组合物优选地是无菌的。
[0119]
用于制备具有式(ii)的络合物的方法
[0120]
本发明还涉及一种用于制备具有式(ii)的络合物的方法,其包括以下连续步骤:
[0121]
a)将具有下式(iii)的六酸:
[0122][0123]
与钆络合以获得具有如前所定义的式(i)的六酸钆络合物,
[0124]
b)通过加热ph为2与4之间的水溶液中的具有式(i)的六酸钆络合物以进行异构化,以获得由至少80%的非对映异构体过量构成的非对映异构体富集的络合物,该非对映异构体过量包含所述具有式(i)的六酸钆络合物的异构体i
‑
rrr和i
‑
sss的混合物,并且
[0125]
c)从步骤b)中获得的非对映异构体富集的络合物开始,通过与3
‑
氨基
‑
1,2
‑
丙二醇反应,形成具有式(ii)的络合物。
[0126]
在本说明书中,除非另有说明,否则不加区别地使用术语“gd”、“钆”和“gd
3+”来代表gd
3+
离子。经过引申,它也可能是游离钆的来源,例如氯化钆(gdcl3)或氧化钆(gd2o3)。
[0127]
在本发明中,术语“游离gd”代表钆的非络合形式,其优选地可用于络合。典型地是溶解在水中的gd
3+
离子。经过引申,它也可能是游离钆的来源,例如氯化钆(gdcl3)或氧化钆。
[0128]
■
步骤a)
[0129]
在该步骤的过程中,具有式(iii)的六酸与钆之间发生络合反应,这使得能够获得具有如前所定义的式(i)的六酸钆络合物。
[0130]
根据一个具体实施方式,步骤a)包括具有式(iii)的六酸与水中的游离gd的来源之间的反应。
[0131]
在一个优选的实施方式中,游离gd的来源是gdcl3或gd2o3,优选地是gd2o3。
[0132]
优选地,步骤a)中使用的试剂,即钆(典型地为氧化钆)的来源、具有式(iii)的六酸和水尽可能地纯,特别地涉及金属杂质。
[0133]
因此,钆的来源将有利地是氧化钆,其纯度优选地大于99.99%,甚至更优选地大于99.999%。
[0134]
在该方法中使用的水优选地包含小于50ppm的钙、更优选地小于20ppm并且最优选地小于15ppm的钙。通常,该方法中使用的水是去离子水、注射用水(注射级水)或纯净水。
[0135]
有利地,在该步骤a)中使用的试剂(具有式(iii)的六酸和钆)的量对应于或接近于化学计量比例,如在该步骤中发生的络合反应的平衡方程式所决定的。
[0136]
术语“接近化学计量比例”是指引入试剂的摩尔比例与化学计量比例之间的差小于15%、特别地小于10%、优选地小于8%。
[0137]
尤其是,相对于化学计量比例,钆的引入可能会稍微过量。然后,作为钆引入的材料量与作为具有式(iii)的六酸引入的材料量的比率大于1,但典型地小于1.15、特别地小于1.10、有利地小于1.08。换句话说,相对于引入的具有式(iii)的六酸的量(其本身对应于1当量),引入的钆的量大于1当量(eq.),但典型地小于1.15eq.、特别地小于1.10eq.、有利
地小于1.08eq.。在其中游离钆的来源为gd2o3的优选的实施方式中,相对于引入的具有式(iii)的六酸的量(1eq.),引入的gd2o3的量典型地大于0.5eq.,但小于0.575eq.、特别地小于0.55eq.、有利地小于0.54eq.。
[0138]
根据一个具体实施方式,步骤a)包括以下连续步骤:
[0139]
a1)制备具有式(iii)的六酸的水溶液,并且
[0140]
a2)向步骤a1)获得的水溶液中添加游离钆的来源。
[0141]
在该实施方式中,在步骤a1)中制备的水溶液中具有式(iii)的六酸的含量相对于水溶液的总重量典型地按重量计在10%与60%之间、特别地在15%与45%之间、优选地在20%与35%之间、有利地在25%与35%之间并且甚至更有利地在25%与30%之间。
[0142]
优选地,步骤a)和b)是根据一锅法实施方式进行的,即在相同的反应器中进行,而没有分离或纯化的中间步骤。
[0143]
因此,在该优选的实施方式中,将步骤a)中形成的具有式(i)的六酸钆络合物不经分离或纯化而直接进行异构化步骤b),并且在与步骤a)所用的反应器相同的反应器中进行。
[0144]
■
步骤b)
[0145]
通过步骤a)中具有式(iii)的六酸与钆之间的络合反应形成的具有式(i)的六酸钆络合物首先以非对映异构体的混合物形式获得。
[0146]
步骤b)涉及富集异构体i
‑
rrr和i
‑
sss中的非对映异构体的混合物,以获得非对映异构体富集的具有式(i)的六酸钆络合物,其由至少85%、特别地至少90%、具体地至少95%、优选地至少97%、有利地至少98%、更有利地至少99%的包含异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量构成。
[0147]
在本发明的上下文中,术语“非对映异构体过量”旨在代表涉及具有式(i)的六酸钆络合物的以下事实,即所述络合物主要以选自以下的非对映异构体的异构体或异构体组的形式存在:i
‑
rrr、i
‑
sss、i
‑
rrs、i
‑
ssr、i
‑
rss、i
‑
srr、i
‑
rsr和i
‑
srs。所述非对映异构体过量表示为百分比,并对应于由相对于具有式(i)的六酸钆络合物总量的由主要异构体或异构体组表示的量。应当理解,该百分比可以基于摩尔或基于质量,因为根据定义,异构体具有相同的摩尔质量。
[0148]
优选地,所述非对映异构体过量由至少70%、特别地至少80%、有利地至少90%、优选地至少95%的异构体i
‑
rrr和i
‑
sss的混合物构成。
[0149]
有利地,所述非对映异构体过量由异构体i
‑
rrr和i
‑
sss的混合物组成。
[0150]
实际上,发明人发现,在步骤a)结束时获得的具有式(i)的六酸钆络合物的溶液的例如ph和温度等的因素对存在于非对映异构体的混合物中的具有式(i)的络合物的各种异构体的比率具有影响。随着时间推移,混合物趋于富集一组异构体,其包含令人惊讶地在热力学上最稳定但在化学上也最稳定的异构体,在这种情况下为异构体i
‑
rrr和i
‑
sss。
[0151]
经过引申,术语“异构体i
‑
rrr和i
‑
sss的混合物”还涵盖了仅存在一种异构体的情况,无论存在的是i
‑
rrr还是i
‑
sss。
[0152]
然而,在一个优选的实施方式中,异构体i
‑
rrr和i
‑
sss以65/35与35/65之间、特别地60/40与40/60之间、具体地55/45与45/55之间的比率存在于所述混合物中。有利地,异构体i
‑
rrr/i
‑
sss的混合物是外消旋(50/50)混合物。
[0153]
水溶液中具有式(i)的六酸钆络合物的异构化的步骤b)典型地在2与4之间、特别地2与3之间、有利地在2.2与2.8之间的ph下进行。
[0154]
