一种A-D-A型有机半导体材料的合成新方法
一种a-d-a型有机半导体材料的合成新方法
技术领域
1.本发明属于有机半导体领域,具体涉及给-受体(d-a)推拉型有机半导体材料的简单、高效合成新方法。
背景技术:
2.有机太阳能电池具有重量轻、柔性好、加工方式简单、可大面积制备并且成本低等潜在优势,受到学术界和工业界的广泛关注。目前,有机太阳能电池发展迅速,其光电转换效率已突破18%,这显示出有机太阳能电池的巨大应用前景。有机太阳能电池的快速发展主要得益于非富勒烯受体的研制。在这些非富勒烯受体中,a-d-a型受体倍受关注,这主要得益处于分子结构中给体(d)和受体(a)之间的电荷转移能够方便的调控吸收和能级,以及其它性质,进而实现光伏性能的提升。(占肖卫等,nat.rev.mater.2018, 3,18003;占肖卫等nat.photon.2018,12,131-142.)最近研究人员通过在d单元中引入吸收电子单元(例如苯并噻二唑,苯并三氮唑等)进一步调控能级,拓展吸收,合成了以y6 为代表的非富勒烯受体,又将光伏性能提升到了15%以上。
3.关于这些a-d-a非富勒受体的合成,通常是将双醛基修饰的d与含有活泼亚甲基的a 单元通过knoevenagel缩合得到。反应中,使用吡啶、三乙胺等碱性催化剂,由于该反应是可逆反应通常需要加入过量的a单元,促使反应向右进行。该方法时间较长,产率不高,且生成多种副产物使得提纯过程繁杂,也带来合成成本高的问题。因此开发新的knoevenagel缩合方法对于提升受体材料纯度与降低生产成本,实现商业化应用来说非常重要。
技术实现要素:
4.本发明的目的是提供一种非富勒受体的其制备方法。
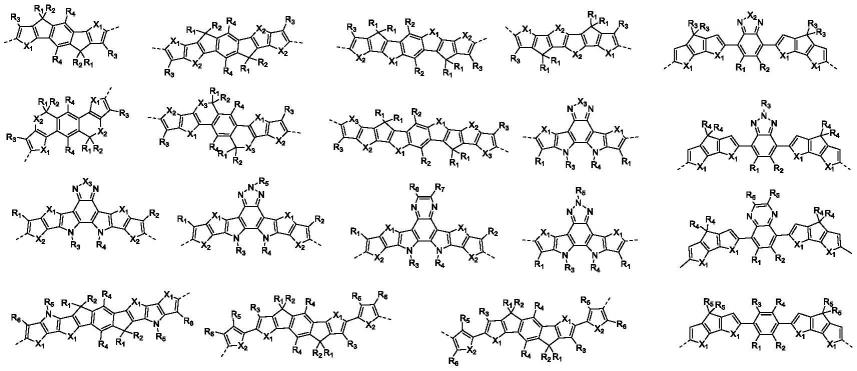
5.本发明所提供的非富勒受体的结构式如式i所示:
[0006][0007]
式i中,基团d选自下述单元中的任一种:
[0008][0009]
其中,式ii中的r1-r6均独立地选自h、卤素、碳原子数为1~30的直链或支链的烷基或烷氧基、烷硫基、硅烷基,以及烷基或烷氧基、烷硫基、硅烷取代的芳基。所述芳基可以为苯环或者噻吩环。所述卤素可为f、br或者cl;
[0010]
式ii-1至式ii-14中的x1-x3均独立地选自o、s、se、te中的一种。
[0011]
a独立地选自下述单元中的任一种:
[0012][0013]
其中,式ⅲ中的r
7-r
10
独立地选自f、cl、br、i、cn、h、三氟甲基、烷氧基、烷硫基础中的一种或几种,烷氧基、烷硫基中烷基碳原子数为1~30直链或者支链;
[0014]
x4和x5选自c、n中的一种或两种。
[0015]
式ⅲ1
‑ⅲ
10中的虚线表示与式i中双键的连接位置。
[0016]
所述非富勒烯受体(式i)具体如式iv所示,但又不仅局限于此:
[0017][0018]
本发明进一步提供了式i所示小分子受体的制备方法,包括如下步骤:
[0019]
式v所示化合物与式
ⅴ
i所示化合物在催化剂的作用下进行缩合反应,得到式i所示小分子受体
[0020][0021]
式
ⅴ
独立地选自下述单元中的任一种。
[0022][0023]
其中,式v中的r
1-r6以及x
1-x3与式ii相同。
[0024]
式v中的双醛基化合物具体如下所示,但又不局限于此:
[0025][0026]
式ⅵ独立地选自下述单元中的任一种,
[0027][0028]
其中,式ⅲ中,r7至r10,x1和x2与式iii中的定义相同。式vi中的化合物具体如下所示,但又不局限于此:
[0029][0030]
上述的制备方法具体可按照下述方法进行:
[0031]
催化剂为路易斯酸,如bf3·
c2h5oc2h5、alcl3、gacl3、incl3、ga(otf)3、gabr3、 gai3中的一种或几种,优选为gacl3、bf3·
c2h5oc2h5、ga(otf)3。
[0032]
所述催化剂的加入量为式ⅳ所示化合物与式
ⅴ
所示化合物的总摩尔量的 0.01%~20%,优选为5-10%。
[0033]
所述酸酐为反应物,如式
