一株大肠杆菌基因工程菌及其发酵生产L-茶氨酸的方法
一株大肠杆菌基因工程菌及其发酵生产l
‑
茶氨酸的方法
技术领域:
1.本发明属于生物工程领域,具体涉及一种利用大肠杆菌基因工程菌发酵生产l
‑
茶氨酸的方法。
技术背景:
2.l
‑
茶氨酸(n
‑
乙基
‑
γ
‑
l
‑
谷氨酰胺)是茶叶中特有的一种非蛋白质氨基酸,于1950年由日本学者酒户弥二郎从玉露茶中分离得到并命名。l
‑
茶氨酸主要存在于茶树类植物中,通常以游离形式存在,不参与合成蛋白质,占茶树体内游离氨基酸总量的40%
‑
70%。l
‑
茶氨酸在茶汤中泡出率可达80%,是茶汤呈现鲜爽味的主要成分,与绿茶等级的正相关系数达0.787
‑
0.876,也是评价其他茶类品质和风味的重要物质。随着对l
‑
茶氨酸生理活性研究的深入,其在食品、保健和医学领域有着广泛的应用前景。
3.目前食品级的l
‑
茶氨酸均采用微生物合成法获得。cn103409475b报道了通过人工合成获得γ
‑
谷氨酰转肽酶的基因,并以大肠杆菌为宿主菌构建一种过量表达γ
‑
谷氨酰转肽酶的基因工程菌,以重组酶作用于不同浓度的谷氨酰胺和乙胺盐酸盐得到l
‑
茶氨酸。cn104087535b报道了使用保藏编号为cctcc m2014254的硝基还原假单胞菌ntlc4.002为出发菌株制备细胞、细胞转化谷氨酰胺和乙胺制备l
‑
茶氨酸。cn106893748b报道了以γ
‑
谷氨酰甲胺合成酶和磷酸激酶为催化剂,以l
‑
谷氨酸钠、乙胺盐酸盐和少量atp为底物合成l
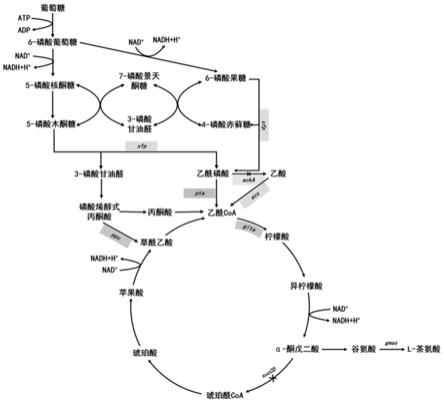
‑
茶氨酸。cn102719367b报道了使用从茶树根际土壤筛选得到的近玫色锁掷孢酵母生产γ
‑
谷氨基甲酰胺合成酶,再用该酶催化l
‑
谷氨酸钠、乙胺盐酸盐和atp生产l
‑
茶氨酸。微生物酶促合成l
‑
茶氨酸的主要问题在于需要以价格较高的谷氨酰胺、谷氨酸盐和atp为原料,因此生产成本极高,产品难以大规模应用。
4.以葡萄糖为原料通过微生物发酵制备l
‑
茶氨酸的方法(cn109370966b、cn109777763b),具有原料简单易得,生产环节简单,成本较低的特点。但发酵法生产l
‑
茶氨酸仍存在产量低、糖酸转化率较低、副产物较多等问题。此外,由于乙胺对于微生物生长存在抑制作用,会造成发酵过程中菌体浓度降低,部分菌体自溶,从而使得发酵液成分复杂,产物难于提取。
5.茶氨酸的分离提取方面,cn105061249b公布了一种通过超滤去除废液中大分子物质,然后添加碱式碳酸铜和稀硫酸,反应生成茶氨酸
‑
硫酸铜,再通过电渗析去除茶氨酸
‑
硫酸铜中的铜离子和硫酸根离子,然后减压浓缩去除淡水室内的水份,再使用无水乙醇分离出硫酸
‑
茶氨酸中的硫酸。cn109851520a公布了一种通过向l
‑
茶氨酸反应液中加入絮凝剂和离子去除剂后,经过超滤、脱色、浓缩、结晶等步骤得到l
‑
茶氨酸。上述提取方法中均用到了大量的酸碱试剂或絮凝剂,收率及产品纯度低,因此,也亟需一种高纯度的l
‑
茶氨酸的分离纯化方法。
技术实现要素:
6.本发明目的是通过基因组改造获得一株糖酸转化率高、生产性能稳定的用于l
‑
茶
氨酸生产的基因工程菌并提供其发酵、提纯方法。
7.针对上述存在的问题,本发明技术方案如下:
8.本发明提供的技术方案之一,是一株无质粒、以葡萄糖等廉价碳源为底物从头高效合成l
‑
茶氨酸的基因工程菌。该基因工程菌株是以zl 201811215068.6(cn 109370966)构建的l
‑
茶氨酸生产基因工程菌为出发菌,通过以下改造获得:在基因组上整合6
‑
磷酸果糖解酮酶基因xfp、磷酸乙酰基转移酶基因pta、乙酰辅酶a合成酶基因acs、柠檬酸合酶基因glta、磷酸烯醇丙酮酸羧化酶基因ppc,敲除乙酸激酶基因acka。
9.进一步地,所述6
‑
磷酸果糖解酮酶基因xfp来源于双歧杆菌(b.adolescentis)atcc 15703,核苷酸序列如序列表seq id no.1所示;
10.进一步地,所述磷酸乙酰基转移酶基因pta来源于大肠杆菌(e.coli)atcc 27325,核苷酸序列如序列表seq id no.2所示;
11.进一步地,所述乙酰辅酶a合成酶基因acs来源于大肠杆菌(e.coli)atcc 27325,核苷酸序列如序列表seq id no.3所示;
