基于蒽衍生物的激基缔合物发光材料及其制备方法和应用
1.本发明专利涉及有机光电材料领域,尤其是指基于蒽衍生物的激基缔合物发光材料及其制备方法和应用。
背景技术:
2.蒽衍生物具有结构简单、易合成以及具有优异的激基缔合物发光性能等特点,激基缔合物发光是指一个激发态分子与另一个相同种类的基态分子发生相互作用,形成能量相对稳定的二聚体,通过辐射跃迁产生的发射光,相比于单分子发光,其表现出光谱红移、增宽等特点,在有机发光二极管(oled)、安全防伪、光催化反应以及细胞成像等领域都得到广泛的应用。然而,双分子在形成激基缔合物的同时,往往会因为严重的分子间π
‑
π作用而导致荧光淬灭,从而影响其发光效率,因此,在激基缔合物形成的同时,保证较高的发光量子产率显得尤为重要。1954年,和kasper首次报道了芘溶液体系的激基缔合物发光。随后,在芘的晶体中也发现了激基缔合物发光,通过晶体结构分析,推测其激基缔合物是激发态的二聚体行为。经过科学家们的不断探索发现,与芘类似,蒽的激基缔合物发光现象也陆续被报道。杨兵教授课题组发现了蒽衍生物2
‑
ta
‑
an晶体体现出526nm的绿光发射,其量子产率达80%,寿命达163.75ns,完全不同于其稀溶液的深蓝光发射(量子产率为26%和荧光寿命为2.27ns)。通过单晶结构分析,发现其呈现出离散的蒽二聚体堆积,蒽单元的距离为既有利于激基缔合物的形成,又避免了分子间相互作用带来的荧光淬灭效应。随后,多种蒽衍生物都被发现具有高效率、较长寿命的激基缔合物发光性能。
3.但是,这种侧边取代的蒽衍生物往往体现出对浓度的高度依赖性,只有在高浓度或者在晶态中才能体现出激基缔合物的性质,大大限制了此类发光的实际应用。若能实现单分子的激基缔合物发光,将大大拓展其实际应用的范围,提高其发光性能的可控性与预见性。
4.因此,研发一种具有单分子激基缔合物发光性能的化合物显得很有必要。
技术实现要素:
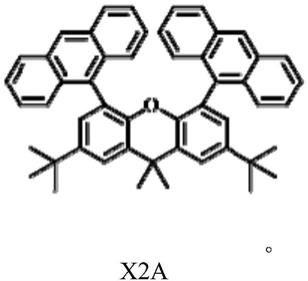
5.本发明旨在至少在一定程度上解决现有技术中存在的技术问题之一,由此,在本发明的第一方面,本发明提供一种基于蒽衍生物的激基缔合物发光材料,所述基于蒽衍生物的激基缔合物发光材料的结构式如下式x2a所示:
[0006][0007]
在本发明的一个或多个实施例中,所述基于蒽衍生物的激基缔合物发光材料溶解在有机溶剂中或掺杂在pmma薄膜中表现出红移的激基缔合物绿光发射;
[0008]
优选地,所述基于蒽衍生物的激基缔合物发光材料为固态时的荧光量子产率大于99.9%;
[0009]
更优选地,所述基于蒽衍生物的激基缔合物发光材料以浓度为10
‑6~10
‑2mol/l溶解在有机溶剂中;所述基于蒽衍生物的激基缔合物发光材料以0.5~2wt%掺杂在pmma薄膜中;
[0010]
更优选地,所述有机溶剂选自四氢呋喃、二氯甲烷、三氯甲烷、甲醇、乙醇中的一种或多种。
[0011]
在本发明的第二方面,本发明提供一种本发明第一方面所述的基于蒽衍生物的激基缔合物发光材料的制备方法,所述式x2a所示化合物由式2所示化合物和式3所示化合物制备得到。
[0012]
在本发明的一个或多个实施例中,式2所示化合物和式3所示化合物制备式x2a所示化合物的反应式如下所示:
[0013][0014]
式2所示化合物和式3所示化合物制备式x2a所示化合物包括如下步骤:
[0015]
在惰性气氛下,式2所示化合物、式3所示化合物、碳酸钾、二氯二叔丁基
‑
(4
‑
二甲基氨基苯基)磷钯(ii)放入反应容器中,加入四氢呋喃和水,加热回流反应5~15小时,纯化,得到式x2a所示化合物;
