一种烟草NtMAB1基因沉默植株的制备方法及该植株的应用
一种烟草ntmab1基因沉默植株的制备方法及该植株的应用
技术领域
1.本发明涉及植物生物技术领域,尤其涉及一种烟草ntmab1基因沉默植株的制备方法及该植株的应用。
背景技术:
2.分枝是植株形态建成的重要构成要素,也是影响作物产量的决定因素之一。烟草是一种以收获叶片为主的经济作物,分枝增多会导致叶片的营养物质减少,进而影响烟叶的产量和质量,因此减少烟草分枝对于烟草生产具有十分重要的意义。在烟草生育期中的现蕾开花期,通常利用打顶措施终止生殖发育过程,以使养分向叶片集中供给;但是打顶后植株顶端优势丧失,烟草腋芽会快速生长成为侧枝,这个过程会消耗大量的营养物质,造成烟叶片营养物质外流并减少,导致烟叶小而薄,油分减少,香气降低,产量和质量严重下降。针对这个问题,目前生产中常规方法是采取人工抹杈或者使用抑芽剂的方法来控制烟草的腋芽生长,但这些方法存在着生产成本升高和环境污染、农药残留等问题。
3.在目前分枝发育研究领域中,各种因子的互作以及复杂的激素调控网络使分枝发育机理的解析存在一定困难,但也为分枝发育的调控提供了更多潜在可行的实际运用手段。rav家族成员在花发育和开花控制方面的功能得到了较深入的解析,但是其参与植物分枝发育过程的分子机制研究甚少。
4.因此,鉴定和分离出烟草腋芽发育相关基因,通过基因工程的方法构建烟草腋芽发育相关基因的基因沉默植株,可减少或者避免人工抹杈或使用抑芽剂,为烟叶生产的减工增效、降低农药残留、提升烟叶质量等方面提供新的方法和研究方向。
技术实现要素:
5.针对目前烟草品种生产中使用人工抹杈或抑芽剂方法控制腋芽,具有高成本、不环保和农残超标等问题,申请人分离并确定了一个新的调控腋芽发育的基因ntmab1,并通过基因沉默的方法构建了一种烟草ntmab1基因沉默植株,提供了该植株的具体制备方法及其应用。这种基因沉默植株在打顶后,不需要人工抹杈或使用抑芽剂也能达到控制腋芽生长的效果,为烟叶生产的减工增效、降低农药残留、提升烟叶质量提供新的方法和研究方向。
6.本发明的技术方案具体如下:
7.本发明利用烟草t-dna激活标签插入突变体库,在突变群体中发现了一个多分枝、矮化的突变植株,对其命名为mab1(more axillary branches 1),并对这个突变株系进行分子生物学研究。通过tail-pcr、基因型与表型的共分离分析、qrt-pcr分析确定了候选基因,并对该基因进行基因克隆和蛋白结构分析,确认该基因是rav家族的一个转录因子,通过ntmab1基因功能研究,确认ntmab1基因是烟草腋芽生长发育的一个正调控因子,并对该基因进行具体研究,通过基因工程的方法,构建了一株烟草ntmab1基因沉默植株。
8.上述基因为序列为seq id no:1和seq id no:2的核苷酸序列(ntmab1-s1和
ntmab1-t1)或者其互补序列。其中,本技术的序列ntmab1-s1表达的蛋白序列如seq id n0:7。
9.第一方面,本发明提供上述烟草ntmab1基因沉默植株的制备方法,该方法包括以下步骤:
10.s1、以具有ntmab1-s1基因外显子基因的序列为模板,设计引物通过pcr扩增得到如seq id no:8所示的m1ri1片段;对m1ri1片段和pucc-rnai载体分别进行双酶切、片段纯化后进行连接,并将连接产物转化大肠杆菌感受态,挑取阳性克隆,对阳性克隆进行pcr检测并将扩增产物进行测序,从测序正确的阳性克隆中提取得到pucc-ri1中间载体;
11.s2、对pucc-ri1载体进行双酶切和片段纯化,将纯化后pucc-ri1中间载体片段与双酶切纯化后的m1ri1片段进行连接,得到pucc-m1ri1沉默载体,并对阳性克隆进行pcr鉴定;
12.s3、以具有ntmab1-s1基因外显子基因的序列(如基因组)为模板,设计引物,通过pcr扩增得到如seq id no:11所示的m1ri2片段;其他步骤同s1和s2,构建得到pucc-ri2载体和pucc-m1ri2沉默载体,转化感受态细胞后,对阳性克隆进行pcr鉴定;
13.s4、对前述步骤制备得到的pucc-m1ri1、pucc-m1ri2和pcambia1300-35s载体分别进行单酶切和纯化;将酶切纯化后的pucc-m1ri1和pucc-m1ri2载体片段分别与单酶切后的pcambia1300-35s载体混合并连接,将连接产物转化大肠杆菌感受态,于抗性培养基上过夜培养,对阳性克隆进行pcr鉴定;阳性克隆携带的载体为ntmab1基因的双元沉默载体;
14.s5、将前一步构建成功的ntmab1基因的两个双元沉默载体,pcambia1300-35s-m1ri1和pcambia1300-35s-m1ri2转化农杆菌,通过叶盘法侵染,获得烟草ntmab1基因沉默植株。