12.进一步地,所述柠檬酸合酶基因glta来源于大肠杆菌(e.coli)atcc 27325,核苷酸序列如序列表seq id no.4所示;
13.进一步地,所述磷酸烯醇丙酮酸羧化酶基因ppc来源于大肠杆菌(e.coli)atcc 27325,核苷酸序列如序列表seq id no.5所示;
14.进一步地,所述乙酸激酶基因acka的核苷酸序列如序列表seq id no.6所示;
15.优选地,xfp、pta、acs、glta、ppc基因均分别由trc启动子控制;
16.所述zl 201811215068.6构建的l
‑
茶氨酸生产基因工程菌,是在大肠杆菌(escherichia coli)w3110基因组上单拷贝来源于t7噬菌体的rna聚合酶基因t7rnap,该基因由木糖启动子控制;双拷贝来源于methylovorus mays的γ
‑
谷氨酰甲胺合成酶基因gmas,该基因由t7启动子控制;敲除木糖操纵子阻遏蛋白基因xylr;敲除琥珀酰coa合成酶基因succd。
17.本发明提供的技术方案之二,是上述基因工程菌的构建方法,本发明中采用crispr/cas9介导的基因编辑技术对大肠杆菌进行定向改造,具体包括如下步骤:
18.(1)为了加强碳循环,减少碳源损失,在出发菌株基因组gapc位点单拷贝来源于双歧杆菌(b.adolescentis)atcc 15703的6
‑
磷酸果糖解酮酶基因xfp(seq id no.1所示),该基因由trc启动子控制;
19.(2)为了加强乙酰磷酸到乙酰辅酶a的代谢,在出发菌株基因组yjit位点单拷贝来源于大肠杆菌atcc 27325的乙酰基转移酶基因pta(seq id no.2所示);
20.(3)为了加强乙酸到乙酰辅酶a的代谢,在出发菌株基因组yghe位点单拷贝来源于大肠杆菌atcc 27325的乙酰辅酶a合成酶基因acs(seq id no.3所示);
21.(4)为了加强乙酰辅酶a到柠檬酸的代谢,在出发菌株基因组ylbe位点单拷贝来源于大肠杆菌atcc 27325的磷酸乙酰基转移酶glta(seq id no.4所示);
22.(5)为了加强磷酸烯醇式丙酮酸到草酰乙酸的代谢,在出发菌株基因组yeel位点单拷贝来源于大肠杆菌atcc 27325的磷酸烯醇丙酮酸羧化酶ppc(seq id no.5所示);
23.(6)为了减少乙酸的积累,敲除出发菌株基因组上的乙酸激酶基因acka(seq id no.6所示);
24.其中,步骤(1)
‑
(6)的构建顺序不分先后,可根据需求进行调整。将最终获得的基因工程菌株命名为e.coli thee。
25.本发明提供的技术方案之三,是上述基因工程菌的应用,特别是在采用发酵法生产l
‑
茶氨酸中的应用;
26.进一步地,采用上述基因工程菌e.coli thee发酵生产l
‑
茶氨酸的方法具体如下:
27.发酵培养:将种子液按照10
‑
15%接种量接入新鲜的发酵培养基,发酵过程中控制ph在6.7
‑
7.2,温度维持在28
‑
36℃,溶氧在10
‑
30%之间;当培养基中的葡萄糖消耗完之后,流加葡萄糖溶液并维持发酵培养基中的葡萄糖浓度<1g/l;
28.进一步地,流加600g/l的葡萄糖溶液维持发酵培养基中的葡萄糖浓度<1g/l;
29.进一步地,为了减少乙胺对于菌体生长的影响,发酵过程中采用od联动的乙胺补加策略,当od
600nm
达到8
‑
12以上时开始添加乙胺,每0.8
‑
1.2小时调节一次乙胺流加速率;乙胺流加速率(g
·
l
‑1·
h
‑1)=0.5
×
od
600nm
值/(发酵液体积(l)
×
发酵时间(h));
30.5l发酵罐发酵20
‑
25h后l
‑
茶氨酸的产量可达75
‑
80g/l,糖酸转化率达到52
‑
55%。
31.进一步地,种子培养方法如下:取适量无菌水于茄形瓶中,将菌悬液接入种子培养基中,ph稳定在7.0左右,温度恒定在37℃,溶氧在25
‑
35%之间,培养至细胞干重达到5
‑
6g/l;
32.进一步地,斜面培养方法如下:从
‑
80℃冰箱保菌管中刮一环菌种,均匀涂布于活化斜面,37℃培养12
‑
16h,转接茄形瓶继续培养12
‑
16h;
33.优选地,斜面培养基组成为:葡萄糖1
‑
5g/l,蛋白胨5
‑
10g/l,牛肉膏5
‑
10g/l,酵母粉1
‑
5g/l,氯化钠1
‑
2.5g/l,琼脂15
‑
20g/l,ph 7.0
‑
7.2;
34.种子培养基组成为:葡萄糖20
‑
30g/l,酵母提取物5
‑
10g/l,蛋白胨10