[0016]
优选地,所述式2所示化合物和式3所示化合物的摩尔比为1:(2~5);更优选地,碳酸钾与式2所示化合物的摩尔比为(2~6):1;更优选地,所述二氯二叔丁基
‑
(4
‑
二甲基氨基苯基)磷钯(ii)与式2所示化合物的摩尔比为(2~8):100;
[0017]
在本发明的一个或多个实施例中,式2所示化合物和式3所示化合物制备式x2a所示化合物过程中,所述纯化包括如下步骤:将反应液冷却至15~35℃,向反应液中加入饱和氯化钠水溶液,用二氯甲烷萃取得到有机相,用无水硫酸钠干燥有机相,除去溶剂,利用硅胶柱层析以石油醚和二氯甲烷的混合溶剂为淋洗剂分离,干燥;
[0018]
优选地,所述混合溶剂中,石油醚和二氯甲烷的体积比为(4~6):1。
[0019]
在本发明的一个或多个实施例中,所述式2所示化合物由式1所示化合物制备得到;
[0020]
式1所示化合物制备式2所示化合物的反应式如下所示:
[0021][0022]
式1所示化合物制备式2所示化合物包括如下步骤:
[0023]
冰浴下,将式1所示化合物溶于三氯甲烷中,缓慢滴加经三氯甲烷稀释的液溴溶液,15~35摄氏度下搅拌反应10~15小时,纯化,得到式2所示化合物;
[0024]
优选地,所述式1所示化合物和液溴的摩尔比为1:(2~4);
[0025]
在本发明的一个或多个实施例中,式1所示化合物制备式2所示化合物过程中,所述纯化包括如下步骤:向反应液中加入亚硫酸钠水溶液淬灭反应,用二氯甲烷萃取得到有机相,用无水硫酸钠干燥有机相,除去溶剂,以石油醚为淋洗剂硅胶柱层析分离,干燥。
[0026]
在本发明的一个或多个实施例中,所述式1所示化合物的合成路线如下所示:
[0027][0028]
所述式1所示化合物通过如下方法制备得到:冰浴下,将9,9
‑
二甲基氧杂蒽与氯化铁溶于二氯甲烷中,加入经二氯甲烷稀释的氯代叔丁烷溶液,15~35摄氏度下搅拌10~15小时,纯化,得到式1所示化合物;
[0029]
优选地,所述9,9
‑
二甲基氧杂蒽和氯代叔丁烷的摩尔比为1:(2~3);更优选地,所述氯化铁与所述9,9
‑
二甲基氧杂蒽的摩尔量为(1~2):20;;
[0030]
在本发明的一个或多个实施例中,式1所示化合物制备过程中,所述纯化包括如下步骤:向反应液中加入水,用二氯甲烷萃取得到有机相,用无水硫酸钠干燥有机相,去除有机溶剂,以石油醚为淋洗剂硅胶柱层析分离,干燥。
[0031]
在本发明的一个或多个实施例中,所述式3所示化合物的合成路线如下所示:
[0032][0033]
所述式3所示化合物通过如下方法制备得到:在惰性气氛下,将9
‑
溴蒽、联硼酸频那醇酯、乙酸钾、[1,1'
‑
双(二苯基膦基)二茂铁]二氯化钯放入反应容器中,加入1,4
‑
二氧六环,加热回流反应8~12小时,纯化,得到式3所示化合物;
[0034]
优选地,所述9
‑
溴蒽与联硼酸频那醇酯的摩尔比为1:(1.5~3);更优选地,所述乙
酸钾与9
‑
溴蒽的摩尔量比为2~4:1;更优选地,所述1,1'
‑
双(二苯基膦基)二茂铁]二氯化钯与9
‑
溴蒽的摩尔量为(1~2):20。
[0035]
在本发明的一个或多个实施例中,式3所示化合物的制备过程中,所述纯化包括如下步骤:将反应液冷却至15~35℃,过滤收集滤液,除去溶剂利用硅胶柱层析以石油醚和二氯甲烷的混合溶剂为淋洗剂分离,干燥;
[0036]
优选地,所述混合溶剂中,石油醚和二氯甲烷的体积比为(3~6):1。
[0037]