15.优选地,s1步骤中,利用如seq id no:9和seq id no:10所示的m1ri-f1和m1ri-r1引物对扩增得到m1ri1片段;s3步骤中,利用seq id no:12和seq id no:13所示的m1ri-f2和m1ri-r2引物对扩增得到m1ri2片段。
16.进一步优选地,上述制备方法具体包括以下步骤:
17.s1、利用seq id no:9和seq id no:10的m1ri-f1和m1ri-r1引物对,通过pcr扩增得到m1ri1片段;利用内切酶bamhi和sali对m1ri1片段和pucc-rnai载体分别进行双酶切和片段纯化;将纯化后的两个片段等比例混合,连接酶连接后转化大肠杆菌感受态,于amp抗性lb培养基上过夜培养,通过seq id no:10和seq id no:14的m1ri-r1和rintron-r1引物对鉴定阳性克隆送测序,得到pucc-ri1中间载体。
18.s2、利用内切酶xhoi和bglii对pucc-ri1载体进行双酶切和片段纯化。将纯化后pucc-ri1中间载体片段与bamhi和sali双酶切纯化后的m1ri1片段进行连接,得到pucc-m1ri1沉默载体。通过m1ri-f1和m1ri-r1的无条带引物对、m1ri-r1和rintron-r1有条带引物对进行鉴定。
19.s3、利用seq id no:12和seq id no:13的m1ri-f2和m1ri-r2引物对,扩增得到m1ri2片段。其他步骤同s1和s2,构建得到pucc-ri2载体和pucc-m1ri2沉默载体。通过m1ri-f2和m1ri-r2的无条带引物对、m1ri-r2和rintron-r1的有条带引物对进行鉴定。
20.s4、利用内切酶psti或者sbfi对pucc-m1ri1、pucc-m1ri2和pcambia1300-35s载体分别进行单酶切和纯化。将酶切纯化后的pucc-m1ri1和pucc-m1ri2载体片段分别与单酶切
后的pcambia1300-35s载体按比例混合,连接酶连接后转化大肠杆菌感受态,于kana抗性lb培养基上过夜培养,分别通过m1ri-r1、m1ri-r2和rintron-r1两对引物进行鉴定。
21.s5、将构建成功的ntmab1基因的两个双元沉默载体,pcambia1300-35s-m1ri1和pcambia1300-35s-m1ri2转化农杆菌,通过叶盘法侵染,获得转基因植株。
22.第二方面,本发明还提供一种烟草ntmab1基因沉默重组载体,为前述制备方法制备得到的pcambia1300-35s-m1ri1和pcambia1300-35s-m1ri2双元沉默载体。
23.第三方面,本发明还提供一种重组宿主细胞,其转化有上述的烟草ntmab1基因沉默重组载体pcambia1300-35s-m1ri1和/或pcambia1300-35s-m1ri2双元沉默载体。所述宿主细胞可以选自植物细胞或者微生物细胞,优选为植物细胞,最优选为烟草细胞。所述细胞可以是分离的、离体的、培养的、或者是植物的一部分。优选地,所述细胞为大肠杆菌或农杆菌细胞。
24.第四方面,本发明还提供上述烟草ntmab1基因的沉默植株在培育腋芽生长受抑制的高效烟草品种中的应用。
25.第五方面,本发明还提供用于鉴定如权利要求1所述的烟草ntmab1基因沉默植株的引物,所述引物为如seq id no:10和seq id no:14所示的m1ri-r1和rintron-r1,或者如seq id no:13和seq id no:14所示的m1ri-r2和rintron-r1。
26.第六方面,本发明还提供一种上述烟草ntmab1基因的沉默植株的鉴定方法,该方法为:以提取的待鉴定烟草的基因组为模板,以如seq id no:10和seq id no:14所示的引物对m1ri-r1和rintron-r1或如seq id no:13和seq id no:14所示的引物对m1ri-r2和rintron-r1分别对两个沉默载体进行pcr扩增,对扩增产物进行测序,并将该测序结果与ntmab1基因的序列比对,即可得到鉴定结果。
27.本发明的技术方案具有以下有益效果:
28.1、本发明通过筛选到的一株多分枝、晚花的突变体植株,并进一步在烟草中确定了一个新的调控腋芽发育的基因ntmab1,在克隆并验证了基因ntmab1的功能后,进一步通过基因工程的方法,制备了ntmab1的基因沉默植株。该植株的形状稳定,相对于野生型,这株植株在打顶前各叶位处腋芽及分枝生长趋势较弱,打顶后各叶位的腋芽的生长也受到明显抑制,在实际应用中可减少或避免使用抑芽剂和进行人工抹杈,达到烟叶生产减工增效、降低农药残留等效果,具有重要的理论参考价值和应用价值。