ⅸ
所示。
[0034][0035]
式
ⅸ
中r为碳原子数为1~30的直链或支链的烷基或者烯烷基;式ix-5中r
12
、 r
13
为酯基、烷氧基或者f、cl、br、i、cn、h、oh中的一种,当为酯基和烷氧基时,碳原子数为1~30的直链或支链的烷基。
[0036]
式ix中的酸酐具体如下所示,但又不局限于此:
[0037][0038]
所述酸酐的加入量为式ⅳ所示化合物与式
ⅴ
所示化合物的总摩尔量的 0.01%~20%,优选为10-30%。
[0039]
所述的溶剂为苯、甲苯、邻二甲苯、对二甲苯、均三甲苯、氯苯。优选为甲苯、二甲苯或者二氯。
[0040]
式ⅶ所示化合物与式
ⅷ
所示化合物的摩尔比为1:2~2.5,如1:2;
[0041]
反应温度为30℃~120℃,优选为50℃-100℃。
[0042]
反应时间为15分钟~24小时,优选为30-60分钟。
[0043]
本发明具有如下有益效果:
[0044]
本发明提供了一种小分子受体材料的合成方法,所述合成方法具有产率高,产物纯净等特点,具有较好的底物适应性,能够广泛地应用于小分子受体的合成。所合成的产物应用于非富勒烯聚合物太阳能电池器件。
[0045]
本发明的另一个目的是提供一种光活性层。所述光活性层由式i所示的非富勒烯受体和p-型电子给体材料(聚合物或者小分子)组成,所述非富勒烯受体与所述p-型给体材料的质量比为1:0.1~10,如1:1;
[0046]
本发明所述p-型电子给体聚合物适用于本领域技术人员可选择的任意种类的p-型电子给体聚合物,如pbdb-t。
[0047]
所述光活性层可采用溶剂甲苯、二甲苯、三甲苯、氯仿、氯苯、二氯苯和三氯苯中至少一种进行混合,所得到的混合物中,所述非富勒烯受体的浓度可为0.5 mg/ml~50mg/ml,优选为4mg/ml~20mg/ml,所述p-型电子给体聚合物的浓度可为 0.5mg/ml~50mg/ml,优选为3mg/ml~20mg/ml。
[0048]
本发明还提供了一种聚合物太阳能电池器件,包括第一电极、与所述第一电极间隔开的第二电极、以及在所述的第一电极和第二电极之间设置的至少一层半导体层,所述半导体层包含所述非富勒烯受体或所述光活性层。
[0049]
所述非富勒受体以及所述光活性层在制备下述功能性能量器件中的应用也属于本发明的保护范围:薄膜半导体器件、光探测器件、聚合物太阳能电池器件和光电器件。
[0050]
针对现有技术的缺陷,本发明的发明人发展了一种快速定量转化,后处理更简便的合成方法。本发明提供了一种大规模和低成本制备受体材料的方法,这是有机太阳能电池商业化所需要的。除了在有机太阳能电池中的应用外,该方法还将在发光二极管、场效应晶体管和存储器件等方面得到应用。
附图说明
[0051]
图1:代表结构iv-1核磁共振氢谱图。
[0052]
图2:代表结构iv-2核磁共振氢谱图。
[0053]
图3:代表结构iv-3核磁共振氢谱图。
[0054]
图4:代表结构iv-4核磁共振氢谱图。
[0055]
图5:代表结构iv-5核磁共振氢谱图。
[0056]
图6:代表结构iv-6核磁共振氢谱图。
[0057]
图7:代表结构iv-7核磁共振氢谱图。
[0058]
图8:代表结构iv-8核磁共振氢谱图。
[0059]
图9:代表结构iv-9核磁共振氢谱图。
[0060]
图10:代表结构iv-10核磁共振氢谱图。
[0061]
图11:通用的合成方法简式
具体实施方式
[0062]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
[0063]
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0064]
在以下实施例中,努力确保所用数字(包括量、温度、反应时间等的准确性),但应考虑到一些实验误差和偏差。在以下实施例中所用的压力以大气压或接近大气压。所用溶剂都是以hplc级购得,除非另外指出,否则所有试剂和原料均是商业上获得的。
[0065]