‑
20g/l,氯化钠10
‑
20g/l,其余为水,ph 7.0
‑
7.2;
35.发酵培养基组成为:葡萄糖10
‑
40g/l,酵母粉2
‑
8g/l,玉米浆2
‑
20ml/l,柠檬酸0.2
‑
2.0g/l,磷酸二氢钾0.5
‑
3.2g/l,磷酸氢二钾0.5
‑
2.4g/l,硫酸镁0.2
‑
1.2g/l,其余为水,ph 7.0
‑
7.2。
36.本发明提供的技术方案之四,是从上述发酵液中分离提取l
‑
茶氨酸的方法,具体如下:
37.(1)在发酵罐中将l
‑
茶氨酸发酵液加热升温至55
‑
60℃维持20
‑
30min后降温至35
‑
40℃,过50
‑
70nm陶瓷膜进行微滤除菌并收集滤出液,0.2
‑
0.3mpa压力下,当滤出液流速低于5ml/min时,补加0.5
‑
1倍发酵液体积的水,直至截留液中l
‑
茶氨酸浓度小于2g/l时,结束陶瓷膜微滤;
38.(2)将陶瓷膜滤液过阳离子树脂吸附l
‑
茶氨酸,然后用质量分数为0.5%
‑
1%的氨水进行洗脱收集;将洗脱液过阴离子树脂吸附色素,收集树脂流出液;
39.优选地,所述阳离子树脂为001
×
7苯乙烯系强酸阳离子交换树脂;
40.优选地,所述的阴离子树脂为d213丙烯酸系强碱阴离子交换树脂;
41.(3)将树脂流出液泵入脱色罐中,加入l
‑
茶氨酸质量1%
‑
3%的药用活性炭进行脱色处理,至料液透光率达到96%以上;将脱色液泵入蒸发器中进行减压浓缩至体积浓缩倍数为6
‑
9倍;
42.进一步地,减压浓缩维持真空度
‑
0.08mpa,温度60℃;
43.(4)将浓缩液泵入结晶罐中,加入浓缩液体积30%
‑
50%的乙醇,真空冷却结晶,离心分离收集湿晶体,烘干后得到l
‑
茶氨酸成品。
44.本发明的有益效果:
45.(1)由于丙酮酸脱氢酶催化丙酮酸形成乙酰coa和co2,所以大肠杆菌经糖酵解途径最多将一分子葡萄糖转化为两分子乙酰coa,剩余部分以co2形式损失,严重限制了葡萄糖到l
‑
茶氨酸的转化率。本发明通过引入6
‑
磷酸果糖解酮酶,可最多将一分子葡萄糖转化为三分子乙酰coa。将6
‑
磷酸果糖解酮酶基因整合到大肠杆菌基因组上,构建出和6
‑
磷酸果糖和5
‑
磷酸木酮糖到乙酰磷酸的代谢途径,再通过强化内源磷酸乙酰基转移酶的表达,使乙酰磷酸转变为乙酰coa。上述代谢改造策略可以有效减少丙酮酸到乙酰coa的代谢,从而减少碳损,提高了葡萄糖到l
‑
茶氨酸的转化率。
46.(2)大肠杆菌发酵过程中会产生一定量的副产物乙酸,影响菌体正常生长。乙酰磷酸的过量合成会进一步加剧乙酸的积累。为了减少副产物乙酸的含量,本技术敲除了乙酸激酶,阻断了乙酰磷酸到乙酸的代谢,同时强化内源乙酰辅酶a合成酶的表达,使乙酸转变为乙酰coa。上述代谢改造策略可以有效减少乙酸对菌体生长的抑制作用,提升了单位菌体的产酸率。
47.(3)异型发酵途径的引入会造成三磷酸甘油醛和乙酰coa的积累。为了增强三羧酸循环的代谢通量,本技术强化内源柠檬酸合酶和磷酸烯醇丙酮酸羧化酶的表达,为三羧酸循环起始反应即柠檬酸的合成提供了充足的酶和底物草酰乙酸。
48.(4)乙胺是l
‑
茶氨酸发酵的前体物,但大量的乙胺对菌体细胞具有较强的毒害作用。在发酵过程采用恒速流加乙胺策略可以有效控制发酵液中乙胺的浓度,但是菌体不同生长阶段对乙胺的消耗不同,恒定补料会影响菌体的产酸能力。本技术根据菌体生长不同周期对于乙胺的耐受程度,开发了od联动的乙胺补料技术,将乙胺流加速率和菌体生物量偶联,拟合出经验方程,使得菌体能够最大程度的代谢乙胺生产l
‑
茶氨酸,从而大幅度提升了生物量和产酸量。
49.(5)本发明不使用传统的沉淀法提纯l
‑
茶氨酸技术,采用膜分离结合阴阳离子树脂串联技术,减少了活性炭使用,提高了产品收率及产品质量,一次结晶收率72.3%,成品纯度为99%。本发明只需一次结晶即可拿到合格产品,不需要精制,缩短了纯化路线,降低了污染风险,增加了生产的稳定性。
附图说明:
50.图1基因工程菌e.coli thee代谢改造策略图
51.其中灰色填充基因为本技术改造点;
52.图2gapc::p
trc
‑
xfp整合电泳图
53.其中,m—1kb maker;1—上游同源臂;2—目的基因;3—下游同源臂;4—重叠片段;5—原菌pcr片段;6—目的菌pcr片段;
54.图3yjit::p
trc
‑
pta整合电泳图