29.2、本发明还提供了上述基因沉默植株的制备方法,并进一步提供了该植株的鉴定方法及采用的鉴定引物,便于在实际应用中对烟草ntmab1基因沉默植株进行快速、准确的鉴别,从而判断是否需要对目标植株进行喷涂抑芽剂或进行人工抹杈。
附图说明
30.图1为mab1突变体的各生育期表型特征及腋芽性状定量分析;a:开花期分枝的表型特征(去除叶片):hd-fp,处于开花期(flowering period)的野生型红花大金元(hd);mab1,与hd-fp同期的突变体;mab1-fp,处于开花期的mab1突变体,温室条件下开花期晚于野生型20天;b和c分别为团棵期的野生型和突变体腋生分枝表型;d:团棵期野生型和突变体各对应叶位的分枝长度比较;e和f分别为旺长期的野生型和突变体腋生分枝表型;g:旺长期野生型和突变体各对应叶位的分枝长度比较;
31.图2为目标插入位点及候选基因的确定;a:t-dna激活标签插入位点及可能激活的插入位点上下游基因,u(upstream)和d(downstream)分别是插入位点上游(与35s增强子序列方向相反)和下游(与35s增强子序列方向一致);b-e:候选基因在hd和mab1突变体不同组织中的表达情况,1s,成苗期茎样品;1l,成苗期叶样品;3ab1u,旺长期打顶前上部3个腋芽混样;3ab1d,旺长期打顶前下部3个腋芽混样;
32.图3为f1代杂合株系f1001的表型特征及ntmab1基因在f1001植株中的表达情况分析;a:f1001的表型特征;b:f1001中腋芽性状定量分析;c,d:ntmab1和ntbrc1基因在f1001中上下部腋芽的表达情况分析;
33.图4为ntmab1基因在绒毛状烟草和林烟草基因组dna和cdna中的pcr扩增;t:绒毛状烟草;s:林烟草;m:marker;
34.图5为ntmab1-s1/t1蛋白和rav家族成员蛋白结构分析;黑线标注为ap2功能域;红线标注的是9个成员共有的b3功能域;蓝线线标注的是在烟草和拟南芥之间高度保守的b3抑制功能域;
35.图6为ntmab1超表达植株表型特征分析;a,b:ntmab1超表达植株照片;c:ntmab1超表达植株腋芽性状定量分析;d:ntmab1超表达植株叶片照片;e,f:ntmab1基因在超表达植株上下部腋芽中的表达水平。oe:over expression,超表达;u:上部腋芽或分枝;d:下部腋芽或分枝;
36.图7为超表达株系中ntbrc1基因的表达分析;ab-u:上部腋芽;ab-d:下部腋芽;
37.图8为mab1突变体背景的rnai转基因植株表型特征分析;a-b:mkd腋芽表型特征;c:mkd腋芽或分枝长度统计;d-f:ntmab1基因在mkd及对照株系中的表达水平检测,1u为打顶前烟株上部腋芽;1d为打顶前烟株下部腋芽;l为叶片;
38.图9为hd背景的rnai沉默植株筛选;a:rnai转基因阳性植株pcr鉴定;b:rnai转基因阳性植株中ntmab1表达水平检测;
39.图10为野生型背景的rnai沉默植株表型特征分析;a:kd植株腋芽表型特征;b:kd植株腋芽长度统计;c-e:ntmab1基因在mkd及对照株系中的表达水平,1u为打顶前烟株上部腋芽;1d为打顶前烟株下部腋芽;l为叶片;
40.图11为旺长期不同类型烟株打顶前和打顶后腋芽长度定量分析;a,b:打顶前不同类型烟草植株各叶位处腋芽或分枝长度统计;c,d:打顶后3天不同类型烟草植株各叶位处腋芽长度统计;e,f:打顶后7天不同类型烟草植株各叶位处腋芽长度统计;
41.图12为打顶后hd、mab1及各转基因株系腋芽生长长度定量分析;
42.图13为ntmab1基因在打顶后3天烟草中表达水平分析;2u:烟株打顶后上部腋芽或分枝;2d:打顶后下部腋芽或分枝。
具体实施方式
43.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。在本发明中,若非特指,所采用的设备和原料等均可从市场购得或是本领域常用的。下述实施例中的方法,如无特别说明,均为本领域的常规方法。
44.实施例一烟草分枝突变体mab1的筛选
45.1、植物材料:
46.选用供体亲本红花大金元(简称红大,hd)和t-dna激活标签t1代突变体株系mab1作为试验材料,野生型材料hd、t-dna激活标签插入突变体mab1种子由国家农作物种质资源平台烟草种质资源子平台和中国农业科学院烟草研究所烟草突变体库提供。该突变体库是通过t-dna激活标签载体pski015插入方式所构建。其他实验材料均以hd为背景通过载体构建、烟草遗传转化获得。