在本发明的第三方面,本发明提供一种薄膜,包括本发明第一方面所述的基于蒽衍生物的激基缔合物发光材料,优选地,所述薄膜为将所述基于蒽衍生物的激基缔合物发光材料掺杂在1wt%的聚甲基丙烯酸甲酯高分子中制备得到。
[0038]
在本发明的第四方面,本发明提供一种本发明第一方面所述的基于蒽衍生物的激基缔合物发光材料或本发明第三方面所述的薄膜在制备有机发光二极管中的应用。
[0039]
在本发明的第五方面,本发明提供一种本发明第一方面所述的基于蒽衍生物的激基缔合物发光材料在制备防伪标识中的应用;优选地,所述防伪标识为防伪膜。
[0040]
相比于现有技术,本发明的有益效果在于:
[0041]
1、本发明提供一种基于蒽衍生物的激基缔合物发光材料相比于常见的激基缔合物材料,该化合物对浓度没有依赖性,在稀溶液中就能实现激基缔合物发光。此外,该基于蒽衍生物的激基缔合物发光材料发光性质稳定且热稳定性较好,在空气中无需任何特殊范围保护即可稳定存放。
[0042]
2、本发明还提供上述基于蒽衍生物的激基缔合物发光材料的制备方法,其通过简单的反应,反应条件较温和,反应产率较高,采用了一种刚性又合适的底座单元9,9
‑
二甲基氧杂蒽,将两个具有深蓝色发光的蒽单元固定住,使其呈现二聚体的模式,得到的化合物实现了红移的480nm左右的激基缔合物绿光发射,其量子产率接近100%,寿命可达48ns。
[0043]
3、本发明还提供上述基于蒽衍生物的激基缔合物发光材料的应用,该基于蒽衍生物的激基缔合物发光材料可以用于防伪标识的制备。
[0044]
4、本发明还提供上述基于蒽衍生物的激基缔合物发光材料的另一应用,该材料在薄膜状态下也能实现激基缔合物发光,可作为有机发光二极管器件中的发光材料应用。
附图说明
[0045]
图1为本发明的基于蒽衍生物的激基缔合物发光材料的合成路线图;
[0046]
图2为本发明制备的基于蒽衍生物的激基缔合物发光材料与单体蒽对比的溶液态荧光发射光谱图;其中,图2左为蒽单体溶液的荧光发射光谱图,图2右为式x2a所示化合物溶液的荧光发射光谱图;
[0047]
图3为本发明制备的基于蒽衍生物的激基缔合物发光材料单晶的荧光光谱图和荧光寿命图;其中,图3左为在固态下测试式x2a所示化合物单晶的荧光光谱图,图3右为在固态下测试式x2a所示化合物单晶的荧光寿命图;
[0048]
图4为本发明制备的基于蒽衍生物的激基缔合物发光材料的10
‑5mol/l四氢呋喃(thf)溶液和1wt%掺杂的聚甲基丙烯酸甲酯(pmma)薄膜在365nm紫外光照射下的绿色荧光图。
[0049]
图5为本发明制备的基于蒽衍生物的激基缔合物发光材料的防伪图案。
具体实施方式
[0050]
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。以下实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行,使用的方法如无特别说明,均为本领域公知的常规方法,使用的耗材和试剂如无特别说明,均为市场购得。除非另有说明,本文中所用的专业与科学术语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法或材料也可应用于本发明中。
[0051]
实施例1:基于蒽衍生物的激基缔合物发光材料化合物x2a的合成
[0052]
合成路线如下:
[0053][0054]
步骤1):式1所示化合物的合成
[0055]
合成路线如下所示:
[0056][0057]
具体步骤如下所示:在冰浴下,将9,9