优选地用酸,优选地无机酸,例如盐酸、氢溴酸、硫酸、硝酸或磷酸,例如用盐酸调节ph。
[0155]
完全令人惊讶的是,在这种ph条件下,发生了混合物,具体地异构体的富集,在这种情况下是异构体i
‑
rrr和i
‑
sss,因为本领域已知钆螯合物的特征在于酸性介质中的低动力学惯性。具体地,介质中h
+
离子的浓度越高,质子转移到配体的一个供体原子的一上的可能性就越大,从而导致络合物解离。因此,本领域技术人员将期望将具有式(i)的六酸钆络合物置于ph为2与4之间的水溶液中导致所述络合物解离,而不是异构化为i
‑
rrr和i
‑
sss。
[0156]
应当注意,ep 1 931 673针对具有式(iii)的六酸的络合推荐的ph范围,即5.0
‑
6.5,不能获得富集其异构体i
‑
rrr和i
‑
sss的具有式(i)的络合物。
[0157]
步骤b)典型地在80℃与130℃之间、特别地在90℃与125℃之间、优选地在98℃与122℃之间、有利地在100℃与120℃之间的温度下进行,典型地进行10小时与72小时、特别地10小时与60小时、有利地12小时与48小时之间的时间。
[0158]
与所有预期相反,与上述ph条件结合使用的此类温度条件应有利于钆螯合物的不稳定性,不会导致其解络合或形成任何其他杂质,而是使其异构化为i
‑
rrr和i
‑
sss。
[0159]
在一个具体实施方式中,步骤b)的水溶液包含乙酸。步骤b)然后有利地在100℃与120℃之间、特别地在110℃与118℃之间的温度下进行,典型地进行12小时与48小时之间、特别地20小时与30小时之间、具体地24小时与26小时之间的时间。
[0160]
优选地在加热步骤a)中获得的具有式(i)的六酸钆络合物的溶液之前加入一定量的乙酸,使得乙酸的含量相对于步骤a)中使用的具有式(iii)的六酸的质量按质量计在25%与75%之间、特别地在40%与50%之间。
[0161]
当将水溶液加热到有利地在100℃与120℃之间,典型地在110℃与118℃之间的温度时,随着水的蒸发逐渐添加乙酸,以保持恒定体积的溶液。
[0162]
根据一个优选的实施方式,在步骤b)结束时,通过结晶,优选地通过晶种进行的结晶来分离非对映异构体富集的络合物。
[0163]
在该实施方式中,步骤b)包括以下连续步骤:
[0164]
b1)通过加热ph为2与4之间的水溶液中的具有式(i)的六酸钆络合物以进行异构化,以获得由至少80%的非对映异构体过量构成的非对映异构体富集的络合物,该非对映异构体过量包含所述具有式(i)的六酸钆络合物的异构体i
‑
rrr和i
‑
sss的混合物,并且
[0165]
b2)通过结晶分离所述非对映异构体富集的络合物,优选地通过晶种进行的结晶分离。
[0166]
结晶步骤b2)首先涉及除去水溶液中存在的任何杂质,这些杂质可能是由先前的步骤导致的,从而以晶体的形式获得了较高纯度的脱色产物,其次涉及继续进行具有式(i)的六酸钆络合物的非对映异构体富集,以获得包含所述络合物的异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量,其比在步骤b1)结束时获得的高。具体地,具有式(i)的六酸络合物的异构体i
‑
rrr和i
‑
sss从水中结晶。另一方面,未富集所述异构体的具有式(i)的六酸钆络合物不会结晶。
[0167]
其中在步骤b)(并且与所有预期相反,根据进行其的条件)的过程中络合物趋于富
集的异构体i
‑
rrr和i
‑
sss是从水中结晶的络合物的唯一异构体这一事实是完全出乎意料的结果。因此,异构化和结晶协同作用有助于异构体i
‑
rrr和i
‑
sss的富集,并因此有助于根据本发明的方法的总效率。
[0168]
此外,应当注意,具有式(i)的六酸钆络合物的目标异构体从水中的结晶使得能够避免如ep 1 931 673的实施方式7中所述的溶剂添加,其涉及从乙醇中沉淀出所述络合物的三钠盐的步骤。
[0169]
步骤b2)有利地在10℃与70℃之间、特别地在30℃与65℃之间、具体地在35℃与60℃之间的温度下进行。
[0170]
根据一种变型,在降低水溶液的温度,这样使得其在上述范围内的后,通过晶种诱导结晶过程。“通过晶种进行结晶”,也称为“通过引晶进行结晶”,包括将已知量的晶体(被称为“种子”或“引物”)引入其中进行结晶的反应器(也称为结晶容器)中。这使得能够减少结晶时间。通过晶种进行结晶是本领域技术人员熟知的。在根据本发明的方法中,使用引物,在当前情况下为添加到预先降低了温度的非对映异构体富集的络合物的水溶液中的非对映异构体富集的具有式(i)的六酸钆络合物的晶体,进行结晶使得能够获得成核作用,从而引发结晶。通过晶种进行结晶的持续时间有利地在2小时与20小时之间,并且优选地在6小时与18小时之间;典型地是16个小时。
[0171]
然后典型地借助于本领域技术人员熟知的任何技术,通过过滤和干燥来分离具有式(i)的非对映异构体富集的六酸钆络合物的晶体。
[0172]
有利地,在步骤b2)结束时分离的具有式(i)的非对映异构体富集的六酸钆络合物的纯度大于95%、特别地大于98%、有利地大于99%,所述纯度表示为具有式(i)的络合物相对于步骤b2)结束时获得的总质量的质量百分比。
[0173]
在一个具体实施方式中,将通过结晶分离的来自步骤b)的非对映异构体富集的络合物再次通过重结晶纯化,以获得非对映异构体富集和纯化的络合物。
[0174]
在该实施方式中,除了先前所述的连续步骤b1)和b2)之外,步骤b)还包括通过重结晶分离的具有式(i)的非对映异构体富集的六酸钆络合物进行纯化的步骤b3)。
[0175]
重结晶步骤b3)与结晶步骤b2)一样,首先涉及获得更高纯度的产物,其次涉及继续进行具有式(i)的六酸钆络合物的非对映异构体富集,以获得包含所述络合物的异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量,其比在步骤b2)结束时获得的高。
[0176]
步骤b3)典型地包括以下连续的子步骤:
[0177]
●
将步骤b2)中分离的具有式(i)的非对映异构体富集的六酸钆络合物悬浮在水溶液中,优选地在水中,
[0178]
●
通过加热至有利地在80℃与120℃之间的温度,例如至100℃的温度,来溶解所述络合物,
[0179]
●
重结晶,优选地通过晶种进行重结晶,有利地在10℃与90℃之间、特别地在20℃与87℃之间、具体地在55℃与85℃之间的温度下进行,典型地持续2小时与20小时之间、特别地在6小时与18小时之间的时间,并且
[0180]
●
分离非对映异构体富集和纯化的具有式(i)的六酸钆络合物的晶体,例如通过过滤和干燥。
[0181]
在步骤b3)结束时分离的具有式(i)的纯化的非对映异构体富集的六酸钆络合物