47.表1 实验所用菌株及载体
[0048][0049]
2、仪器与设备:花盆,育苗基质,佳能5ds照相机,直尺
[0050]
3、试验方法
[0051]
观察红花大金元和t1代mab1突变体在整个生育期内的腋芽表型,利用佳能5ds照相机对植株生长情况进行拍照。在团棵期和旺长期,统计mab1纯合突变体植株5个株系,用直尺测量并记录红花大金元和mab1突变体的腋芽或分枝长度。
[0052]
4、结果与分析
[0053]
实验室在t1代激活标签突变体库中筛选得到一个多分枝晚花的突变体株系,在团棵期,整个植株各叶位的腋生分枝显著可见,并且突变体各叶位分枝长度显著高于野生型(图1b,c,d),我们将该功能获得型突变体命名为mab1。
[0054]
在旺长期,mab1突变体植株各叶位的腋生分枝仍然显著可见(图1e,f),该突变体各叶位分枝长度仍显著高于野生型(图1g)。突变体腋芽表现为持续增长,在后期均生长成为分枝;并且表现为晚花,温室条件下该突变体的开花期比野生型晚20天(图1a),且开花进度缓慢,花朵小且少;最上部2-3个分枝会出现花序,但均比主茎花序小;在植株高度上,团棵期和旺长期该突变体略矮于野生型hd,但是进入生殖生长时期,突变体的株高与hd一致,甚至高于hd(图1a)。
[0055]
实施例二目标插入位点和候选基因ntmab1的确定
[0056]
试剂及试剂盒:
[0057]
cwbio植物基因组dna提取试剂盒、植物rna提取试剂盒、反转录试剂盒、rtaq酶、kod酶、takara荧光定量反应聚合酶试剂盒。
[0058]
1、实验方法
[0059]
(1)侧翼序列扩增
[0060]
本试验使用tail-pcr(thermal asymmetric interlaced pcr)的方法对t-dna激活标签两侧的片段进行扩增。以t1代阳性植株dna作为模板,根据pski015特异性嵌套引物sp1、sp2、sp3,及随机引物ad,进行三次pcr扩增,每次反应体系如下:
[0061]
设定pcr反应程序:
[0062][0063]
将经过三步pcr反应扩增所获得的产物,进行电泳试验,因为小于500bp的片段多为载体序列,很难分析出有效的侧翼序列信息,所以将条带大于500bp的产物进行切胶回收并送测序,将测序结果在烟草基因组数据库中进行blast比对分析,确定t-dna插入位置。
[0064]
(2)共分离分析
[0065]
根据插入位置上游和下游基因组1000bp序列设计一对基因组特异性引物gf/gr,根据t-dna插入序列设计一个特异性引物tp并与gr组成一对引物(表2),分别进行两次pcr反应,判断植株是纯合突变还是杂合突变以及证明插入位置的准确性。pcr反应体系与程序参照常规pcr反应。
[0066]
表2 共分离实验引物
[0067][0068]
(3)候选基因的确定
[0069]
通过侧翼序列分析和共分离确定目标插入位置,利用gbrows工具对其插入位点上下游100kb内的基因进行检索,获取插入位点旁侧候选基因。
[0070]
(4)候选基因的表达
[0071]
rna提取:在温室以相同的栽培方法与管理条件种植纯合突变体与红花大金元,在旺长期分别取突变体与红花大金元的新鲜叶片和腋芽,并用锡箔纸包好迅速置于液氮中保存。使用takara植物rna小量提取试剂盒提取总rna,具体步骤依据说明书进行。
[0072]
cdna链的合成:本试验采用takara primescript
tm ii 1st strand cdna synthesis kit反转录试剂盒对提取的rna进行反转录,体系步骤参照说明书。反转录后的cdna保存至-20℃冰箱。
[0073]
荧光定量pcr确定目的基因:参照实时荧光定量pcr引物设计原则,根据候选基因
序列设计引物(表3)。将cdna浓度稀释4倍后作为qrt-pcr(real-time quantitative reverse transcription-pcr)反应模板,以烟草管家基因ubiquitin-conjugating enzyme e2(ntubc2)作为内参,使用takara公司的荧光定量试剂盒(sybr premix ex taq tm)进行qrt-pcr扩增,每个反应设计三个技术重复,体系和反应程序完全参照说明书。利用abi 7500荧光定量分析软件分析试验数据。
[0074]
表3 荧光定量pcr引物
[0075][0076]
(5)纯合mab1突变体与hd的杂交一代的分析
[0077]
2、实验结果及分析
[0078]