‑
二甲基氧杂蒽(3.00g,14.30mmol)和无水氯化铁(0.12g,0.72mmol)溶于15ml二氯甲烷中,再将氯代叔丁烷(3.68ml,35.75mmol)用15ml二氯甲烷稀释于恒压滴液漏斗中,冰浴下滴加至上述9,9
‑
二甲基氧杂蒽溶液中,滴加完毕后反应液15~35摄氏度下搅拌15小时,待反应完成后,向反应液中加水淬灭反应,用二氯甲烷萃取,收集有机相并用无水硫酸钠干燥,得到粗产品用硅胶柱层析以石油醚为淋洗剂分离纯化,用真空干燥箱干燥后得到白色固体(3.20g,产率70%),并用1h nmr对其结构进行表征,证实该化合物为化合物1。1h nmr(400mhz,cdcl3,δ):7.40(d,j=2.2hz,2h,arh),7.21(d,j=2.2hz,1h,arh),7.20(d,j=2.2hz,1h,arh),6.96(s,1h,arh),6.94(s,1h,arh),
1.65(s,6h,
‑
ch3),1.33(s,18h,
‑
ch3)。
[0058]
步骤2):化合物2的合成:
[0059]
合成路线如下所示:
[0060][0061]
具体步骤如下所示:冰浴下,将式1所示化合物(1.00g,3.10mmol)溶于15ml三氯甲烷中,将液溴(0.34ml,6.50mmol)稀释于三氯甲烷中,采用恒压滴液漏斗滴加至上述式1所示化合物溶液中,滴加完毕后搅拌反应2小时,再次重复上述滴加过程,将经三氯甲烷稀释后的液溴(0.34ml,6.50mmol)滴加入反应液中,滴加完毕后,在15~35摄氏度下搅拌反应12小时,向反应液中加亚硫酸钠水溶液淬灭反应,用二氯甲烷萃取,收集有机相并用无水硫酸钠干燥,得到粗产品用硅胶柱层析以石油醚为淋洗剂分离纯化,用真空干燥箱干燥后得到白色固体(1.40g,收率95%),并用1h nmr对其结构进行表征,证实该化合物为化合物2。1h nmr(400mhz,cdcl3,δ):7.47(d,j=2.2hz,2h,arh),7.33(d,j=2.2hz,2h,arh),1.62(s,6h,
‑
ch3),1.32(s,18h,
‑
ch3)。
[0062]
步骤3):化合物3的合成:
[0063]
合成路线如下所示:
[0064][0065]
具体步骤如下所示:在氮气的氛围下,将9
‑
溴蒽(3.00g,11.67mmol),联硼酸频那醇酯(4.44g,17.52mmol),乙酸钾(2.28g,23.34mmol),[1,1'
‑
双(二苯基膦基)二茂铁]二氯化钯(0.6g,0.81mmol)加入反应瓶中,加入提前除氧的1,4
‑
二氧六环35ml,回流反应12小时。对反应液进行抽滤收集滤液,得到粗产品用硅胶柱层析以石油醚和二氯甲烷体积比为(4:1)的混合溶剂作为淋洗剂分离纯化,用真空干燥箱干燥后得到白色固体(2.49g,70%),并用1h nmr对其结构进行表征,证实该化合物为式3所示化合物。1h nmr(400mhz,cdcl3,δ):8.48
‑
8.43(m,3h,arh),7.99(d,j=8.2hz,2h,arh),7.50
‑
7.42(m,4h,arh),1.58(s,12h,
‑
ch3)。
[0066]
步骤4):化合物x2a的合成:
[0067]
合成路线如下所示:
[0068][0069]
具体步骤如下所示:在氮气的氛围下,将式2化合物所示(1.00g,2.08mmol),式3化合物所示化合物(1.90g,6.24mmol),无水碳酸钾(1.15g,8.32mmol),1,1'
‑
