一株高产Xcn1的嗜线虫致病杆菌及其应用
一株高产xcn1的嗜线虫致病杆菌及其应用
技术领域
1.本发明涉及微生物技术领域,具体涉及一株高产xcn1的嗜线虫致病杆菌菌株及其应用。
背景技术:
2.昆虫病原线虫共生菌是存在于昆虫病原线虫肠道内的一类细菌,属肠杆菌科(enterobacteriaceae),包括与斯氏线虫属(steinernema)线虫共生的致病杆菌属(xenorhabdus)和与异小杆线虫属(heterorhabditis)线虫共生的发光杆菌属(photorhabdus)。共生菌分泌多种酶,降解昆虫组织,释放营养物质,并产生抑菌物质抑制其它杂菌生长,有助于线虫的生长繁殖。共生菌来源于特殊的生境,其代谢物中存在多种具有生物活性的新化合物,目前已分离鉴定的主要有抗菌物质、杀虫蛋白、抗癌物质等。
3.研究者前期分离收集到多株昆虫病原线虫共生细菌,通过对不同共生细菌的抑菌活性评价,发现在北京郊区分离到的嗜线虫致病杆菌cb6菌株(cgmcc no.1173,本研究室保存)的代谢产物具有很高的抑菌活性,对马铃薯晚疫病、番茄晚疫病和黄瓜白粉病等多种植物病害具有显著的防治效果。通过对cb6菌株发酵产物中的抗菌成份进行分离纯化和结构鉴定,确定xenocoumacin1(xcn1)是拮抗植物病害的主要活性物质。xcn1是由精氨酸残基、亮氨酸残基和四个乙酸酯单元形成的异香豆素类衍生物,该化合物具有拮抗马铃薯晚疫病、番茄晚疫病、黄瓜白粉病和辣椒疫霉病等植物真菌病害的生物学功能。xcn1是嗜线虫致病杆菌cb6产生的主要活性物质,但是在菌株代谢过程中,过量积累的xcn1会激活xcn1的降解代谢途径,xcn1会被转化成xcn2等低活性的衍生物,发酵生产得到的xcn1随着时间的推进反而产量进一步下降,导致xcn1无法大量积累。此外,野生型cb6菌株发酵产生xcn1的水平较低,推测xcn1合成基因簇中各种酶的表达水平较低是关键限制因素。
技术实现要素:
4.基于以上技术缺陷,本发明的技术目的是提供一株高产xenocoumacin1(xcn1)的嗜线虫致病杆菌,基于同源重组技术,对嗜线虫致病杆菌cb6野生型菌株中与xcn1合成代谢相关的基因进行工程改造,获得不产xcn2、高产xcn1的菌株,经技术优化后,最终获得一株命名为cb6
‑
t2的xcn1产量最高的菌株。经研究发现所述cb6
‑
t2菌株内介导xcn1降解转化的酶基因xcnm缺失突变,xcn1合成基因簇的第一个基因xcna启动子替换为来源于嗜线虫致病杆菌自身表达强度更高的启动子promoter
‑
g3509。
5.本发明先提供一株嗜线虫致病杆菌,其中所述酶基因xcnm首先以同源重组双交换的方式被缺失突变。具体以大肠杆菌
‑
嗜线虫致病杆菌穿梭克隆载体pjq200sk为骨架构建基因敲除载体,利用同源重组原理,通过双交换过程获得xcn1降解途径被阻断,xcn1高产的菌株。
6.在此基础上,进一步本发明还发现,在用同源重组基因敲除改造技术将嗜线虫致病杆菌cb6野生型菌株内介导xcn1转化为xcn2的酶基因xcnm以同源重组双交换的方式缺失
突变后,再同样采用同源重组双交换方式,将xcn1合成基因簇的第一个基因xcna的启动子替换为来源于嗜线虫致病杆菌自身表达强度更高的启动子promoter
‑
g3509,可以获得更好的基因表达效果,xcn1产量更高的技术效果。经试验发现,通过上述手段最终获得的嗜线虫致病杆菌cb6
‑
t2,在lb培养基中发酵得到的xcn1产量至少在782mg/l以上,远高于嗜线虫致病杆菌cb6野生型菌株的发酵产量71mg/l。可以有效降低xcn1产业化开发的生产成本。
7.本发明还提供所述的高产xcn1的嗜线虫致病杆菌的构建方法,优选是采用大肠杆菌
‑