的纯度典型地大于98%、特别地大于99%、有利地大于99.5%,所述纯度表示为具有式(i)的络合物相对于步骤b2)结束时获得的总质量的质量百分比。
[0182]
在另一个实施方式中,通过除非对映异构体i
‑
rrr和i
‑
sss以外的具有式(i)的络合物的非对映异构体的选择性解络合,即通过非对映异构体i
‑
rss、i
‑
srr、i
‑
rsr、i
‑
srs、i
‑
rrs和i
‑
ssr的选择性解络合,进一步富集来自步骤b)的非对映异构体富集的络合物。
[0183]
在该实施方式中,除了先前所述的连续步骤b1)和b2)之外,步骤b)还包括除非对映异构体i
‑
rrr和i
‑
sss以外的具有式(i)的络合物的非对映异构体的选择性解络合的步骤b4)。在该变型中,步骤b)还可以包括先前所述的步骤b3),所述步骤b3)在步骤b2)与b4)之间或在b4)的后进行。
[0184]
选择性解络合步骤b4)涉及继续进行具有式(i)的六酸钆络合物的非对映异构体富集,从而获得非对映异构体过量,其包含所述络合物的异构体i
‑
rrr和i
‑
sss的混合物,当所述步骤在步骤b4)之前进行时,该非对映异构体过量高于在步骤b2)结束时或在步骤b3)结束时获得的非对映异构体过量。
[0185]
步骤b4)典型地包括以下连续的子步骤:
[0186]
●
将步骤b2)或步骤b3)中分离的具有式(i)的非对映异构体富集的六酸钆络合物悬浮在水中,
[0187]
●
添加碱,例如氢氧化钠,
[0188]
●
加热至有利地在30℃与60℃之间、特别地在35℃与55℃之间的温度,例如40℃,典型地持续2小时与20小时之间、特别地10小时与18小时之间的时间,
[0189]
●
冷却至有利地在10℃与30℃之间的温度,例如至30℃,并且
[0190]
●
分离非对映异构体富集和纯化的具有式(i)的六酸钆络合物,例如通过过滤和干燥。
[0191]
异构体i
‑
rrr和i
‑
sss在碱性介质中最稳定的事实使得步骤b4)成为可能。此类碱性条件促进了氢氧化钆的形成,因此促进了最不稳定的异构体的解络合。因此,应当注意,令人惊讶的是,异构体i
‑
rrr和i
‑
sss在允许进行异构化步骤b1)的酸性介质和在允许进行选择性解络合步骤b4)的碱性介质中都更稳定。
[0192]
在一个优选的实施方式中,根据上述任一变型在步骤b)结束时获得的非对映异构体富集的络合物具有至少85%、特别地至少90%、具体地至少95%、优选地至少97%、有利地至少98%、更有利地至少99%的包含异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量。
[0193]
优选地,所述非对映异构体过量由至少70%、特别地至少80%、有利地至少90%、优选地至少95%的异构体i
‑
rrr和i
‑
sss的混合物构成。
[0194]
有利地,所述非对映异构体过量由异构体i
‑
rrr和i
‑
sss的混合物组成。
[0195]
经过引申,术语“异构体i
‑
rrr和i
‑
sss的混合物”还涵盖了仅存在一种异构体的情况,无论存在的是i
‑
rrr还是i
‑
sss。然而,术语“异构体i
‑
rrr和i
‑
sss的混合物”优选地代表异构体i
‑
rrr和i
‑
sss中的每个以可变但非零的量存在的所有情况。
[0196]
在一个优选的实施方式中,异构体i
‑
rrr和i
‑
sss以65/35与35/65之间、特别地60/40与40/60之间、具体地55/45与45/55之间的比率存在于所述混合物中。有利地,异构体i
‑
rrr/i
‑
sss的混合物是外消旋(50/50)混合物。
[0197]
■
步骤c)
[0198]
步骤c)涉及从其前体,即在步骤b)中获得的非对映异构体富集的具有式(i)的六酸钆络合物形成具有式(ii)的络合物。
[0199]
在该步骤中,相对于在其上接枝有侧链的大环的氮原子,位于络合物侧链上γ位置的碳原子携带的具有式(i)的六酸络合物的三个羧酸官能团,通过与外消旋或对映体纯形式、优选地外消旋形式的3
‑
氨基
‑
1,2
‑
丙二醇的酰胺化反应,转化为酰胺官能团。
[0200]
该酰胺化反应不会改变相对于接枝有侧链的大环的氮原子位于所述侧链上α位的三个不对称碳原子的绝对构型。因此,步骤c)使得能够获得具有非对映异构体过量的具有式(ii)的络合物,该非对映异构体过量包含异构体ii
‑
rrr和ii
‑
sss的混合物,其与包含异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量相同,用至少80%的该包含异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量获得在步骤b)结束时获得的具有式(i)的非对映异构体富集的六酸钆络合物。
[0201]
在一个优选的实施方式中,在步骤c)结束时获得的具有式(ii)的络合物具有至少85%、特别地至少90%、具体地至少92%、优选地至少94%、有利地至少97%、更有利地至少99%的包含异构体ii
‑
rrr和ii
‑
sss的混合物的非对映异构体过量。
[0202]
优选地,所述非对映异构体过量由至少70%、特别地至少80%、有利地至少90%、优选地至少95%的异构体ii
‑
rrr和ii
‑
sss的混合物构成。
[0203]
有利地,所述非对映异构体过量由异构体ii
‑
rrr和ii
‑
sss的混合物组成。
[0204]
经过引申,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”还涵盖了仅存在一种异构体的情况,无论存在的是ii
‑
rrr还是ii
‑
sss。然而,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”优选地代表异构体ii
‑
rrr和ii
‑
sss中的每个以可变但非零的量存在的所有情况。
[0205]
在一个优选的实施方式中,异构体ii
‑
rrr和ii
‑
sss以65/35与35/65之间、特别地60/40与40/60之间、具体地55/45与45/55之间的比率存在于所述混合物中。有利地,异构体ii
‑
rrr和ii
‑
sss以50/50的比率存在于混合物中。
[0206]
酰胺化反应可以根据本领域技术人员熟知的任何方法进行,特别地在存在用于活化羧酸官能团的试剂和/或通过酸催化的情况下。
[0207]
可以特别地根据ep 1 931 673中描述的方法来进行,特别地在所述专利的第[0027]段中。
[0208]
在一个具体实施方式中,步骤c)包括活化相对于在其上接枝有侧链的大环的氮原子,位于具有式(i)的六酸络合物的所述侧链上γ位置的碳原子携带的羧酸(
‑