双(二苯基膦基)二茂铁]二氯化钯(0.08g,0.11mmol)加入反应瓶中,加入提纯除氧的四氢呋喃25ml和5ml去离子水,回流反应12小时。加入饱和氯化钠水溶液淬灭反应,用二氯甲烷萃取收集有机层,用无水硫酸钠干燥后得到粗产品,用硅胶柱层析以石油醚和二氯甲烷混合溶剂(石油醚和二氯甲烷体积比为6:1)作为淋洗剂分离纯化,用真空干燥箱干燥后得到浅黄色固体(1.12g,收率60%),并用1h nmr、
13
c nmr、ms和ea对其结构进行表征,证实该化合物为x2a。1h nmr(400mhz,cdcl3,δ):7.90(s,2h,arh),7.59(d,j=8.4hz,4h,arh),7.56(d,j=2.2hz,2h,arh),7.04
‑
6.97(m,10h,arh),6.74(t,j=8.0hz,4h,arh),1.90(s,6h,
‑
ch3),1.30(s,18h,
‑
ch3);
13
c nmr(100mhz,cdcl3)δ(ppm):132.11,130.19,129.23,129.01,127.86,125.87,125.36,123.95,123.88,123.44,113.57,100.27,99.99,99.86,35.18,34.57,32.62,31.62.ms(ei,m/z):[m]
+
calcd for:c
51
h
46
o,674.35;found,674.18.anal.cacld for c
51
h
46
o:c,90.76;h,6.87.found:c,90.67;h,7.08。
[0070]
步骤5):式x2a所示化合物单晶的制备:取15mg式x2a所示化合物溶于3ml二氯甲烷溶剂,再滴加入少量的石油醚溶剂,常温下静置培养单晶,待溶剂挥发干后得到块状的绿色发光晶体。
[0071]
实施例2:基于蒽衍生物的激基缔合物发光材料化合物x2a的性能测试
[0072]
将实施例1得到的式x2a所示化合物与蒽分别溶于适量的二氯甲烷中分别制备成1
×
10
‑5mol/l溶液,测试其荧光发射光谱图,结果如图2所示,其中,图2左为蒽单体溶液的荧光发射光谱图,图2右为式x2a所示化合物溶液的荧光发射光谱图。在固态下测试式x2a所示化合物单晶的荧光光谱和荧光寿命,结果如图3所示,其中,图3左为在固态下测试式x2a所示化合物单晶的荧光光谱图,图3右为在固态下测试式x2a所示化合物单晶的荧光寿命图。式x2a所示化合物配制成浓度为10
‑5mol/l的四氢呋喃(thf)溶液,用365nm紫外光照射,结果如图4左所示,由结果可知,其具有绿色荧光,将式x2a所示化合物单晶以1wt%的比例掺杂在聚甲基丙烯酸甲酯(pmma)中制备成薄膜,用365nm紫外光照射,其依然表现出明亮的绿色荧光,结果如图4右所示,x2a所示化合物这种高量子产率、寿命较长的激基缔合物发光有望进一步应用于有机发光二极管领域中。
[0073]
实施例3:基于蒽衍生物的激基缔合物发光材料在防伪领域的应用
[0074]
将式x2a所示化合物的白色粉末在白色滤纸上平铺成武汉大学的缩写“whu”字样,用365nm紫外灯照射下滤纸上出现明亮的绿色“whu”字样。结果如图5所示,结果可以说明,该化合物可用于制备防伪标识,用于信息存储、信息传递等领域。
[0075]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述
实施例进行变化、修改、替换和变型,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1