下面通过具体实施例,对本发明的技术方案作进一步的具体说明。应当理解,本发明的实施并不局限于下面的实施例,对本发明所做的任何形式上的变通或改变都将落入本发明保护范围。在本发明中,若非特指,所有的份、百分比均为物质的量单位,所采用的设备和原料等均可从市场购得或是本领域常用的。
[0066]
对实施例中的测试方法进行以下说明:
[0067]
本文采用核磁共振波谱仪和激光辅助飞行时间质谱仪来表征所合成的结构。
[0068]
实施例1
[0069]
ⅳ‑
1的合成
[0070]
合成过程见下式:
[0071][0072]
在100ml单口瓶内加入1.026g式
ⅳ‑
4、0.460g式
ⅷ‑
3、4mg无水三氯化镓溶于 20.0ml甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物式
ⅳ‑
1 1.421g,产率98%。产物结构经过1h nmr和
madil-tof得到确认。
[0073]1h nmr(400mhz,cdcl3)δ9.15(s,2h),8.56(t,j=6.4hz,2h),7.73(t,j=7.6hz,2h), 4.81(d,j=7.2hz,4h),3.24(d,j=7.6hz,4h),2.20-2.01(m,2h),1.95-1.83 (m,4h),1.36-1.16(m,36h),1.12-1.00(m,12h),0.87(t,j=6.8hz,6h),0.79(t,j=4.8hz, 6h),0.69(t,j=7.2hz,6h)..ms(madil-tof)m/z:[m]
+
:calcd for c
82h86
f4n8o2s5; found[m+1]
+
:1451.589.
[0074]
实施例2
[0075]
ⅳ‑
2的合成
[0076]
合成过程见下式:
[0077][0078]
在100ml单口瓶内加入1.026g
ⅵ‑
4、0.364g式
ⅷ‑
6、4mg无水三氯化镓溶于 20.0ml甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物1.291g,产率96%。产物结构经过1h nmr和madil-tof得到确认。
[0079]1h nmr(400mhz,cdcl3)δ8.24(s,2h),7.80-7.76(m,4h),4.80(d,j=6.0hz,4h), 3.25(t,j=7.6hz,4h),2.21-2.12(m,2h),1.94-1.86(m,4h),1.35-1.15 (m,36h),1.15-0.99(m,12h),0.88(t,j=6.8hz,6h),0.80(t,j=4.8hz,6h),0.70(t,j=7.2 hz,6h).ms(madil-tof)m/z:[m]
+
:calcd for c
76h86
f4n4o4s5;found[m+1]
+
: 1354.627.
[0080]
实施例3
[0081]
ⅳ‑
3的合成
[0082]
合成过程见下式:
[0083][0084]
在100ml单口瓶内加入1.026g
ⅶ‑
1、0.388g
ⅷ‑
1、4mg无水三氯化镓溶于20.0ml 甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离
心得到并烘干黑色粉末产物
ⅳ‑
31.336g,产率97%。产物结构经过1hnmr和madil-tof得到确认。
[0085]1hnmr(400mhz,cdcl3)δ8.89(s,2h),8.71(d,j=7.2hz,2h),8.25(s,2h),7.94(d,j=6.8hz,2h),7.81-7.14(m,4h),7.66(s,2h),7.24(d,j=8.4hz,4h),7.16(d,j=8.4hz,4h),2.60(t,j=8.0hz,8h),1.68-1.60(m,8h),1.39-1.27(m,24h),0.88(t,j=6.8hz,12h).ms(madil-tof)m/z:[m]
+
:calcdforc
90h82
n4o6s2;found[m+1]
+
:1426.635.