55.其中,m—1kb maker;1—上游同源臂;2—目的基因;3—下游同源臂;4—重叠片段;5—原菌pcr片段;6—目的菌pcr片段;
56.图4yghe::p
trc
‑
acs整合电泳图
57.其中,m—1kb maker;1—上游同源臂;2—目的基因;3—下游同源臂;4—重叠片段;5—原菌pcr片段;6—目的菌pcr片段;
58.图5ylbe::p
trc
‑
glta整合电泳图
59.其中,m—1kb maker;1—上游同源臂;2—目的基因;3—下游同源臂;4—重叠片段;5—原菌pcr片段;6—目的菌pcr片段;
60.图6yeel::p
trc
‑
ppc整合电泳图
61.其中,m—1kb maker;1—上游同源臂;2—目的基因;3—下游同源臂;4—重叠片段;5—原菌pcr片段;6—目的菌pcr片段;
62.图7acka敲除电泳图
63.其中,m—1kb maker;1—上游同源臂;2—下游同源臂;3—重叠片段;4—原菌pcr片段;5—目的菌pcr片段;
64.图8基因工程菌e.coli thee 5l罐发酵进程图;
65.图9乙胺流速线性拟合图。
具体实施方式:
66.为了使本专利的目的、技术方案及优点更加清楚明白,以下结合具体实施例,对本专利进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本专利,并不用于限定本发明。
67.本发明以zl 201811215068.6(cn 109370966)构建的l
‑
茶氨酸生产基因工程菌为出发菌,该菌株是在大肠杆菌w3110基因组上laci
‑
lacz位点单拷贝来源于t7噬菌体的rna聚合酶基因t7rnap(核苷酸序列见cn 109370966 b序列表1),该基因由木糖启动子p
xylf
控制;在w3110基因组上yghx及yeep位点双拷贝来源于methylovorus mays的γ
‑
谷氨酰甲胺合成酶基因gmas(核苷酸序列见cn 109370966 b序列表2),该基因经过密码子优化并由t7启动子控制;敲除w3110的木糖操纵子阻遏蛋白基因xylr;敲除w3110的琥珀酰coa合成酶基因succd获得。详细构建过程可参考cn 109370966 b。
68.本发明所用trc启动子序列如下:ttgacaattaatcatccggctcgtataatgtgtggaattgtgagcggataacaatttcacacaggaaacagacc。
69.本发明实施例中,大肠杆菌e.coli thee构建过程涉及的xfp、pta、acs、glta、ppc、acka基因序列见序列表seq id no.1
‑
6所示。
70.以下将结合附图,通过具体实施方式对本发明作进一步地解释说明。
71.实施例1:基因工程菌株大肠杆菌e.coli thee的构建(改造策略见图1):
72.1、基因编辑的方法
73.本发明中采用crispr/cas 9介导的基因编辑方法参照文献(metabolic engineering,2015,31:13
‑
21.)进行,该方法所用的两个质粒分别为pgrb与predcas9。其中predcas9携带grna质粒消除系统,λ噬菌体的red重组系统及cas9蛋白表达系统,奇霉素抗性(工作浓度:100mg/l),32℃培养;pgrb质粒,以puc18为骨架,包括启动子j23100,grna
‑
cas9结合区域序列和终止子序列,氨苄青霉素抗性(工作浓度:100mg/l),37℃培养。
74.2、菌株构建的具体过程
75.以zl 201811215068.6构建的l
‑
茶氨酸生产基因工程菌(在大肠杆菌w3110基因组
上laci
‑
lacz位点单拷贝来源于t7噬菌体的rna聚合酶基因t7rnap(核苷酸序列见cn 109370966b序列表1),该基因由木糖启动子p
xylf
控制;在基因组上yghx及yeep位点双拷贝γ
‑
谷氨酰甲胺合成酶基因gmas(核苷酸序列见cn 109370966 b序列表2),该基因经过密码子优化并由t7启动子控制;敲除木糖操纵子阻遏蛋白基因xylr;敲除w3110的琥珀酰coa合成酶基因succd获得)为出发菌,并进行如下改造:
76.2.1将p
trc
‑
xfp(含有trc启动子和xfp基因的片段)整合在假基因gapc位点处
77.以e.coli atcc27325基因组为模板,根据其gapc基因的上、下游序列设计上游同源臂引物up
‑
gapc
‑
s(seq id no.7)、up