cooh)官能团,其呈衍生官能团的形式,包括羰基(c=o)基团,使得该羰基基团的碳原子比羧酸官能团的羰基基团的碳原子更具亲电性。因此,根据该具体实施方式,所述羧酸官能团特别地可以酯、酰氯或酸酐官能团的形式、或者以可以产生酰胺键的任何活化形式被活化。可以产生酰胺键的活化形式是本领域技术人员熟知的,并且可以例如通过肽化学中已知的用于形成肽键的一组方法获得。此类方法的实例在出版物synthesis of peptides and peptidomimetics第e22a卷,第425
‑
588页,houben
‑
weyl等人,goodman编辑,thieme
‑
stuttgart
‑
纽约(2004)中给出,并且在这些实例中可以特别地提及通过迭氮化物(酰基迭氮化物)活化羧酸的方法,例如通过试剂(例如二苯基磷酰基迭氮化物(通常缩写为dppa))的作用、单独或在催化剂(例如n
‑
羟基琥珀酰亚胺及其衍生物)存在下使用碳二亚胺、使用
羰基二咪唑(1,1
’‑
羰基二咪唑,cdi)、使用鏻盐(例如苯并三唑
‑1‑
基氧基三(二甲基氨基)鏻六氟磷酸盐(通常缩写为bop))、或其他脲鎓,例如2
‑
(1h
‑
苯并三唑
‑1‑
基)
‑
1,1,3,3
‑
四甲基脲鎓六氟磷酸盐(通常缩写为hbtu)。
[0209]
优选地,步骤c)包括以酯、酰氯或酸酐官能团的形式活化上述羧酸(
‑
cooh)官能团。
[0210]
该实施方式比ep 1 931 673中所述的使用偶合剂(例如edci/hobt)通过活化羧酸官能团以进行肽偶合更优选的。具体地,这种偶合导致形成一当量的1
‑
乙基
‑3‑
[3
‑
(二甲基氨基)丙基]脲,必须将其除去,特别地通过硅胶层析法或通过添加溶剂进行液/液萃取来除去。如先前所讨论的,独立于由这样的附加步骤导致的方法的复杂性增加,不期望使用此类纯化方法。此外,hobt的使用本身是有问题的,因为其为易爆产品。
[0211]
在本发明的含义内,术语“酯官能团”旨在代表
‑
c(o)o
‑
基团。它具体地可以是基团
‑
c(o)o
‑
r1,其中r1对应于(c1‑
c6)烷基基团。
[0212]
在本发明的含义内,术语“(c1‑
c6)烷基基团”是指含有1至6个,优选地1至4个碳原子的直链或支链的饱和烃基链。可以提及的实例包括甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基和己基基团。
[0213]
在本发明的含义内,术语“酰氯官能团(acyl chloride function)”,也称为“酰氯官能团(acid chloride function)”旨在代表
‑
co
‑
cl基团。
[0214]
在本发明的含义内,术语“酸酐官能团”旨在代表
‑
co
‑
o
‑
co
‑
基团。它具体地可以是基团
‑
co
‑
o
‑
co
‑
r2,其中r2对应于(c1‑
c6)烷基基团。
[0215]
用于将羧酸官能团转化成酯、酰氯或酸酐官能团的反应是本领域技术人员熟知的,他们将能够根据他熟悉的任何常规方法来进行反应。
[0216]
然后,通过以酯、酰氯或酸酐官能团,特别地酯或酸酐、优选地酯形式的活化的羧酸官能团,通过与外消旋或对映体纯形式、优选地外消旋形式的3
‑
氨基
‑
1,2
‑
丙二醇反应进行的胺解获得具有式(ii)的络合物。
[0217]
优选地,根据一锅法实施方式进行活化羧酸官能团和胺解的步骤,该一锅法实施方式即在同一反应器中,没有分离或纯化的中间体的中间步骤,该中间体包括以酯、酰氯或酸酐官能团,特别地酯或酸酐,优选地酯的形式活化的羧酸官能团。
[0218]
根据一个具体实施方式,步骤c)包括以下连续步骤:
[0219]
c1)形成具有式(vii)的活化络合物,
[0220][0221]
其中y表示氯原子、
‑
or1或
‑
o
‑
c(o)
‑
r2基团;优选地,y表示
‑
or1或
‑
o
‑
c(o)
‑
r2基团,
其中r1和r2彼此独立地对应于(c1‑
c6)烷基基团,以及
[0222]
c2)具有式(vii)的活化络合物与3
‑
氨基
‑
1,2
‑
丙二醇进行的胺解。
[0223]
对于本领域技术人员而言非常明显的是,形成具有式(vii)的活化络合物的反应不会改变相对于接枝有侧链的大环的氮原子位于所述侧链上α位的三个不对称碳原子的绝对构型。因此,步骤c1)使得能够获得具有非对映异构体过量的具有式(vii)的活化的络合物,该非对映异构体过量包含具有以下表示的式(vii
‑
rrr)和(vii
‑
sss)的异构体vii
‑
rrr和vii
‑
sss的混合物,其与包含异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量相同,在步骤b)结束时获得的具有式(i)的非对映异构体富集的六酸钆络合物具有至少80%的该包含异构体i
‑
rrr和i
‑
sss的混合物的非对映异构体过量。
[0224][0225]
在y表示氯原子的情况下,步骤c1)典型地通过步骤b)中获得的具有式(i)的非对映异构体富集的六酸钆络合物与亚硫酰氯(socl2)之间的反应来进行。
[0226]
在y表示
‑
o
‑
c(o)
‑
ch3基团的情况下,步骤c1)典型地通过步骤b)中获得的具有式(i)的非对映异构体富集的六酸钆络合物与乙酰氯之间的反应来进行。
[0227]
在有利的实施方式中,步骤c)包括以酯官能团的形式活化上述羧酸(
‑
cooh)官能团。
[0228]
根据该实施方式,步骤c)可以更具体地包括以下连续步骤:
[0229]
c1)形成具有式(viii)的三酯,
[0230][0231][0232]
其中r1表示(c1‑
c6)烷基基团,以及
[0233]
c2)具有式(viii)的三酯与3
‑
氨基
‑
1,2
‑
丙二醇进行的胺解。
[0234]
步骤c1)典型地在酸(例如盐酸)的存在下,在充当溶剂和试剂两者的具有式r1oh的醇中进行。
[0235]
步骤c2)还典型地在酸(例如盐酸)的存在下,在具有式r1oh的醇中进行。
[0236]
在第一阶段,将具有式(i)的六酸钆络合物和醇r1oh放入反应器中。然后将反应介质冷却至低于10℃、特别地低于5℃、典型地至0℃的温度,然后逐渐加入醇r1oh的酸性溶液,典型地为盐酸在r1oh中的溶液。将反应介质在室温下(即在20℃与25℃之间的温度)保持搅拌典型地大于5小时,优选地在10小时与20小时之间的时间。在步骤c2)之前,将反应介质冷却至低于10℃,特别地在0℃与5℃之间的温度。
[0237]
因此,根据一锅法实施方式,可以容易地进行步骤c1)和c2)。有利地,在步骤c1)与c2)之间不分离具有式(vii)的三酯。
[0238]
然而,为了促进胺解反应,在步骤c2)中,优选地通过真空蒸馏除去具有式r1oh的醇。
[0239]