[0086]
实施例4
[0087]
ⅳ‑
4的合成
[0088]
合成过程见下式:
[0089][0090]
在100ml单口瓶内加入1.026g
ⅶ‑
1、0.388g
ⅷ‑
1、4mg无水三氯化镓溶于20.0ml甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物
ⅳ‑
41.26g,产率95%。产物结构经过1hnmr和madil-tof得到确认。
[0091]1hnmr(400mhz,cdcl3)δ7.98-7.96(m,4h),δ7.94(s,2h)δ7.81-7.78(m,4h),δ7.64(s,2h),7.25(t,j=7.2hz,8h),7.17(d,j=8.4hz,h),2.60(t,j=8.0hz,8h),1.68-1.60(m,8h),1.39-1.27(m,24h),0.88(t,j=6.8hz,12h).ms(madil-tof)m/z:[m]
+
:calcdforc
88h82
o4s4;found[m+1]
+
:1331.635.
[0092]
实施例5
[0093]
ⅳ‑
5的合成
[0094]
合成过程见下式:
[0095][0096]
在100ml单口瓶内加入1.074g
ⅶ‑
2、0.426g
ⅷ‑
5、4mg无水三氯化镓溶于20.0ml甲
苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物
ⅳ‑
51.392g,产率95%。产物结构经过1hnmr和madil-tof得到确认。
[0097]1hnmr(400mhz,cdcl3)δ8.02(s,4h),δ7.94(s,2h),δ7.63(s,2h),7.25(t,j=7.2hz,8h),7.17(d,j=8.4hz,h),2.60(t,j=8.0hz,8h),1.68-1.60(m,8h),1.39-1.27(m,24h),0.88(t,j=6.8hz,12h).ms(madil-tof)m/z:[m]
+
:calcdforc
88h78
cl4o4s4;found[m+1]
+
:1467.481.
[0098]
实施例6
[0099]
ⅳ‑
6的合成
[0100]
合成过程见下式:
[0101][0102]
在100ml单口瓶内加入1.074g
ⅶ‑
2、0.522g
ⅷ‑
2、4mg无水三氯化镓溶于20.0ml甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物式
ⅳ‑
61.530g,产率98%。产物结构经过1hnmr和madil-tof得到确认。
[0103]1hnmr(400mhz,cdcl3)δ8.90(s,2h),8.79(s,2h),8.26(s,2h),7.97(s,2h),7.68(s,2h),7.23(d,j=8.0hz,4h),7.17(d,j=8.0hz,4h),2.60(t,j=7.6hz,8h),1.65-1.54(m,8h),1.39-1.25(m,24h),0.88(t,j=6.8hz,12h).ms(madil-tof)m/z:[m]
+
:calcdforc
94h78
cl4n4o2s4;found[m+1]
+
:1563.421.
[0104]
实施例7
[0105]
ⅳ‑
7的合成
[0106]
合成过程见下式:
[0107][0108]
在100ml单口瓶内加入0.770g
ⅶ‑
2、0.388g
ⅷ‑
1、4mg无水三氯化镓溶于20.0ml 甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物
ⅳ‑
7 1.088g,产率97%。产物结构经过1h nmr和madil-tof得到确认。
[0109]1h nmr(400mhz,cdcl3)δ9.01(s,2h),8.74(dd,j=1.2hz,j=6.0hz 2h),7.97(s,dd, j=0.8hz,j=6.4hz 2h),7.83-7.78(m,4h),7.75(s,2h),7.62(s,2h),2.10(dt,j=3.6hz, j=13.6hz,4h),1.98(dt,j=3.6hz,j=13.6hz,4h).1.20-1.07(m,24h),0.96-0.85 (m,8h),0.81(t,j=7.2hz,12h).ms(madil-tof)m/z:[m]
+
:calcd for c
70h66
n4o2s4; found[m+1]
+
:1123.583.