‑
gapc
‑
a(seq id no.8)和下游同源臂引物dn
‑
gapc
‑
s(seq id no.9)、dn
‑
gapc
‑
a(seq id no.10),并扩增gapc基因的上、下游同源臂;根据xfp基因设计引物xfp
‑
s(seq id no.11)、xfp
‑
a(seq id no.12),并扩增xfp基因片段。启动子p
trc
则设计在gapc基因上游同源臂的下游引物和xfp基因的上游引物中。上述片段通过重叠pcr的方法获得xfp基因的整合片段(gapc基因上游同源臂
‑
p
trc
‑
xfp
‑
gapc基因下游同源臂),构建pgrb
‑
gapc使用的含靶序列的dna片段通过引物grna
‑
gapc
‑
s(seq id no.13)和grna
‑
gapc
‑
a(seq id no.14)的退火制得,与线性化的pgrb载体重组后获得重组的pgrb
‑
gapc。将整合片段和pgrb
‑
gapc电转化至含有predcas9载体的大肠杆菌出发菌株感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
gapc。验证图如图2所示。
78.2.2将p
trc
‑
pta(含有trc启动子和pta基因的片段)整合在假基因yjit位点处
79.以e.coli atcc27325基因组为模板,根据其yjit基因的上、下游序列设计上游同源臂引物up
‑
yjit
‑
s(seq id no.15)、up
‑
yjit
‑
a(seq id no.16)和下游同源臂引物dn
‑
yjit
‑
s(seq id no.17)、dn
‑
yjit
‑
a(seq id no.18),并扩增yjit基因的上、下游同源臂;根据pta基因设计引物pta
‑
s(seq id no.19)、pta
‑
a(seq id no.20),并扩增pta基因片段。启动子p
trc
则设计在yjit基因上游同源臂的下游引物和pta基因的上游引物中。上述片段通过重叠pcr的方法获得pta基因的整合片段(yjit基因上游同源臂
‑
p
trc
‑
pta
‑
yjit基因下游同源臂),构建pgrb
‑
yjit使用的含靶序列的dna片段通过引物grna
‑
yjit
‑
s(seq id no.21)和grna
‑
yjit
‑
a(seq id no.22)的退火制得,与线性化的pgrb载体重组后获得重组的pgrb
‑
yjit。将整合片段和pgrb
‑
yjit电转化至含有predcas9载体的上步构建的大肠杆菌感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
yjit。验证图如图3所示。
80.2.3将p
trc
‑
acs(含有trc启动子和acs基因的片段)整合在假基因yghe位点处
81.以e.coli atcc27325基因组为模板,根据其yghe基因的上、下游序列设计上游同源臂引物up
‑
yghe
‑
s(seq id no.23)、up
‑
yghe
‑
a(seq id no.24)和下游同源臂引物dn
‑
yghe
‑
s(seq id no.25)、dn
‑
yghe
‑
a(seq id no.26),并扩增yghe基因的上、下游同源臂;根据acs基因设计引物acs
‑
s(seq id no.27)、acs
‑
a(seq id no.28),并扩增acs基因片段。启动子p
trc
则设计在yghe基因上游同源臂的下游引物和acs基因的上游引物中。上述片段通过重叠pcr的方法获得acs基因的整合片段(yghe基因上游同源臂
‑
p
trc
‑
acs
‑
yghe基因下游同源臂),构建pgrb
‑
yghe使用的含靶序列的dna片段通过引物grna
‑
yghe
‑
s(seq id no.29)和grna
‑
yghe
‑
a(seq id no.30)的退火制得,与线性化的pgrb载体重组后获得重组的pgrb
‑
yghe。将整合片段和pgrb
‑
yghe电转化至含有predcas9载体的上步构建的大肠杆菌感受态
细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
yghe。验证图如图4所示。
82.2.4将p
trc
‑
glta(含有trc启动子和glta基因的片段)整合在假基因ylbe位点处