(1)利用tail-pcr的方法扩增突变体dna得到2条侧翼序列,说明该突变体至少含有2个插入位点。根据插入位点两侧的基因序列和激活标签载体序列分别设计1对基因组特异性引物和1条载体特异性引物,3条引物组成2对引物组合,对突变体分离世代进行基因分型,获得了分离株系中每个单株的基因型,而后和单株表型对应。如果突变纯合和杂合基因型均对应突变表型,野生型基因型对应野生型表型,则该插入位点就是目标插入位点,即基因型与表型共分离。基因型和表型的共分离分析显示,只有插入位点1(c110)符合基因型与表型的共分离规律,因此c110就是目标插入位点。
[0079]
(2)由于35s增强子的作用没有方向性,获得目标插入位点之后,我们对目标插入位点上下游100kb内存在的基因进行分析(图2a),如图2所示,插入位点上下游100kb内共有6个基因,其中上游4个,命名为u1-u4;下游2个,命名为d1-d2,此范围内的6个基因都有可能被激活表达。
[0080]
以旺长期hd和突变体mab1植株上下部腋芽,成苗期hd和mab1的茎和叶为样品,针对以上6个旁侧基因进行qrt-pcr分析(图2.2b-e),结果表明:基因d1在成苗期突变体mab1的茎和叶中的表达水平较hd分别升高45和32倍;基因d1在旺长期上部腋芽ab1u和下部腋芽ab1d中的表达水平分别较hd上下部腋芽中该基因的表达水平高32和116倍,基因d1在下部
腋芽中的表达水平比在上部腋芽中高3-4倍,而其他5个基因的表达量较hd没有显著性差异,说明d1基因为本研究的候选基因,将其命名为ntmab1。
[0081]
(3)为了进一步确认mab1突变体的表型为ntmab1基因激活所致,我们将纯合mab1突变体与hd进行杂交,发现:f1代(命名为f1001)腋生分枝的表型介于野生型与纯合mab1突变体之间,而且f1001植株上下部腋芽中ntmab1基因表达量也介于hd和纯合mab1之间(图3a,b)。ntbrc1在很多物种中是一个维持腋芽休眠的基因,ntbrc1表达量越高,对腋芽的活性抑制作用越强,腋芽生长也就越慢。对烟草中维持腋芽休眠的ntbrc1基因进行表达量检测,结果发现,ntbrc1基因在f1001植株上下部腋芽中的表达均在hd和mab1之间,且mab1中ntbrc1基因的表达水平最低,ntbrc1与ntmab1在三类植株中的表达情况呈相反的趋势(图3c,d)。
[0082]
实施例三ntmab1基因的克隆及生物学信息学分析
[0083]
1、实验方法
[0084]
(1)基因目的片段扩增纯化
[0085]
普通烟草为异源四倍体,两个祖先种分别为林烟草(n.sylvestris)和绒毛状烟草(n.tomentosiformis)。同理,ntmab1基因有两个分别来自林烟草和绒毛状烟草的拷贝,根据特异性分别设计一对引物(表4),从普通烟草基因组dna和cdna中分别扩增ntmab1-s1和ntmab1-t1的基因组dna(gdna)和cdna序列,使用takara dna片段纯化试剂盒纯化pcr反应产物。
[0086]
表4 ntmab1基因扩增引物
[0087][0088]
(2)ta克隆与大肠杆菌的转化
[0089]
将纯化后的dna片段进行末端加“a”反应后,与pmd
tm
19t载体按照3:1的比例加入离心管并混合均匀,加入等体积的solutioni混合均匀,16℃连接1-2h。将连接好的载体转化进入大肠杆菌中,待lb培养基上长出大肠杆菌,在1.5ml离心管中加入1ml液体lb培养基和1μl氨苄青霉素,在超净台中挑取单菌落,置于此离心管中,依次编号,放在37℃摇床震荡5-6h,做菌液pcr检测目的片段。pcr体系和程序参照rtaq酶反应体系。选取阳性菌株送华大基因测序。
[0090]
2、实验结果
[0091]
如图4所示,ntmab1-s1的gdna序列全长为2294bp,ntmab1-t1的gdna片段长度为2368bp,同时在普通烟草cdna中扩增得到ntmab1-s1和ntmab1-t1的cdna序列,全长分别为1002bp和1187bp。对基因序列分析时发现,绒毛状烟草的基因拷贝出现了终止密码子,该基因拷贝是假基因。
[0092]
将ntmab1-s1基因在ncbi中进行blast,发现:该基因与拟南芥中b3超家族rav亚家族中ngal1基因相似性较高,属于rav家族。
[0093]
rav家族成员除了包含b3结构域,还可能包含一个ap2结构域。将ntmab1-s1与拟南芥中几个已经有过报道的rav蛋白进行序列比对,发现ntmab1-s1不包含ap2功能域和核定位信号nls;但是rav家族成员在b3功能域是高度保守的;烟草和拟南芥之间含有高度保守的转录抑制功能域(brd,b3 repression domain),其关键抑制区序列为“r/klfgv”(如图5所示),表明ntmab1基因很可能作为转录抑制子起作用。