[0110]
实施例8
[0111]
ⅳ‑
8的合成
[0112]
合成过程见下式:
[0113][0114]
在100ml单口瓶内加入0.770g
ⅶ‑
2、0.292g
ⅷ‑
4、4mg无水三氯化镓溶于20.0ml 甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物
ⅳ‑
8 0.974g,产率95%。产物结构经过1h nmr和madil-tof得到确认。
[0115]1h nmr(400mhz,cdcl3)δ8.05(s,2h),8.02-7.80(m,6h),7.83-7.78(m,4h),7.58(s, 2h),7.62(s,2h),2.10(dt,j=3.6hz,j=13.6hz,4h),1.96(dt,j=3.6hz,j=13.6hz, 4h).1.20-1.07(m,24h),0.96-0.85(m,8h),0.81(t,j=7.2hz,12h).ms(madil-tof)m/z: [m]
+
:calcd for c
64h66
o4s4;found[m+1]
+
:1027.415.
[0116]
实施例9
[0117]
ⅳ‑
9的合成
[0118]
合成过程见下式:
[0119][0120]
在100ml单口瓶内加入1.074g
ⅶ‑
1、0.460g
ⅷ‑
3、4mg无水三氯化镓溶于20.0ml 甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物
ⅳ‑
9 1.468g,产率98%。产物结构经过1h nmr和madil-tof得到确认。
[0121]1h nmr(400mhz,cdcl3)δ8.87(s,2h),7.45(dd,j=6.4hz,j=9.6hz,2h),8.26(s, 2h),7.70(t,j=7.6hz 2h),7.67(s,2h),7.23(d,j=8.0hz,4h),7.17(d,j=8.0hz,4h), 2.56(t,j=7.6hz,8h),1.65-1.59(m,8h),1.40-1.25(m,24h),0.88(t,j=6.8hz,12h).ms (madil-tof)m/z:[m]
+
:calcd for c
94h78
f4n4o2s4;found[m+1]
+
:1499.554.
[0122]
实施例10
[0123]
ⅳ‑
10的合成
[0124]
合成过程见下式:
[0125][0126]
在100ml单口瓶内加入1.074g
ⅶ‑
1、0.460g
ⅷ‑
6、4mg无水三氯化镓溶于20.0ml 甲苯,加热至60℃。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,重新滴加至100ml甲醇中,离心得到并烘干黑色粉末产物式
ⅳ‑
10 1.359g,产率97%。产物结构经过1h nmr和madil-tof得到确认。
[0127]1h nmr(400mhz,cdcl3)δ8.45(s,2h),δ7.91(s,2h),7.73(t,j=7.2hz,4h),7.66
(s,2h),7.24(d,j=8.4hz,8h),7.16(d,j=8.4hz,8h),2.60(t,j=8.0hz,8h),1.68-1.60(m,8h),1.39-1.27(m,24h),0.88(t,j=6.8hz,12h).ms(madil-tof)m/z:[m]
+
:calcdforc
88h78
f4o4s4;found[m+1]
+
:1403.531.
[0128]
上述实施例中,
ⅳ‑
1至
ⅳ‑
10的核磁谱图如图1-图10所示
[0129]
对比例1吡啶催化合成式
ⅳ‑
2的合成
[0130]
合成过程见下式:
[0131][0132]
在100ml单口瓶内加入1.026gitic-cho、0.460g
ⅷ‑
3、0.5ml吡啶溶于30.0ml三氯甲烷,加热至回流。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,经过硅胶固定相(200-300目)柱层析法提纯(流动相v
dcm
:v
pe
=3:2)得到黑色粉末产物产物sma11.015g,产率70%。产物结构经过1hnmr和madil-tof得到确认。
[0133]1hnmrspectrumof2f-y6.1hnmr(400mhz,cdcl3)δ9.15(s,2h),8.56(t,j=6.4hz,2h),7.73(t,j=7.6hz,2h),4.81(d,j=7.2hz,4h),3.24(d,j=7.6hz,4h),2.20-2.01(m,2h),1.95-1.83(m,4h),1.36-1.16(m,36h),1.12-1.00(m,12h),0.87(t,j=6.8hz,6h),0.79(t,j=4.8hz,6h),0.69(t,j=7.2hz,6h).ms(madil-tof)m/z:[m]
+
:calcdforc
76h86
f4n4o4s5;found[m+1]
+
:1451.589.