83.以e.coli(atcc27325)基因组为模板,根据其ylbe基因的上、下游序列设计上游同源臂引物up
‑
ylbe
‑
s(seq id no.31)、up
‑
ylbe
‑
a(seq id no.32)和下游同源臂引物dn
‑
ylbe
‑
s(seq id no.33)、dn
‑
ylbe
‑
a(seq id no.34),并扩增ylbe基因的上、下游同源臂;根据glta基因设计引物glta
‑
s(seq id no.35)、glta
‑
a(seq id no.36),并扩增glta基因片段。启动子p
trc
则设计在ylbe基因上游同源臂的下游引物和glta基因的上游引物中。上述片段通过重叠pcr的方法获得glta基因的整合片段(ylbe基因上游同源臂
‑
p
trc
‑
glta
‑
ylbe基因下游同源臂),构建pgrb
‑
ylbe使用的含靶序列的dna片段通过引物grna
‑
ylbe
‑
s(seq id no.37)和grna
‑
ylbe
‑
a(seq id no.38)的退火制得,与线性化的pgrb载体重组后获得重组的pgrb
‑
ylbe。将整合片段和pgrb
‑
ylbe电转化至含有predcas9载体的上步构建的大肠杆菌感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
ylbe。验证图如图5所示。
84.2.5将p
trc
‑
ppc(含有trc启动子和ppc基因的片段)整合在假基因yeel位点处
85.以e.coli atcc27325基因组为模板,根据其yeel基因的上、下游序列设计上游同源臂引物up
‑
yeel
‑
s(seq id no.39)、up
‑
yeel
‑
a(seq id no.40)和下游同源臂引物dn
‑
yeel
‑
s(seq id no.41)、dn
‑
yeel
‑
a(seq id no.42),并扩增yeel基因的上、下游同源臂;根据ppc基因设计引物ppc
‑
s(seq id no.43)、ppc
‑
a(seq id no.44),并扩增ppc基因片段。启动子p
trc
则设计在yeel基因上游同源臂的下游引物和ppc基因的上游引物中。上述片段通过重叠pcr的方法获得ppc基因的整合片段(yeel基因上游同源臂
‑
p
trc
‑
ppc
‑
yeel基因下游同源臂),构建pgrb
‑
yeel使用的含靶序列的dna片段通过引物grna
‑
yeel
‑
s(seq id no.45)和grna
‑
yeel
‑
a(seq id no.46)的退火制得,与线性化的pgrb载体重组后获得重组的pgrb
‑
yeel。将整合片段和pgrb
‑
yeel电转化至含有predcas9载体的上步构建的大肠杆菌感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
yeel。验证图如图6所示。
86.2.6 acka基因的敲除
87.根据acka基因的上、下游序列设计上游同源臂引物up
‑
acka
‑
s(seq id no.47)、up
‑
acka
‑
a(seq id no.48)和下游同源臂引物dn
‑
acka
‑
s(seq id no.49)、dn
‑
acka
‑
a(seq id no.50),并pcr扩增其上、下游同源臂片段;上述片段通过重叠pcr的方法获得acka基因的敲除片段(acka上游同源臂
‑
acka下游同源臂)。设计引物grna
‑
acka
‑
s(seq id no.51)和grna
‑
acka
‑
a(seq id no.52)扩增包含靶序列的dna片段,与线性化的pgrb载体重组后获得重组的pgrb
‑
acka。将敲除片段和pgrb
‑
acka电转化至含有predcas9载体的上步构建的大肠杆菌感受态细胞中,将电转化后复苏培养的菌体涂布于含氨苄青霉素和奇霉素的lb平板上,32℃过夜培养后利用pcr验证阳性重组子,再消除用于基因编辑的pgrb
‑
acka和predcas9。验证图如图7所示。
88.上述步骤的顺序可根据实际情况进行调整,最终获得菌株e.coli thee。
89.3.菌株构建过程中用到的引物
90.菌株构建过程中所涉及的所有引物见下表:
91.92.[0093][0094]
实施例2:菌株e.coli thee 5l发酵罐发酵生产l