[0094]
实施例四烟草ntmab1基因过表达载体构建及遗传转化
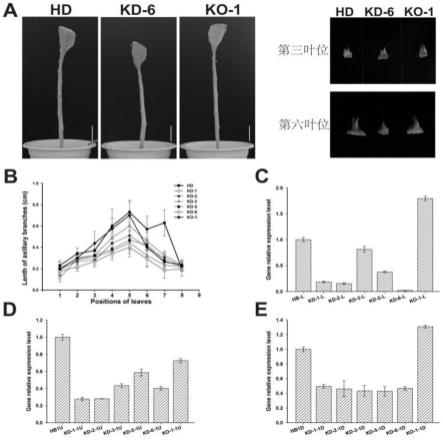
[0095]
1、实验方法
[0096]
(1)过表达载体构建
[0097]
将测序结果正确的菌株放在15ml的无菌离心管扩繁,利用天根公司高纯度质粒提取试剂盒进行质粒提取。将提取出的质粒进行双酶切。综合考虑pmd
tm
19t酶切位点以及过表达载体pcambia1300-35s上的酶切位点,确定酶切位点为sal i、bamh i,分别对连有目的基因的pmd
tm
19t载体和过表达载体pcambia1300-35s进行双酶切,反应体系参照nebcutsmart反应体系。酶切产物进行电泳分析,回收正确条带,并测得其浓度。
[0098]
将获得的含有目的基因的双酶切dna片段及过表达载体pcambia1300-35s双酶切dna片段进行酶连反应。反应结束后将反应产物转化进大肠杆菌感受态将转化后的大肠杆菌进行阳性检测和测序,确定转化是否成功。
[0099]
(2)普通烟草无菌苗的培养
[0100]
农杆菌介导的烟草叶盘遗传转化需要无菌的烟草叶片,因此在转化之前需要培养烟草无菌苗。在超净工作台中,对红花大金元的种子进行灭菌处理,在ms培养基上25℃恒温培养40天左右,选择叶色浓绿、生长健壮的无菌苗待用。
[0101]
(3)农杆菌介导的烟草叶盘转化
[0102]
利用叶盘转化法使重组载体转化烟草,在分化培养基中应加入潮霉素作为抗性筛选。操作步骤完全参照现有的烟草叶盘转化法进行。
[0103]
(4)烟草转基因苗的阳性鉴定及表型分析
[0104]
使用剪过的枪头作为打孔器取直径5-7mm叶片组织到100μl离心管中。向离心管中加入50μlbufferp1。盖好离心管盖,将其置于pcr仪中,95℃裂解10min。加入50μlbufferp2,用移液器吸打混匀。所得裂解混合液可直接作为模板进行pcr反应。根据载体片段与连入的基因片段设计阳性鉴定引物jc-f、jc-r(表5)。
[0105]
表5 转基因苗阳性鉴定引物
[0106][0107]
pcr反应体系:
[0108][0109]
pcr反应程序同常规pcr程序。
[0110]
2、实验结果及分析
[0111]
(1)与野生型hd相比,35spro:ntmab1超表达株系表现为腋生分枝多且长、叶型狭窄、植株矮及晚花等表型,其中oe-1类型的超表达植株与激活标签插入突变体mab1表型非常一致。为了定量分析ntmab1过表达植株的腋芽表型特征,对hd和35spro:ntmab1的各叶位叶腋处的腋芽或者分枝长度进行测量,结果表明,35spro:ntmab1超表达oe-1和mab1在分枝上的表型非常相似,分枝数目多且腋生分枝长,而对照组hd各叶位处的腋芽较短,腋芽处于休眠状态(图6a-c)。对不同类型烟草植株株高、叶宽进行观察,发现ntmab1过表达植株oe-1与mab1激活标签突变体表型类似,较对照hd,植株矮、叶型狭窄。而在35spro:ntmab1超表达株系中发现一些类似oe-2类型植株的表型,植株表现为极度矮化,每个叶位处腋生分枝较oe-1更长,叶片更窄,而且在后续生长过程中一直处于矮化多分枝状态(图6c)。
[0112]
(2)在转录水平上对hd和两个35spro:ntmab1超表达株系的上下部腋生分枝(或腋芽)的ntmab1的表达量进行检测,发现在oe-1和oe-2两个超表达株系上部腋芽中,ntmab1的表达量较对照hd升高120倍,在oe-1和oe-2的下部腋芽中,ntmab1的表达量较对照hd分别升高200和120倍(图6e,f),同时也发现在类型于mab1激活标签突变体表型的oe-1超表达株系中,ntmab1在下部腋芽中的表达水平是上部腋芽中表达水平的两倍,这与mab1上下部腋芽中ntmab1的表达趋势是一致的,而在oe-2株系上下部腋芽中ntmab1基因的表达水平接近,没有出现出下部腋芽中ntmab1基因表达水平较上部腋芽高的情况(图6e,f)。
[0113]
brc1是维持腋芽休眠的关键基因。对hd和超表达株系表型调查过程中发现,超表达株系中腋芽的活性很强,在植株成苗期就开始出现腋芽,而且在烟株生长过程中腋芽也快速生长,这可能是由于brc1基因表达受到抑制,其维持休眠的作用减弱导致。因此,对上述三种株系腋芽中brc1基因的表达量进行检测,发现在ntmab1超表达烟草植株中,ntbrc1基因的表达量较低于对照hd显著性降低,而且在腋芽长度最长的oe-2株系中,ntbrc1基因的表达最低(图7)。
[0114]