在本发明的含义内,术语“真空蒸馏”是指在10与500毫巴之间、特别地在10与350毫巴之间、优选地在10与150毫巴之间、特别是在50与100毫巴之间的压力下进行的混合物的蒸馏。
[0240]
类似地,为了促进胺解反应,在步骤c2)中,大量引入3
‑
氨基
‑
1,2
‑
丙二醇。典型地,相对于步骤c)中最初引入的具有式(i)的非对映异构体富集的六酸钆络合物的材料量(其本身对应于1当量),引入的3
‑
氨基
‑
1,2
‑
丙二醇的材料量大于4eq.、特别地大于7eq.、有利地大于10eq.。
[0241]
令人惊讶地,尽管步骤c1)和c2)中典型地采用的酸性条件会增加钆络合物的动力学不稳定性,但未观察到具有式(viii)的三酯的解络合或异构化。获得具有非常良好的转化度的所需三酰胺,并且相对于大环的氮原子,位于侧链上α位的三个不对称碳原子的绝对构型得以保留。
[0242]
此外,应当注意,通常在文献中很少描述通过酯与胺之间直接反应进行的酰胺化反应(涉及该主题参见,k.c.nadimpally等人,tetrahedron letters,2011,52,2579
‑
2582)。
[0243]
在一个优选的实施方式中,步骤c)包括以下连续步骤:
[0244]
c1)形成具有式(iv)的甲基三酯,
[0245][0246]
尤其是在酸(例如盐酸)存在下,在甲醇中进行反应,以及
[0247]
c2)具有式(iv)的甲基三酯与3
‑
氨基
‑
1,2
‑
丙二醇的胺解,尤其是在酸(例如盐酸)的存在下,在甲醇中进行胺解。
[0248]
有利地,在步骤c1)与c2)之间不分离具有式(iv)的甲基三酯。
[0249]
在一个优选的实施方式中,在步骤c2)中,通过真空蒸馏除去甲醇,直至达到典型地高于55℃,特别地在60℃与65℃之间的温度,并且将反应介质在真空下保持在该温度下典型地大于5小时,尤其是在10与20小时之间的时间,的后冷却至室温并用水稀释。
[0250]
本发明包括与该方法的每个步骤相关的上述特定的、有利的或优选的实施方式的所有组合。
[0251]
■
制备具有式(iii)的六酸
[0252]
可以根据已知的任何方法,特别地根据ep 1 931 673中所述的方法来制备具有式(iii)的六酸,其参与制备根据本发明的具有式(ii)的络合物的方法的步骤a)。
[0253]
然而,根据一个优选的实施方式,具有式(iii)的六酸通过以下获得:具有式(v)的pyclene:
[0254][0255]
与具有式r3ooc
‑
chg
p
‑
(ch2)2‑
coor4(ix)的化合物烷基化,
[0256]
其中:
[0257]
‑
r3和r4彼此独立地表示(c3‑
c6)烷基基团,尤其是(c4‑
c6)烷基基团,例如丁基、异丁基、仲丁基、叔丁基、戊基或己基基团,并且
[0258]
‑
gp表示脱离基,例如甲苯磺酸酯或三氟甲磺酸酯基团、或卤素原子,优选地为溴原子,
[0259]
以获得具有式(x)的六酯
[0260][0261]
然后进行水解步骤,得到所述具有式(iii)的六酸。
[0262]
在一个优选的实施方式中,r3和r4是相同的。
[0263]
根据一个有利的实施方式,具有式(iii)的六酸通过以下获得:具有式(v)的pyclene:
[0264][0265]
与2
‑
溴戊二酸二丁酯烷基化以得到具有式(vi)的丁基六酯:
[0266][0267]
然后进行水解步骤,得到所述具有式(iii)的六酸。
[0268]
所用的2
‑
溴戊二酸二丁酯为外消旋或对映体纯形式,优选地为外消旋形式。
[0269]
与使用ep 1 931 673中所述的2
‑
溴戊二酸乙酯相比,使用2
‑
溴戊二酸二丁酯特别有利。具体地,商购的2
‑
溴戊二酸二乙酯是一种相对不稳定的化合物,它会随着时间的推移并在温度的作用下降解。更精确地,该酯具有被水解或环化并因此失去其溴原子的趋势。纯化商购的2
‑
溴戊二酸二乙酯、或开发新的合成途径以得到纯度更高的该酯并且因此防止其降解的尝试均未成功。
[0270]
烷基化反应典型地在极性溶剂中、优选地在水中、具体地在去离子水中、有利地在碱例如碳酸钾或碳酸钠的存在下进行。
[0271]
出于明显的原因,优选地使用水,特别优选地使用乙腈,如ep 1 931 673中所述。
[0272]
该反应有利地在40℃与80℃之间,典型地在50℃与70℃之间并且特别地在55℃与60℃之间的温度下进行,持续5小时与20小时之间,具体地8小时与15小时之间的时间。
[0273]
水解步骤有利地在酸或碱,有利地在碱(例如氢氧化钠)的存在下进行。水解溶剂
可以是水、醇(例如乙醇)或水/醇混合物。该步骤有利地在40℃与80℃之间,典型地在40℃与70℃之间并且特别地在50℃与60℃之间的温度下进行,典型地持续10小时与30小时之间,具体地15小时与25小时之间的时间。
[0274]
用于纯化具有式(ii)的络合物的方法
[0275]
本发明还涉及一种用于纯化以下具有式(ii)的络合物的方法:
[0276][0277]
该络合物具有至少80%的非对映异构体过量,其包含具有下式的异构体ii
‑
rrr和ii
‑
sss的混合物:
[0278]
[0279]
该方法包括:
[0280]
1)以下两个步骤的组合:
[0281]
1b)通过一种或多种离子交换树脂,以及
[0282]
1c)超滤所述络合物,以及
[0283]
2)分离如此获得的呈固体形式的纯化的络合物。
[0284]
有利地,根据先前所述的制备方法在之前获得该具有式(ii)的络合物,其具有至少80%、优选地至少85%、特别地至少90%、具体地至少95%、更具体地至少97%、优选地至少98%并且有利地至少99%的包含异构体ii
‑
rrr和ii
‑
sss的混合物的非对映异构体过量。
[0285]
在一个优选的实施方式中,在其上进行纯化过程的非对映异构体富集的复合物具有至少85%、特别地至少90%、具体地至少92%、优选地至少94%、有利地至少97%、更有利地至少99%的包含异构体ii
‑
rrr和ii
‑
sss的混合物的非对映异构体过量。
[0286]
优选地,所述非对映异构体过量由至少70%、特别地至少80%、有利地至少90%、优选地至少95%的异构体ii
‑
rrr和ii
‑
sss的混合物构成。
[0287]
有利地,所述非对映异构体过量由异构体ii
‑
rrr和ii
‑
sss的混合物组成。
[0288]
经过引申,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”还涵盖了仅存在一种异构体的情况,无论存在的是ii
‑
rrr还是ii
‑
sss。然而,术语“异构体ii
‑
rrr和ii
‑
sss的混合物”优选地代表异构体ii
‑
rrr和ii
‑
sss中的每个以可变但非零的量存在的所有情况。
[0289]
在一个优选的实施方式中,异构体ii
‑
rrr和ii
‑
