一种阿帕鲁胺的合成方法与流程
1.本发明属于医药化学领域的制备方法,具体涉及一种阿帕鲁胺及其中间体的合成方法。
背景技术:
2.阿帕鲁胺(apalutamide)是由美国加利福尼亚大学研制的一种雄激素受体(ar)抑制剂,该药于2009年被授权给了美国aragon制药公司。2018年2月,阿帕鲁胺获美国fda批准上市,用于治疗非转移性去势抵抗性前列腺癌(nm-crpc),已经于2019年在中国上市。其中文化学名为4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫代-5 ,7-二氮杂螺[3 .4]-辛烷-5-基)-2-氟-n-甲基苯甲酰,其化学结构如下结构式所示:目前合成阿帕鲁胺的方法主要有以下几种:(1)文献wo2007126765a2首先报道以化合物1为原料,与环丁酮、氰化钠缩合制得苯甲酰胺中间体2;以化合物3为原料,与硫光气反应制得硫代异氰酸基吡啶中间体4;最后两个中间体在微波促进下制得阿帕鲁胺。该路线需要在酸性条件下使用氰化钠以及硫光气,最后的环合反应采用微波,工业化生产较为困难。
[0003]
(2)pct专利wo2016100645简化了阿帕鲁胺的新合成方法,利用3-氟-4-碘苯胺与环丁酮、氰化物的strecker反应得到的环丁氨腈中间体2与2-氰基-3-三氟甲基-5-氨基吡啶化合物3在硫代羰基化合物作用下缩合环化,再完成贵金属钯的催化下的羰基插入反应得到羧酸酯中间体或经格氏交换后与干冰反应得到羧酸中间体,最后再酰胺化得到最终产品阿帕鲁胺。路线虽然步骤较短,钯催化的羰基插入反应或格氏交换反应路线成本较高,工艺生产实验条件较苛刻,不适宜工业化生产的方法合成阿帕鲁胺。
[0004]
(3)专利cn104211683a用tmscn替代nacn,避免了剧毒试剂的使用,但在最后一步缩合反应中,采用剧毒试剂硫光气一锅法合成阿帕鲁胺,工艺生产实验条件较苛刻,不适宜工业化生产的方法合成阿帕鲁胺。
技术实现要素:
[0005]
针对现有技术的不足,本发明提供了一种新的阿帕鲁胺的合成方法,该方法原料廉价易得、反应条件温和、操作简便、适宜工业化生产。
[0006]
本发明提供的一种阿帕鲁胺的合成方法,包括如下步骤:步骤1,将6-氨基-2-氰基-3-三氟甲基吡啶(i)和1-氨基-1-环丁基甲酸甲酯或其盐(ii)在tcdi和碱a的作用下,在溶剂a中反应得到中间体(iii);步骤2,将中间体(iii)与n-甲基-2-氟-4-卤代苯甲酰胺(iv)在催化剂和碱b的作用下,在溶剂b中反应得到阿帕鲁胺,用反应式表示如下:其中,x为氟、氯、溴或碘。
[0007]
作为优选,所述的盐为盐酸盐或硫酸盐。
[0008]
步骤1中所述的碱a为三乙胺、吡啶、二异丙基乙胺或dbu;所述的溶剂a为二氯甲烷、thf、甲苯或乙腈;反应温度为20-30℃。
[0009]
化合物i与化合物ii的投料摩尔比为1:1,tcdi与化合物1的投料摩尔比为1.2-1.5:1,碱a与化合物i的投料摩尔比为2-5:1。
[0010]
步骤2中所述的催化剂为氯化亚铜、溴化亚铜或碘化亚铜。所述的催化剂配合助剂2-乙酰胺环己酮、n1,n2-双(2,6-二甲基苯基)草酰胺、n1-苄基-n2-(2,6-二甲基苯基)草酰胺或n1-苄基-n2-(2-甲基萘-1-基)草酰胺催化反应。
[0011]
n1,n2-双(2,6-二甲基苯基)草酰胺的结构式为:。
[0012]
n1-苄基-n2-(2,6-二甲基苯基)草酰胺的结构式为:。
[0013]
n1-苄基-n2-(2-甲基萘-1-基)草酰胺的结构式为:。
[0014]