实施例五 ntmab1基因rnai沉默对mab1腋芽表型的影响
[0115]
1、实验方法
[0116]
(1)rnai沉默载体构建
[0117]
原理为:将m1ri1、m1ri2两段序列分别分两次连接到puccrnai载体上,形成两个具有发夹结构的rnai沉默载体。将这两个沉默载体与pcambia1300-35s通过酶切连接的方式构建rnai双元沉默载体。
[0118]
a.pucc-ri1中间载体的制备:
[0119]
分别利用m1ri-f1/r1和m1ri-f2/r2这两引物对从ntmab1-s1基因外显子中扩增到长度分别为323bp的序列m1ri1和545bp的序列m1ri2;
[0120]
利用内切酶bamhi和sali对m1ri1片段和pucc-rnai载体分别进行双酶切和片段纯化;将纯化后的两个片段等比例混合,连接酶连接后转化大肠杆菌感受态,于amp抗性lb培养基上过夜培养,通过m1ri-r1和rintron-r1引物对鉴定阳性克隆送测序,得到pucc-ri1中间载体。
[0121]
b.pucc-m1ri1沉默载体的制备:
[0122]
利用内切酶xhoi和bglii对pucc-ri1载体进行双酶切和片段纯化。将纯化后pucc-ri1中间载体片段与bamhi和sali双酶切纯化后的m1ri1片段进行连接,得到pucc-m1ri1沉默载体。通过m1ri-f1和m1ri-r1的无条带引物对、m1ri-r1和rintron-r1有条带引物对进行鉴定。
[0123]
c.pucc-m1ri2沉默载体的制备:
[0124]
采用以上的相同步骤针对m1ri2片段构建得到pucc-ri2载体和pucc-m1ri2沉默载体。通过m1ri-f2和m1ri-r2的无条带引物对、m1ri-r2和rintron-r1有条带引物对进行鉴定。
[0125]
d.双元沉默载体的构建:
[0126]
利用内切酶psti或者sbfi对pucc-m1ri1、pucc-m1ri2和pcambia1300-35s载体分别进行单酶切和纯化。将酶切纯化后的pucc-m1ri1和pucc-m1ri2载体片段分别与单酶切后的pcambia1300-35s载体按比例混合,连接酶连接后转化大肠杆菌感受态,于kana抗性lb培养基上过夜培养,分别通过m1ri-r1、m1ri-r2和rintron-r1两对引物进行鉴定。通过以上方法构建获得ntmab1基因的两个双元沉默载体,即pcambia1300-35s-m1ri1和pcambia1300-35s-m1ri2。
[0127]
(2)转基因沉默植株的创制
[0128]
同上,通过农杆菌介导的叶盘法创制转基因沉默株系。两个rnai载体分别对突变体mab1材料进行转基因,对mab1突变体中ntmab1基因进行rnai沉默,此类转基因植株类型分别命名为m1ri1m-1,2,
…
,n(323bp)和m1ri2m-1,2,
…
,n(545bp)。
[0129]
表6 rnai沉默片段扩增和鉴定引物
[0130][0131]
2、实验结果及分析
[0132]
(1)经过转基因阳性鉴定及ntmab1基因表达水平检测,共筛选到10个ntmab1基因表达水平低于mab1突变体株系,其中8个m1ri1m株系,2个m1ri2m株系。我们选择表达量较低的m1ri1m-29、35和m1ri2m-27三个株系进行表型调查,在后续的研究中分别命名为mkd-1、mkd-2和mkd-3。
[0133]
(2)mkd-1、mkd-2和mkd-3三个株系不同叶位处腋芽的长度较mab1短很多,甚至可
以恢复到对照组hd的水平(图8a-c)。对hd、mab1以及mkd-1、mkd-2和mkd-3三个rnai株系中ntmab1基因表达水平进行检测,结果表明mkd-1、mkd-2两个株系腋芽中ntmab1基因的表达水平略高于hd,与mab1相比下降40多倍,而mkd-3株系中ntmab1基因的表达水平低于mab1,比hd高8倍(图8e,f)。这些结果表明,腋芽生长受ntmab1基因的表达影响。对几个mkd株系叶片中ntmab1基因表达水平进行检测,结果表明,三个mkd株系叶片中ntmab1基因的表达水平较hd的变化趋势与在腋芽中的检测结果一致,mkd-3中ntmab1基因的表达水平较野生型高10倍左右,其他两个株系ntmab1基因的表达比hd略高(图8d),这个结果表明mab1在腋芽生长及叶片发育的表型都受ntmab1基因表达的影响。
[0134]
实施例六 ntmab1基因rnai沉默对野生型腋芽表型的影响
[0135]
1、实验方法
[0136]
将实施例五构建的两个rnai载体(即双元沉默载体)分别转入hd,对hd中ntmab1基因进行沉默,转基因株系分别命名为m1ri1-1,2,