sss以65/35与35/65之间、特别地60/40与40/60之间、具体地55/45与45/55之间的比率存在于所述混合物中。有利地,异构体ii
‑
rrr和ii
‑
sss以50/50的比率存在于混合物中。
[0290]
■
步骤1b)和1c)的组合
[0291]
步骤1b)和1c)涉及通过除去由于其生产过程而可能存在的杂质来纯化具有式(ii)的络合物。
[0292]
所述杂质特别地可能包括3
‑
氨基
‑
1,2
‑
丙二醇和/或双偶合杂质。
[0293]
具体地,3
‑
氨基
‑
1,2
‑
丙二醇可能存在于实施具有式(ii)的络合物的制备方法的过程中获得的最终产物中,典型地当以具有式(i)的络合物和3
‑
氨基
‑
1,2
‑
丙二醇开始通过酰胺化来获得具有式(ii)的络合物时。对于根据本发明的制备具有式(ii)的络合物的方法特别地如此。如前所详述,酰胺化反应可以包括活化相对于在其上接枝有侧链的大环的氮原子,由位于具有式(i)的络合物的所述侧链上γ位置的碳原子携带的三个羧酸官能团,然后通过与3
‑
氨基
‑
1,2
‑
丙二醇反应使活化的羧酸官能团胺解。然后有利地过量使用3
‑
氨基
‑
1,2
‑
丙二醇,以确保良好地转化为三个活化的羧酸官能团的酰胺官能团。
[0294]
术语“双偶合杂质”旨在代表具有以下所示的式(ii
‑
dc
‑
a)、(ii
‑
dc
‑
b)、(ii
‑
dc
‑
c)的络合物,或其混合物:
[0295][0296]
双偶合杂质特别地可能由具有式(ii)的络合物的酰胺官能团的水解反应引起。当制备具有式(ii)的络合物涉及此类步骤时,这也可能是由于具有式(i)的络合物的羧酸官能团活化不完全(三个官能团中的两个被活化)或被活化的羧酸官能团的胺解不完全(三个官能团中的两个被胺解)所导致的。对于根据本发明的制备具有式(ii)的络合物的方法特别地如此。
[0297]
■
步骤1b)对应于将如先前所述的非对映异构体富集的具有式(ii)的络合物通过一种或多种离子交换树脂。
[0298]
在本发明的含义内,术语“离子交换树脂”是指通常为由聚合物基体构成的珠粒形
式的固体材料,其上接枝了带正电的官能团(阴离子树脂)或带负电的官能团(阳离子树脂),这将使其有可能通过吸附分别捕获阴离子或阳离子。阴离子或阳离子在树脂上的吸附通过最初存在的官能团的抗衡离子之间的离子交换进行,以确保树脂和拟被捕获的阴离子或阳离子的电中性。
[0299]
步骤1b)涉及使非对映异构体富集的具有式(ii)的络合物的水溶液与强阴离子树脂接触。所使用的水优选地纯净水。
[0300]
所述强阴离子树脂典型地包括作为交换官能团的铵基团(n(rr’r”)
+
,其中r、r’和r”是相同或不同的(c1‑
c6)烷基基团)。可以特别提及的是陶氏化学公司(dow chemical)出售的树脂fpa900,优选地为ho
‑
形式。
[0301]
通过强阴离子树脂使得能够至少部分除去双偶合杂质。
[0302]
步骤1b)还可以涉及使非对映异构体富集的具有式(ii)的络合物的水溶液与弱阳离子树脂接触。所使用的水优选地纯净水。
[0303]
所述弱阳离子树脂典型地包括作为交换官能团的羧酸酯基团(co2‑
)。可以特别提及的是陶氏化学公司(dow chemical)出售的树脂hp336,有利地为h
+
形式。
[0304]
通过弱阳离子树脂使得能够至少部分除去3
‑
氨基
‑
1,2
‑
丙二醇和可能的gd
3+
残基。
[0305]
应当注意,通过提高根据本发明的非对映异构体富集的具有式(ii)的络合物的稳定性使通过一种或多种离子交换树脂的步骤1b)成为可能,因此在该步骤中保持了该络合物的完整性。
[0306]
■
步骤1c)对应于如先前所述的非对映异构体富集具有式(ii)的络合物的超滤。
[0307]
在本发明中,术语“超滤”是指通过中孔半透膜过滤的方法,在力(例如典型地介于1与10巴之间的压力梯度)以及视需要的浓度梯度的作用下,该中孔半透膜的孔通常具有1与100nm之间、具体地2与50nm之间、特别地10与50nm之间的直径(中孔)。因此,这是膜分离的过程,通过该过程,溶液或悬浮液中尺寸大于孔尺寸的颗粒被膜保留,并从包含它们的液体混合物中分离出来。
[0308]
在根据本发明的纯化方法的背景下,超滤对于除去内毒素特别有利。
[0309]
有利地,在步骤1c)中使用的超滤膜具有小于100kd、特别地小于50kd、具体地小于25kd的截止阈值,典型地10kd的截止阈值。
[0310]
优选地,在步骤1c)中,跨膜压力在1与5巴之间,具体地在2.25与3.25巴之间。
[0311]
■
在一个具体实施方式中,步骤1b)和1c)也与纳米过滤步骤1a)结合。
[0312]
在本发明中,术语“纳米过滤”是指通过多孔半透膜过滤的方法,在力(例如典型地介于1与50巴之间的压力梯度)以及视需要的浓度梯度的作用下,该多孔半透膜的孔通常具有在0.1与100nm之间、具体地在0.1与20nm之间、特别地在1与10nm之间的直径。因此,这是膜分离的过程,通过该过程,溶液或悬浮液中尺寸大于孔尺寸的颗粒被膜保留,并从包含它们的液体混合物中分离出来。
[0313]
纳米过滤步骤1a)使得能够除去大部分过量3
‑
氨基
‑
1,2
‑
丙二醇(视需要以盐的形式(具体地盐酸盐),或以衍生物的形式,特别地乙酰胺衍生物)和矿物盐。
[0314]
在该具体实施方式中,可以对根据先前所述的制备方法获得的粗非对映异构体富集的具有式(ii)的络合物直接进行纳米过滤步骤。特别地,不需要通过添加溶剂来沉淀先前制备的非对映异构体富集的具有式(ii)的络合物。
[0315]
有利地,在步骤1a)中使用的纳米过滤膜具有小于1kd、特别地小于500道耳顿、具体地小于300道耳顿的截止阈值,典型地200道耳顿的截止阈值。
[0316]
优选地,在步骤1a)中,跨膜压力在10与40巴之间,具体地在2与30巴之间。
[0317]
特别地,在步骤1a)中进行超滤的具有式(ii)的络合物的溶液的温度在20℃与40℃之间,特别地在25℃与35℃之间。
[0318]
在该具体实施方式的一种替代方案中,步骤1b)不涉及使非对映异构体富集的具有式(ii)的络合物的水溶液与弱阳离子树脂接触。
[0319]
在一个具体实施方式中,按此顺序进行步骤1a)(当它存在时)、1b)和1c)。该有利的实施方式特别使得能够最小化树脂的使用量并因此最小化工业制造成本。
[0320]
■
步骤2)
[0321]
步骤2)涉及以固体形式分离在步骤1b)和1c)的组合(以及视需要也与步骤1a组合)结束时获得的具有式(ii)的纯化的络合物。
[0322]
可以根据本领域技术人员熟知的任何方法来进行该固体形式的分离步骤,特别地通过雾化、通过沉淀、通过冻干或通过离心,有利地是通过雾化。
[0323]
在一个优选的实施方式中,步骤2)包括雾化。