[0134]
对比例2
[0135]
吡啶催化合成
ⅳ‑
1的合成
[0136]
合成过程见下式:
[0137][0138]
在100ml单口瓶内加入1.026gitic-cho、0.460gic、0.5ml吡啶溶于30.0ml三氯甲烷,加热至回流。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,经过硅胶固定相(200-300目)柱层析法提纯(流动相v
dcm
:v
pe
=3:2)得到黑色粉末产物产物sma11.015g,产率70%。产物结构经过1hnmr和madil-tof得到确认。
[0139]1hnmrspectrumof2f-y6.1hnmr(400mhz,cdcl3)δ9.15(s,2h),8.56(t,j=6.4hz,2h),7.73(t,j=7.6hz,2h),4.81(d,j=7.2hz,4h),3.24(d,j=7.6hz,4h),2.20-2.01(m,2h),1.95-1.83(m,4h),1.36-1.16(m,36h),1.12-1.00(m,12h),0.87(t,j=6.8hz,6h),0.79(t,j=4.8hz,6h),0.69(t,j=7.2hz,6h).ms(madil-tof)m/z:[m]
+
:calcdforc
82h86
f4n8o2s5;found[m+1]
+
:1451.589.
[0140]
对比例3吡啶催化合成
ⅳ‑
4的合成
[0141]
合成过程见下式:
[0142][0143]
在100ml单口瓶内加入1.074g
ⅶ‑
1、1.150g
ⅷ‑
1、0.5ml吡啶溶于30.0ml氯仿,加热至回流。tlc检测至原料点消失。tlc检测至原料点消失,将反应液滴加至100ml甲醇中。过滤得到粗产物,用甲醇多次洗涤得到二次粗产物。将二次粗产物溶于二氯甲烷,经过硅胶固定相(200-300目)柱层析法提纯(流动相v
dcm
:v
pe
=3:2)得到黑色粉末产物9.68g,总产率65%。产物结构经过1hnmr和madil-tof得到确认。
[0144]1hnmr(400mhz,cdcl3)δ7.98-7.96(m,4h),δ7.94(s,2h)δ7.81-7.78(m,4h),δ7.64(s,2h),7.25(t,j=7.2hz,8h),7.17(d,j=8.4hz,h),2.60(t,j=8.0hz,8h),1.68-1.60(m,8h),1.39-1.27(m,24h),0.88(t,j=6.8hz,12h).ms(madil-tof)m/z:[m]+:calcdforc
88h82
o4s4;found[m+1]+:1331.635.
[0145]
对于实施例1-10,可以通过简单的后处理得到纯净的目标产物,避免使用柱层析法,减少了溶剂的使用量,也使得产率得到提高。可以发现经过简单的甲醇沉降,本发明所述的方法可以得到较纯净的产物。对于传统方法,如对比例1、对比例2和对比例3所示,需要经过柱层析提纯,这意味着更多的损耗。
[0146]
实施例11制备常规结构的聚合物光伏器件
[0147]
将本发明实施例1合成的小分子受体与市售聚合物给体pm6以重量比为1:1共混溶解于三氯甲烷制备共混活性层溶液。在透明氧化铟锡(ito)衬底上制备聚合物光伏器件。将常用的阳极修饰层聚3,4-亚乙基二氧噻吩:聚苯乙烯磺酸盐(pedot:pss)旋涂在ito表面进行修饰,使用膜厚仪测试pedot:pss层的厚度为30nm。接着上述共混的活性层溶液旋涂薄层。然后在大约10-4
pa的压力下相继蒸镀银的薄层,得到常规结构的聚合物光伏器件。在填充n2的手套箱中使用aaa级太阳光模拟器am1.5g(100mw/cm2)的强度下队所制备聚合物光伏器件的开路电压、短路电流、填充因子和能量转换效率进行测试。
[0148]
其中开路电压为0.848v,短路电流为25.01ma/cm2,填充因子为76.91%,能量转换效率为16.31%。
[0149]
本发明参照特定的实施方案和实施例进行描述。然而,本发明不局限于仅仅上述
的实施方案和实施例。本领域普通技术人员应认识到,基于本文的教导,在不偏离权利要求书所限定的本发明的范围下可进行许多替代和改变。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1