…
,n和m1ri2-1,2,
…
,n。对这些转基因植株进行ntmab1基因表达量检测、表型分析。
[0137]
2、实验结果及分析
[0138]
(1)对rnai转基因株系进行pcr阳性检测,转基因阳性株系进行ntmab1基因表达量检测,从中挑选出10个表达量低于hd的株系,m1ri1的8个和m1ri2的2个。其中有5个株系表达量较hd低5倍以上,其余5个株系表达量占hd的30-40%(图9a,b)。挑选5个表达量较低的rnai株系用于后续实验,将这5个转基因株系m1ri1-9、m1ri1-23、m1ri1-29、m1ri2-1和m1ri2-4分别命名为kd-1、kd-2、kd-3、kd-5和kd-6。
[0139]
(2)对上述5个rnai株系以及hd旺长期各叶位处腋芽长度测量,发现这5个rnai株系与hd一致,腋芽生长较缓慢。腋芽长度测量统计过程中发现,5个rnai株系各叶位的腋芽长度较hd中相应叶位腋芽长度短(图10a,b,c)。对5个rnai株系以及hd上部以及下部腋芽中ntmab1基因表达水平进行qrt-pcr检测,结果表明在5个rnai株系上下部腋芽中ntmab1基因表达量都低于hd(图10d,e),对叶片中ntmab1基因的表达水平的检测结果与腋芽中ntmab1基因表达量检测结果一致,5个rnai株系中ntmab1基因的表达水平都低于hd,其中kd-6叶片中该基因的表达水平最低(图10c)。这些结果表明,rnai沉默ntmab1基因导致基因表达量下降,进而导致腋芽活性降低。
[0140]
实施例七 烟草ntmab1基因在打顶措施下对分枝发育的影响
[0141]
打顶是烟草种植过程中去除植株顶端优势的一项非常重要的措施。烟草打顶以后,腋芽的生长会影响烟叶的品质。而本研究中的ntmab1基因如何响应烟草打顶措施,打顶以后该基因的表达及对腋芽生长的调控并不清楚。因此,对旺长期ntmab1基因超表达及突变体各株系中腋芽及分枝进行定量分析。
[0142]
1、实验方法
[0143]
(1)自最下部向上数相同叶数后选择适宜叶位打顶。利用直尺直接测量各个材料的腋芽或者分枝长度,每个材料5株取平均值。
[0144]
2、实验结果及分析
[0145]
结果表明,打顶前不同烟草株系各叶位处腋芽及分枝长度呈现出的趋势为mab1>mkd≥hd>ko-1>kd,而在kd各株系中kd-6各叶位处腋芽长度最短(图11a,b)。其中,hd为野生型;mab1为功能获得型突变体;mkd为ntmab1基因rnai沉默mab1突变体;ko-1为利用
crispr-cas9技术对野生型烟草的ntmab1基因进行敲除得到的基因敲除株系;kd为野生型的ntmab1基因rnai沉默植株。
[0146]
我们对打顶后三天和七天的腋芽及分枝进行长度测量及统计分析。结果表明,打顶后3天,mab1的各叶位分枝长度最长,在分析过程中发现hd及其他转基因株系各叶位处的腋芽长度趋势较为复杂,上部腋芽跟下部腋芽生长长度呈相反的趋势,上部腋芽长度在各株系中呈现的趋势为kd/ko-1≥mkd>hd,而在下部腋芽在各株系中呈现的趋势为mkd≥hd≥kd/ko-1,而下部腋芽的生长趋势与打顶前相对各株系不同叶位腋芽长度的趋势较为一致(图11c,d)。打顶后七天腋芽在各株系不同叶位处的定量统计结果与打顶后3天腋芽定量统计结果相似(图11d,e)。
[0147]
表7 打顶后三天不同株系各叶位腋芽长度比较
[0148][0149][0150]
注:加粗方块代表腋芽长度大于对照hd。加阴影的方块代表腋芽长度小于对照hd。
[0151]
上述表7的结果表明:打顶三天以内,各叶位腋芽生长长度趋势为mab1>mkd≥kd/ko-1≥hd,而打顶七天内,中上部腋芽的生长趋势为mab1>mkd≥kd/ko-1>hd,下部腋芽的生长趋势为mab1>mkd≥hd>kd/ko-1,具体见图12。
[0152]
对打顶后各株系上下部腋芽中ntmab1基因进行表达水平检测,结果表明,打顶以后不同烟草株系上下部腋芽中ntmab1基因的表达情况与打顶前趋势一致,表现为mab1中ntmab1基因表达量较hd高,kd株系中ntmab1基因的表达量低于hd(图13)。同时,也发现打顶以后mab1上下部腋芽中ntmab1基因的表达水平是一致的,这与oe-2上下部腋芽中ntmab1基因的表达情况是一致的。
[0153]
可以理解的是,对本领域普通技术人员来说,可以根据本发明的技术方案及本发明构思加以等同替换或改变,而所有这些改变或替换都应属于本发明所附的权利要求的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1