嗜线虫致病杆菌穿梭克隆载体pjq200sk为骨架构建基因敲除载体,先构建获得xcnm敲除质粒,再将敲除质粒转化到大肠杆菌s17
‑
1λpir中,将大肠杆菌s17
‑
1λpir与嗜线虫致病杆菌共培养进行结合转移,涂布在筛选平板上,获得xcnm敲除阳性克隆。具体方案是先将pjq200sk质粒线性化、再将pjq200sk质粒线性化片段、上游同源臂和下游同源臂通过无缝克隆的方式连接在一起,转化到dh5α大肠杆菌感受态中,取100μl涂布含50μg/ml庆大霉素的lb平板上,37℃过夜培养后,获得xcnm敲除阳性克隆质粒。随后将该质粒转化到大肠杆菌s17
‑
1λpir中,将大肠杆菌s17
‑
1λpir与嗜线虫致病杆菌进行接合转移共培养,涂布在筛选平板上,挑取单菌落进行pcr验证,获得xcnm敲除阳性克隆。经试验验证,所述阳性克隆菌株发酵液中只能检测到xcn1,检测不到xcn2,说明xcnm基因缺失突变成功,该基因失去活性功能。
8.所述的构建方法,进一步还包括构建xcna启动子敲除替换质粒的步骤,将xcna启动子替换为来源于嗜线虫致病杆菌自身表达强度更高的启动子promoter
‑
g3509,所述启动子promoter
‑
g3509的序列如seq id no.15所示。为保证启动子顺利发挥功能,在启动子promoter
‑
g3509序列后面接一段核糖体结合位点rbs序列,如seq id no.16所示。经试验验证,所述启动子promoter
‑
g3509因来源于嗜线虫致病杆菌自身,表达强度更高,改良后的菌株在发酵培养到48h时xcn1达到最高水平,xcn1的含量为782mg/l,在后续的培养过程中,xcn1的含量依然维持在600mg/l以上。上述结果表明,本发明最终获得的改良菌株cb6
‑
t2,可以阻断代谢产物xcn1的降解并提高xcn1合成基因簇的转录表达,从而提高了目标代谢产物xcn1的产量。
附图说明
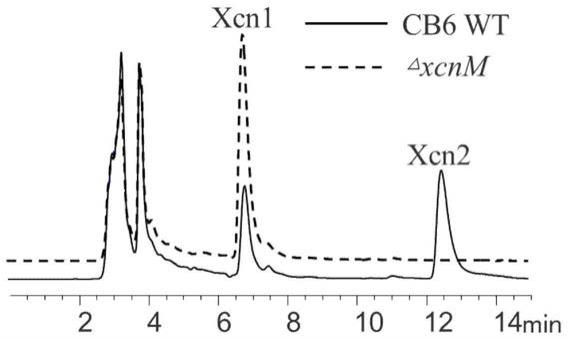
9.图1是hplc检测嗜线虫致病杆菌cb6野生型菌株与
△
xcnm菌株产xcn1及其代谢产物情况;
10.图2是利用hplc检测嗜线虫致病杆菌cb6
‑
t2菌株与cb6野生型菌株xcn1的产量。
具体实施方式
11.为进一步说明本发明,结合以下实施例具体说明:
12.本发明涉及的嗜线虫致病杆菌(xenorhabdus nematophila)cb6菌株是已知的菌种,在中国微生物菌种保藏管理委员会普通微生物中心(cgmcc)有所登记和保藏,地址:北京市朝阳区北辰西路1号院3号,中国科学院微生物研究所,邮政编码:100101,菌株编号cgmcc no.1173。
13.本发明提供一株高产xcn1的嗜线虫致病杆菌cb6
‑
t2,以cgmcc no.1173嗜线虫致病杆菌cb6为出发菌株,先将cb6野生型菌株内合成xcn1的基因簇进行改造,将介导xcn1向
xcn2转化的酶基因xcnm缺失突变,阻断了xcn1的降解转化。再将xcn1合成基因簇的第一个基因xcna的启动子替换为表达强度更高的启动子promoter
‑
g3509。本发明获得的嗜线虫致病杆菌cb6
‑
t2菌株,可以有效阻断xcn1的降解转化,促进菌株产生更多的xcn1,同时有效提高xcn1合成基因的表达水平,显著提高xcn1的产量。
14.本实施例以大肠杆菌
‑
嗜线虫致病杆菌穿梭克隆载体pjq200sk为骨架构建基因敲除载体。首先构建xcnm敲除质粒,采用引物pjq
‑
infusion
‑
f(atcgataccgtcgacctc,如seq id no.1所示)和pjq
‑
infusion
‑
r(gatatcgaattcctgcagcc,如seq id no.2所示)将pjq200sk质粒通过pcr方式进行线性化,上游同源臂pcr扩增引物为xcnm