[0324]
具体地,通过雾化以固体形式分离纯化的具有式(ii)的络合物使得能够特别省去沉淀溶剂的使用。
[0325]
然后,雾化器中的空气进口温度典型地在150℃与180℃之间、特别地在160℃与175℃之间、有利地在165℃与170℃之间。出口温度本身典型地在90℃与120℃之间,优选地在105℃与110℃之间。
[0326]
有利地,在步骤2)结束时分离的非对映异构体富集异构体ii
‑
rrr和ii
‑
sss的混合物的具有式(ii)的纯化的络合物的纯度大于95%、特别地大于97%、优选地大于97.5%、更优选地大于98%、有利地大于99%,所述纯度表示为具有式(ii)的络合物相对于在步骤2)结束时获得的总质量的质量百分比。
[0327]
本发明还涉及非对映异构体富集和纯化的具有式(ii)的络合物,其可以根据本发明的纯化方法获得。
[0328]
优选地,先前描述的根据本发明的组合物中包括的具有式(ii)的络合物是非对映异构体富集和纯化的具有式(ii)的络合物,其可以根据本发明的纯化方法获得。
[0329]
实例
[0330]
以下给出的实例作为本发明的非限制性说明呈现。
[0331]
通过uhplc分离具有式(ii)的络合物的异构体组iso1、iso2、iso3和iso4
[0332]
使用由泵系统、进样器、层析柱、uv检测器和数据站组成的uhplc机器。使用的层析柱为uhplc 150
×
2.1mm
‑
1.6μm柱(watersuplc t3柱)。
[0333]
‑
流动相:
[0334]
途径a:100%乙腈和途径b:0.0005%v/v的h2so4(96%)水溶液
[0335]
‑
制备测试溶液:
[0336]
2mg/ml的具有式(ii)的络合物在纯净水中的溶液
[0337]
‑
分析条件:
[0338][0339][0340]
‑
梯度:
[0341]
时间%acn%h2so40.0005%019935951210901525751619920199
[0342]
获得四个主峰。uhplc图的峰4(即iso4)对应于6.3分钟的保留时间。
[0343]
制备具有式(vi)的丁基六酯
[0344]
在反应器中将184kg(570mol)的2
‑
溴戊二酸二丁酯和89kg(644mol)的碳酸钾混合,并加热到55
‑
60℃。将在24kg水中的29.4kg(143mol)pyclene的水溶液添加到前述制剂中。将反应混合物保持在55
‑
60℃,然后回流约10小时。反应后,将介质冷却,用155kg甲苯稀释,然后用300升水洗涤。用175kg(1340mol)磷酸(75%)将丁基六酯萃取到水相中。然后将其用150kg甲苯洗涤3次。通过用145kg甲苯和165kg水稀释,再将丁基六酯萃取到甲苯相中,然后用30%氢氧化钠(m/m)碱化以达到5
‑
5.5的ph。除去下部水相。通过在60℃真空浓缩至干燥得到丁基六酯,产率约为85%。
[0345]
制备具有式(iii)的六酸
[0346]
将113kg(121mol)的丁基六酯与8kg的乙醇一起放入反应器中。将介质升至55
±
5℃,然后在3小时内加入161kg(1207.5mol)的30%氢氧化钠(m/m)。将反应混合物在该温度下保持约20小时。然后通过倾析反应介质除去丁醇。用水稀释以钠盐形式获得的具有式(iii)的六酸,以获得约10%(m/m)的水溶液。该溶液在酸性阳离子树脂上处理。以约90%的产率和95%的纯度获得水溶液中的具有式(iii)的六酸。
[0347]
制备具有式(i)的六酸钆络合物
[0348]
■
实验方案
[0349]
●
络合和异构化
[0350]
‑
不含乙酸
[0351]
将418kg(117kg具有式(iii)的纯六酸/196mol)的按重量计28%的具有式(iii)的
六酸的水溶液置于反应器中。通过添加盐酸将溶液的ph调节至2.7,然后添加37kg(103.2mol)的氧化钆。将反应介质在100
‑
102℃下加热48小时以实现具有式(iii)的六酸的预期异构体分布。
[0352]
‑
含有乙酸
[0353]
将氧化钆(0.525摩尔当量)以按质量计28.1%悬浮在具有式(iii)的六酸的溶液中。
[0354]
在室温下将99
‑
100%的乙酸(按质量计50%/纯的具有式(iii)的六酸)倒入介质中。
[0355]
将该介质加热至回流,随后通过在除去水时按质量计逐渐用乙酸重新填充该介质来将其蒸馏至高达113℃。一旦达到113℃的温度,就添加足够量的乙酸以达到起始体积。
[0356]
将该介质在113℃下保持过夜。
[0357]
●
结晶,重结晶
[0358]
‑
结晶
[0359]
将溶液中的具有式(i)的六酸钆络合物冷却至40℃,添加引物,并使试剂接触至少2小时。然后通过在40℃过滤分离产物,并用渗透水洗涤。
[0360]
‑
重结晶
[0361]
将180kg先前获得的具有式(i)的六酸钆络合物(固体含量为约72%)悬浮在390kg水中。将介质加热至100℃以溶解产物,然后冷却至80℃以通过添加少量引物进行预处理。冷却至室温后,通过过滤和干燥分离具有式(i)的六酸钆络合物。
[0362]
●
选择性解络合
[0363]
将干燥产物与20℃的渗透水一起放入反应器中。添加的水的质量等于具有式(i)的六酸钆络合物的理论质量的两倍。将30.5%的氢氧化钠(m/m)(6.5当量)倒入20℃的介质中。在添加naoh结束时,将介质在50℃下保持接触16小时。将该介质冷却至25℃,并将产物在clarcel垫上过滤。
[0364]
■
非对映异构体i
‑
rrr和i
‑
sss的混合物的含量
[0365]
在非对映异构体的混合物中存在具有式(i)的络合物的各种异构体的比率取决于进行络合和异构化步骤的条件,如下表3中所见。
[0366][0367]
[表3]:随络合/异构化条件变化的i
‑
rrr和i
‑
sss混合物的含量
[0368]
重结晶和选择性解络合的额外步骤使得能够增加i
‑
rrr和i
‑
sss混合物的非对映
异构体过量(见表4)。
[0369][0370]
[表4]:结晶/重结晶/选择性解络合后i
‑
rrr和i
‑
sss混合物的含量
[0371]
制备具有式(ii)的络合物
[0372]
将90kg(119mol)的具有式(i)的六酸络合物和650kg的甲醇置于反应器中。将混合物冷却至约0℃,然后倒入111kg(252mol)盐酸的甲醇溶液(在甲醇中的8.25%hcl),同时保持温度在0℃。使反应介质达到室温,然后继续搅拌16小时。冷却至0
‑
5℃后,添加120kg(1319mol)的3
‑
氨基
‑
1,2
‑
丙二醇。然后加热反应介质,同时在真空下蒸馏出甲醇,直到达到60
‑