步骤2中所述的碱b为三乙胺、二异丙基乙胺、乙酸钠、丙酸钾、碳酸钾、碳酸钠、碳酸铯或dbu。所述的溶剂b为n ,n-二甲基甲酰胺、n ,n-二甲基乙酰胺、n-甲基吡咯烷酮或二甲基亚砜。步骤2的反应温度为80-90℃。
[0015]
化合物iii与化合物iv的 投料摩尔比为1:1.2,催化剂与助剂的投料摩尔比为1:1,催化剂与化合物iii的投料摩尔比为0.2-0.3:1,碱b与化合物iii的投料摩尔比为2-5:1。
[0016]
本发明的有益效果体现在:该方法起始原料廉价易得,避免了使用剧毒的氰化钠和贵金属催化剂,降低了工艺成本,符合绿色环保的要求,而且减少了副产物的生成,提高了产品的收率和纯度,适合放大生产。
附图说明
[0017]
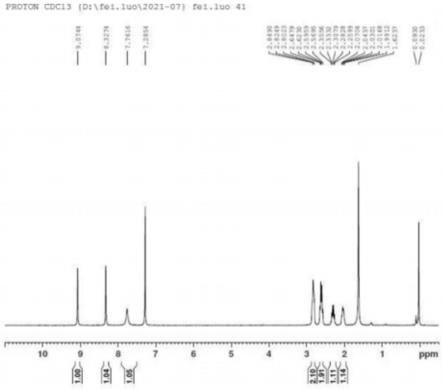
图1是化合物iii的1h-nmr谱图。
[0018]
图2是阿帕鲁胺的1h-nmr谱图。
具体实施方式
[0019]
下面结合具体实施例对本发明进行进一步说明,但本发明所申请保护的内容和范围并不受下述实施例的限制。
[0020]
实施例1三口烧瓶中加入6-氨基-2-氰基-3-三氟甲基吡啶1g(5.34mmol)、tcdi 1.4g(8.01mmol)和甲苯10ml,氮气保护下,降温至0-10℃,滴加三乙胺1.1g(10.68mmol),滴毕,升温至20-30℃反应3-4h,分批加入1-氨基-1-环丁基甲酸甲酯盐酸盐0.88g(5.34mmol),加
毕,继续在20-30℃反应1-2h。反应结束加入水10ml,乙酸乙酯10ml,搅拌分液,有机相用水洗涤二次,分液,有机相减压浓缩得油状物,经柱层析纯化得化合物iii,收率90%。化合物iii的氢谱谱图信息见图1。
[0021]
实施例2三口烧瓶中加入6-氨基-2-氰基-3-三氟甲基吡啶5g(26.7mmol)、tcdi 5.6g(32.04mmol)和甲苯50ml,氮气保护下,降温至0-10℃,滴加吡啶10.5g(132.7mmol),滴毕,升温至20-30℃反应5h,分批加入1-氨基-1-环丁基甲酸甲酯3.45g(26.7mmol),加毕,继续在20-30℃反应2h。反应结束加入水60ml,乙酸乙酯50ml,搅拌分液,有机相用水洗涤二次,分液,有机相减压浓缩得油状物,经柱层析纯化得化合物iii,收率92%。
[0022]
实施例3向三口烧瓶中加入中间体iii10.4g(32mmol)、n-甲基-2-氟-4-溴苯甲酰胺8.8g(38mmol)和二甲基亚砜50g,氮气保护下加入dbu9.8g(64mmol)、碘化亚铜1.2g(6.4mmol)和n1-苄基-n2-(2-甲基萘-1-基)草酰胺2g(6.4mmol),搅拌均匀后加热至80~90℃反应6h。反应结束加入水50ml和乙酸乙酯50g,搅拌分液,有机相用饱和食盐水洗涤二次,分层,收集有机相浓缩干后,加入甲醇50g和水150ml结晶,过滤,滤饼干燥得阿帕鲁胺,收率92%,hplc纯度99.62%,氢谱谱图见图2。
[0023]
实施例4向三口烧瓶中加入中间体iii10.4g(32mmol)、n-甲基-2,4-二氟苯甲酰胺6.5g(38mmol)和二甲基亚砜50g,氮气保护下加入dbu9.8g(64mmol)、碘化亚铜1.2g(6.4mmol)和n1-苄基-n2-(2-甲基萘-1-基)草酰胺2g(6.4mmol),搅拌均匀后加热至80~90℃反应6h。反应结束加入水50ml和乙酸乙酯50g,搅拌分液,有机相用饱和食盐水洗涤二次,分层,收集有机相浓缩干后,加入甲醇50g和水150ml结晶,过滤,滤饼干燥得阿帕鲁胺,收率85%,纯度99.05%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1