‑
5dt(aaatagaacaatttggtggtgaag,如seq id no.3所示)和xcnm
‑
p1(ttctacttgttgatgaccaaaaaaat,如seq id no.4所示),下游同源臂pcr扩增引物为xcnm
‑
3dt(gaaagaataacaatccaataaatgag,如seq id no.5所示)和xcnm
‑
p2(gatctattacagtcggaaggaat,如seq id no.6所示),上游同源臂和下游同源臂pcr扩增模板是嗜线虫致病杆菌cb6基因组。使用se无缝克隆和组装试剂盒(庄盟生物,zc231)将pjq200sk质粒线性化片段、上游同源臂和下游同源臂通过无缝克隆的方式连接在一起,转化到dh5α大肠杆菌感受态中,取100μl涂布lb平板(含50μg/ml的庆大霉素)上,37℃过夜培养后,挑取单菌落培养,送单克隆到测序公司(擎科生物)进行测序,获得阳性克隆。转接阳性克隆至lb液体培养基(含50μg/ml的庆大霉素)中,37℃,200rpm过夜培养,提取质粒,电转至s17
‑
1λpir大肠杆菌感受态中。再次挑取单菌落,送单克隆到测序公司(擎科生物)进行测序,获得阳性克隆。
15.xcnm敲除步骤:将e.coil s17菌株与cb6菌株培养至od600=0.6~0.8,分别吸取1ml e.coil s17菌液与1ml cb6菌液,5000rpm,离心3min,弃上清,收集菌体,使用等体积lb重悬菌体,5000rpm,离心3min,弃上清,收集菌体,重复lb重悬清洗步骤一次。使用200μl lb重悬菌体,并将两种菌液充分混合,5000rpm,离心3min,弃上清,收集菌体。使用50μl lb重悬菌体,点加至lb培养基平板中央,28℃,培养24h。使用无菌枪头刮下lb培养基平板上接合转移的菌体,使用1ml lb培养基重悬菌体。吸取100μl菌液,涂布在lb(含50μg/ml的庆大霉素和100μg/ml的氨苄青霉素)培养基平板上,28℃,培养24h。选取单交换菌株,接种至3ml lbn(lb不含氯化钠)培养基中,28℃,200rpm,培养24h。吸取适量菌液,5000rpm离心3min,收集菌体,使用150μl无菌水重悬菌体,涂布至筛选平板上28℃,培养24h
‑
48h。挑取单菌落至lb液体培养基中培养,28℃,200rpm,培养过夜。吸取200μl菌液,12000rpm离心2min,收集菌体,提取dna,20μl体系pcr验证,验证引物为xcnm
‑
tf(agcaagatccgacacaaatgg,如seq id no.7所示)和xcnm
‑
tr(tgttcagctataccaataatccac,如seq id no.8所示)。pcr扩增验证获得目标突变
△
xcnm菌株后,将嗜线虫致病杆菌cb6和
△
xcnm菌株转接lb液体培养基中,28℃,200rpm,连续培养2天,在48h时结束发酵培养,12000rpm,离心10min,收集发酵液上清,使用0.22μm的过滤器进行过滤,利用hplc检测xcn1及其降解产物的含量。hplc所用的柱子为c18柱(安捷伦,agilent,4.6*150mm),流动相为含有0.1%三氟乙酸的水和乙腈,体积比为70:30。结果:
△
xcnm菌株发酵液中只能检测到xcn1,检测不到xcn2(如图1所示),说明xcnm基因缺失突变成功,该基因失去活性功能。
16.随后构建xcna启动子敲除替换质粒,采用引物pjq
‑
infusion
‑
f(atcgataccgtcgacctc,如seq id no.1所示)和pjq
‑
infusion
‑
r(gatatcgaattcctgcagcc,
如seq id no.2所示)将pjq200sk质粒通过pcr方式进行线性化,上游同源臂pcr扩增引物为xcna