‑
茶氨酸
[0095]
斜面培养基组成为:葡萄糖2g/l,蛋白胨6g/l,牛肉膏6g/l,酵母粉3g/l,氯化钠2g/l,琼脂18g/l,ph 7.0。
[0096]
种子培养基组成为:葡萄糖25g/l,酵母提取物6g/l,蛋白胨15g/l,氯化钠15g/l,其余为水,ph 7.0。
[0097]
发酵培养基组成为:葡萄糖30g/l,酵母粉6g/l,玉米浆8ml/l,柠檬酸1.5g/l,磷酸二氢钾2.5g/l,磷酸氢二钾2.0g/l,硫酸镁1.0g/l,其余为水,ph 7.2。
[0098]
(1)斜面培养:从
‑
80℃冰箱保菌管中刮一环菌种,均匀涂布于活化斜面,37℃培养14h,转接茄形瓶继续培养14h;
[0099]
(2)种子培养:取适量无菌水于茄形瓶中,将菌悬液接入种子培养基中,ph稳定在7.0左右,温度恒定在37℃,溶氧在30
‑
34%之间,培养至细胞干重达到6g/l;
[0100]
(3)发酵培养:将种子液按照12%接种量接入新鲜的发酵培养基,发酵过程中控制ph在7.0,温度维持在36℃,溶氧在25
‑
30%之间;当培养基中的葡萄糖消耗完之后,流加600g/l的葡萄糖溶液并维持发酵培养基中的葡萄糖浓度<1g/l;
[0101]
为了比较不同乙胺补料方式对于发酵结果的影响,设置对照组和实验组:
[0102]
对照组采用本领域常用的乙胺流加方式,即当od
600
=20时开始以30ml/h的速率恒速流加2mol/l的乙胺溶液至发酵结束;
[0103]
实验组采用od联动的乙胺补加策略,乙胺流加速率(g
·
l
‑1·
h
‑1)=0.5
×
od
600nm
值/(发酵液体积(l)
×
发酵时间(h)),当od
600nm
达到10以上时开始添加乙胺,每小时调节一次乙胺流加速率。
[0104]
5l发酵罐发酵22h后l
‑
茶氨酸的产量和糖酸转化率如下表所示,实验组发酵过程曲线如图8所示(本发明糖酸转化率为:(l
‑
茶氨酸产量/葡萄糖消耗量)
×
100%):
[0105]
组别l
‑
茶氨酸产量(g/l)糖酸转化率(%)最高od
600nm
值实验组80
±
2.555
±
1.281
±
2.6对照组72
±
2.150
±
1.353
±
3.5
[0106]
实施例3od联动乙胺补加策略的确定
[0107]
在5l发酵罐中发酵生产l
‑
茶氨酸,当od
600nm
达到10以上时开始添加乙胺,根据菌体消耗葡萄糖的程度,每小时对乙胺流加速度进行不同程度的调整,保证耗糖速率维持在7
‑
8g
·
l
‑1·
h
‑1。收集50批次l
‑
茶氨酸产量达到80g/l的发酵批报,计算不同发酵时间下的平均od
600nm
值、平均发酵液体积以及平均乙胺流加速率,如下表所示:
[0108][0109][0110]
按照上述表格中数据使用origin软件进行线性拟合,y值为平均乙胺流加速率,x值为平均od
600nm
值/(平均发酵液体积(l)
×
发酵时间(h)。拟合结果如图9所示。根据拟合方程获得od联动的乙胺补加策略,即乙胺流加速率(g
·
l
‑1·
h
‑1)=0.5
×
od
600nm
值/(发酵液体积(l)
×
发酵时间(h))。
[0111]
实施例4从发酵液中分离提取l
‑
茶氨酸
[0112]
(1)实施例2发酵结束后共得发酵液体积3.2l,l
‑
茶氨酸含量80g/l,共计256g。将发酵液加热升温至60℃维持30min后降温至35℃。过50nm陶瓷膜进行微滤除菌并收集滤出液,在0.2mpa下当滤出液流速低于5ml/min时,补加0.8倍发酵液体积的水,直至截留液中l
‑
茶氨酸浓度为1.3g/l时,结束陶瓷膜微滤。
[0113]
(2)将5.1l陶瓷膜滤出液(l
‑
茶氨酸含量48.8g/l)过001
×
7阳离子树脂吸附l
‑
茶氨酸,吸附量160g(l
‑
茶氨酸)/l(树脂),然后用0.6%氨水进行洗脱收集,所得洗脱液体积4.6l(l
‑
茶氨酸含量52.9g/l)。将洗脱液过d213阴离子树脂吸附色素,收集4.8l树脂流出液
(l
‑
茶氨酸含量为49.3g/l)。
[0114]
(3)将树脂流出液泵入脱色罐中,加入l
‑
茶氨酸质量2%的药用活性炭(4.7g)进行脱色处理,至料液透光率达到98%。将脱色液泵入蒸发器中进行减压浓缩,维持真空度
‑
0.08mpa,温度60℃,浓缩至体积600ml(l
‑
茶氨酸含量为364.2g/l)。