65℃的温度。将浓缩物在该温度下真空保持16小时。接触结束时,将介质用607kg水稀释,同时冷却至室温。用20%盐酸(m/m)中和具有式(ii)的粗络合物的溶液。由此获得978.6kg的溶液,其浓度为10.3%,表示101kg的材料。得到的产率为86.5%,具有式(ii)的络合物的纯度为92.3%(hplc s/s)。双偶合杂质的量为6.4%(hplc s/s)。
[0373]
纯化具有式(ii)的络合物
[0374]
●
纳米过滤
[0375]
使用的纳米过滤膜具有200道耳顿的截留阈值(koch membran system sr3d)。该处理以以下方式进行:
[0376]
将具有式(ii)的粗络合物的溶液加热至30℃。纳米过滤器填充有所述溶液。首先以低速率打开泵以吹扫系统,然后将纳米过滤器泵的速率逐渐增加至所需的再循环速率(对于2.5
×
40英寸的膜为1.0m3/h)。然后将系统置于30℃的完全循环中至少2个小时,以建立偏振层。然后,将介质在30℃和2.5巴下进行渗滤,同时通过添加纯水保持体积恒定,直到渗余物的电导率小于1000μs。在渗滤结束时,将介质浓缩以获得约40%(m/m)的浓度。
[0377]
●
树脂上的处理
[0378]
将从纳米过滤获得的具有式(ii)的络合物的溶液在搅拌下用纯净水稀释以获得15%的溶液(m/m)。将该溶液在50升oh
‑
形式的强阴离子树脂(fpa900),然后50升h
+
形式的弱阳离子树脂(hp336)上以2v/v/h(2体积溶液/树脂体积/小时)的平均洗脱流速进行系列洗脱。然后用约450升纯净水冲洗树脂,直到获得小于1.3335的折射率。
[0379]
然后通过在20毫巴的真空下加热至50
‑
60℃来浓缩具有式(ii)的络合物溶液,以达到35%(m/m)的浓度。
[0380]
●
超滤
[0381]
超滤膜是uf 10kd koch螺旋膜。
[0382]
向超滤器进料先前的加热至40℃的35%具有式(ii)的络合物溶液。以3m3/h的流速和2.5
‑
3巴的跨膜压力进行超滤。用13升无热原纯净冲洗该系统若干次,直到达到25%(m/m)的具有式(ii)的络合物的最终稀释度。
[0383]
●
雾化
[0384]
通过将先前的浓缩至25%的具有式(ii)的络合物的溶液雾化来获得粉末形式的
具有式(ii)的络合物。
[0385]
雾化以以下方式进行:
[0386]
通过将入口温度设置为165℃
‑
170℃并调整进料速度以使出口温度在105℃与110℃之间,使雾化器与无热原纯水平衡。
[0387]
然后添加具有式(ii)的络合物的浓缩溶液,并调节流速以保留上述参数。
[0388]
在整个雾化过程中保持这些操作条件,同时确保粉末在雾化室中和雾化器出口处的良好表现。特别地应确保没有产品附着。
[0389]
在向雾化器递送溶液结束时,用无热原纯水冲洗该具有式(ii)的络合物的容器和雾化器,直到获得最大的粉末回收率。
[0390]
获得纯度为99.6%的具有式(ii)的络合物。
[0391]
该纯度通过反相液相层析法确定。
[0392]
根据本发明的组合物及其研究结果
[0393]
●
根据本发明的制造方法的实例
[0394]
根据以下步骤进行用于制造根据本发明的组合物的方法:
[0395]
a)将485.1g(即0.5m)的具有式(ii)的络合物溶于水(适量,1升)中,将罐加热至39℃与48℃之间的温度,并剧烈搅拌溶液直至该络合物完全溶解在水中。然后将溶液冷却至约30℃。
[0396]
b)在搅拌下通过10%m/v的dota溶液将0.404g(即,相对于步骤a)中添加的络合物的比例为0.2mol/mol%)的dota(simafex,法国)添加到步骤a)中获得的溶液中。
[0397]
c)在搅拌下将胺丁三醇(tris)添加到步骤b)中获得的溶液中。然后通过在搅拌下添加盐酸溶液将ph调节至7.2与7.7之间的值。
[0398]
d)通过分两步添加注射用水直至达到1.198与1.219g/ml之间的密度值来获得目标浓度(0.5mol/l)。
[0399]
然后将液体组合物通过聚醚砜膜过滤,并放置在其最终容器中,将其最终在121℃下灭菌15分钟。
[0400]
●
根据本发明的组合物的实例
[0401]
通过上述方法获得以下制剂:
[0402][0403][0404]
*用二甲酚橙通过比色法进行测量
[0405]
**以无水纯净形式表示
[0406]
●
进行的制剂测试
[0407]
测试了从0至100mm的各种浓度的胺丁三醇。这些测试的结果表明,10mm(0.12%w/v)的含量足以确保制剂的ph稳定性,同时限制降解杂质的形成。
[0408]
测试了从0至2.5mm的各种浓度的dota。这些测试的结果表明,1mm的含量(对应于0.04%m/v或0.2mol/mol%)使得能够确保在方法期间和产品的使用寿命期间不释放游离gd。
[0409]
●
在加速条件下根据本发明的组合物的稳定性研究
[0410]
在制造后(t0)和制造后在40℃下储存6个月(t+6个月)后,分析前述实例的制剂。
[0411]
在t0:
[0412]
‑
通过层析法评估的纯度*:99.6%
[0413]
‑
gd
‑
dota的浓度:0.007%(m/v)
[0414]
‑
gd的浓度:低于0.0001%(m/v)
[0415]
‑
ph:7.5
[0416]
在t+6个月:
[0417]
‑
通过层析法评估的纯度*:97.2%
[0418]
‑
gd
‑
dota的浓度:0.014%(m/v)
‑
0.25mm
[0419]
‑
gd的浓度:低于0.0001%(m/v)
[0420]
‑
ph:7.5
[0421]
*反相液相层析法
[0422]
这些结果证明该制剂随时间推移具有良好的稳定性。
[0423]
●
比较稳定性研究
[0424]
随时间推移评估以下组合物的稳定性。术语“未优化的ap”代表有效成分,即根据ep 1 931 673中所述的方法获得的具有式(ii)的络合物。术语“优化的ap”代表通过根据本发明的方法获得的具有式(ii)的非对映异构体富集和纯化的络合物。
[0425]
[0426][0427]
*lc甲酸盐:涉及荧光检测的层析方法。在反相c18接枝层析柱上以梯度模式洗脱以进行分离。
[0428]
以上报告的结果表明,不可能用游离dota配制未优化的ap。原因是螯合赋形剂完全被具有式(ii)的络合物与dota之间的反式连接反应所消耗,因此不能再起捕获释放的gd
3+
的作用。
[0429]
另一方面,可以将根据本发明的方法获得的具有式(ii)的非对映异构体富集和纯化的络合物与游离dota一起配制。具体地,观察到在40℃下6个月的组合物中不存在游离gd,无论制剂的ph如何并且缓冲物质存在与否,都是这种情况。此外,螯合赋形剂的消耗非常低,因为它不超过0.08mol/mol%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1