‑
5dt(gttattgctgtttgtatttgtg,如seq id no.9所示)和xcna
‑
p1(ttaagttggtgccataattaatag,如seq id no.10所示),下游同源臂pcr扩增引物为xcna
‑
3dt(aagccccattaccatcttcaa,如seq id no.11所示)和xcna
‑
p2(atgaagaagacgatttttgagttg,如seq id no.12所示),启动子promoter
‑
g3509 pcr扩增引物为pro
‑
f(gtcacaggcagcaacatttc,如seq id no.13所示)和pro
‑
r(ctttatcgatagcaacgacaac,如seq id no.14所示),启动子promoter
‑
g3509、上游同源臂和下游同源臂pcr扩增模板是嗜线虫致病杆菌cb6基因组。为保证启动子顺利发挥功能,在启动子promoter
‑
g3509序列(如seq id no.15所示)后面接一段核糖体结合位点(rbs)序列(taagtaggtgatcactagtaattaaagaggagaaattaagc,如seq id no.16所示)。构建xcna启动子敲除替换质粒过程和xcna启动子敲除替换突变体筛选过程与构建
△
xcnm菌株过程一样,最后xcna启动子敲除替换突变体cb6
‑
t2菌株验证引物为xcna
‑
tf(caggtatagtaaattaatagggg,如seq id no.17所示)和xcna
‑
tr(gtttctcttcactactccaacg,如seq id no.18所示)。pcr扩增验证获得目标突变菌株cb6
‑
t2后,将嗜线虫致病杆菌cb6和cb6
‑
t2接种于nbta(胰蛋白胨10g/l;牛肉粉3g/l;氯化钠5g/l;氯化三苯基四唑0.04g/l;溴百里酚兰0.025g/l,ph=7.8)的平板上,28℃培养48h。挑取单菌落转接到lb(胰蛋白胨10g/l,酵母提取物5g/l,氯化钠10g/l)液体培养基中,28℃,200rpm过夜培养,得到发酵种子液。
17.将嗜线虫致病杆菌cb6和cb6
‑
t2种子液按1%的接种量转接lb液体培养基中,28℃,200rpm,连续培养3天。分别在第12h、16h、20h、24h、36h、42h、48h、60h、72h、84h、96h取样。
18.收集每个时间点的样品以得到其中的代谢产物,通过12000rpm,离心10min,收集发酵液上清,使用0.22μm的过滤器过滤,利用hplc检测xcn1及其降解产物的含量。hplc所用的柱子为c18柱(安捷伦,agilent,4.6*150mm),流动相为含有0.1%三氟乙酸的水和乙腈,体积比为70:30。结果:嗜线虫致病杆菌cb6野生型菌株在发酵培养到48h时xcn1达到最高水平,xcn1的含量为71mg/l;随后随着培养时间的延长,xcn1的含量逐渐降低。嗜线虫致病杆菌cb6
‑
t2菌株在发酵培养到48h时xcn1达到最高水平,xcn1的含量为782mg/l,在后续的培养过程中,xcn1的含量依然维持在600mg/l以上(如图2所示)。上述结果表明,本发明获得的改良菌株cb6
‑
t2,阻断代谢产物xcn1的降解并提高xcn1合成基因簇的转录表达,从而提高了目标代谢产物xcn1的产量。
19.以上所述的实施例仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通工程技术人员对本发明的技术方案作出的各种变形和改进,均应落入本发明的权利要求书确定的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1