[0115]
(4)将浓缩液泵入结晶罐中,加入240ml 95%乙醇,真空冷却结晶,离心分离得到母液820ml,收集湿晶体215.0g,水分13.9%,烘干后得到l
‑
茶氨酸成品185g,一次结晶收率72.3%。经液相检测,成品纯度为99%。
[0116]
实施例5菌株e.coli thee 5l发酵罐发酵生产l
‑
茶氨酸
[0117]
斜面培养基组成为:葡萄糖1g/l,蛋白胨5g/l,牛肉膏5g/l,酵母粉1g/l,氯化钠1g/l,琼脂15g/l,ph 7.0
‑
7.2;
[0118]
种子培养基组成为:葡萄糖20g/l,酵母提取物5g/l,蛋白胨10g/l,氯化钠10g/l,其余为水,ph 7.0
‑
7.2;
[0119]
发酵培养基组成为:葡萄糖10g/l,酵母粉2g/l,玉米浆2ml/l,柠檬酸0.2g/l,磷酸二氢钾0.5g/l,磷酸氢二钾0.5g/l,硫酸镁0.2g/l,其余为水,ph 7.0
‑
7.2。
[0120]
(1)斜面培养:从
‑
80℃冰箱保菌管中刮一环菌种,均匀涂布于活化斜面,37℃培养16h,转接茄形瓶继续培养16h;
[0121]
(2)种子培养:取适量无菌水于茄形瓶中,将菌悬液接入种子培养基中,ph稳定在7.0左右,温度恒定在37℃,溶氧在25
‑
35%之间,培养至细胞干重达到5g/l;
[0122]
(3)发酵培养:将种子液按照15%接种量接入新鲜的发酵培养基,发酵过程中控制ph在6.7
‑
7.2,温度维持在28℃,溶氧在10
‑
30%之间;当培养基中的葡萄糖消耗完之后,流加600g/l的葡萄糖溶液并维持发酵培养基中的葡萄糖浓度<1g/l;
[0123]
实验组采用od联动的乙胺补加策略,乙胺流加速率(g
·
l
‑1·
h
‑1)=0.5
×
od
600nm
值/(发酵液体积(l)
×
发酵时间(h)),当od
600nm
达到10以上时开始添加乙胺,每小时调节一次乙胺流加速率。
[0124]
5l发酵罐发酵20h后l
‑
茶氨酸的产量为75g/l,糖酸转化达到52%。
[0125]
实施例6菌株e.coli thee 5l发酵罐发酵生产l
‑
茶氨酸
[0126]
斜面培养基组成为:葡萄糖5g/l,蛋白胨10g/l,牛肉膏10g/l,酵母粉5g/l,氯化钠2.5g/l,琼脂20g/l,ph 7.0
‑
7.2;
[0127]
种子培养基组成为:葡萄糖30g/l,酵母提取物10g/l,蛋白胨20g/l,氯化钠20g/l,其余为水,ph 7.0
‑
7.2;
[0128]
发酵培养基组成为:葡萄糖40g/l,酵母粉8g/l,玉米浆20ml/l,柠檬酸2.0g/l,磷酸二氢钾3.2g/l,磷酸氢二钾2.4g/l,硫酸镁1.2g/l,其余为水,ph 7.0
‑
7.2;
[0129]
(1)斜面培养:从
‑
80℃冰箱保菌管中刮一环菌种,均匀涂布于活化斜面,37℃培养12h,转接茄形瓶继续培养12h;
[0130]
(2)种子培养:取适量无菌水于茄形瓶中,将菌悬液接入种子培养基中,ph稳定在7.0左右,温度恒定在37℃,溶氧在25
‑
35%之间,培养至细胞干重达到6g/l;
[0131]
(3)发酵培养:将种子液按照12%接种量接入新鲜的发酵培养基,发酵过程中控制ph在6.7
‑
7.2,温度维持在35℃,溶氧在10
‑
30%之间;当培养基中的葡萄糖消耗完之后,流加600g/l的葡萄糖溶液并维持发酵培养基中的葡萄糖浓度<1g/l;
[0132]
实验组采用od联动的乙胺补加策略,乙胺流加速率(g
·
l
‑1·
h
‑1)=0.5
×
od
600nm
值/(发酵液体积(l)
×
发酵时间(h)),当od
600nm
达到10以上时开始添加乙胺,每小时调节一次乙胺流加速率。
[0133]
5l发酵罐发酵25h后l
‑
茶氨酸的产量为77g/l,糖酸转化达到53%。
[0134]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为详细,但并不能因此而理解为对专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本专利构思的前提下,上述各实施方式还可以做出若干变形、组合和改进,这些都属于本专利的保护范围。因此,本专利的保护范围应以权利要求为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1