制备4-烷氧基-3-羟基吡啶甲酸的方法与流程
制备4-烷氧基-3-羟基吡啶甲酸的方法
1.本发明申请是基于申请日为2017年1月23日,申请号为201780007857.8,发明名称为“制备4-烷氧基-3-羟基吡啶甲酸的方法”的专利申请的分案申请。
技术领域
2.本技术涉及制备4-烷氧基-3-羟基吡啶甲酸的方法。更具体地,本技术涉及由糠醛制备4-烷氧基-3-羟基吡啶甲酸的方法。
背景技术:
3.美国专利6,521,622b1和美国申请序列号61/747,723特别描述了以下通式的某些杂环芳族酰胺化合物
[0004][0005]
和这些化合物作为杀真菌剂的用途。
[0006]
这些公开内容也描述了4-烷氧基-3-羟基吡啶甲酸及其衍生物作为在制备这些杂环芳族酰胺化合物时的关键中间体的制备。从廉价的原料获得4-烷氧基-3-羟基吡啶甲酸的有效且可扩展的工艺路线将是有用的。
技术实现要素:
[0007]
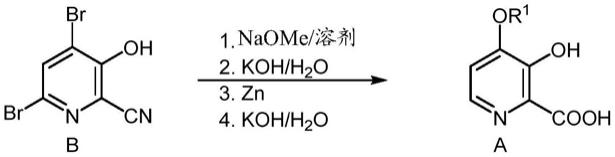
本技术涉及由式b化合物制备式a的4-烷氧基-3-羟基吡啶甲酸的方法
[0008][0009]
其中r1为c
1-c3烷基;
[0010][0011]
式a化合物可以通过包括以下步骤的一锅法制备:
[0012]
a)制备含有式c碱金属醇盐和所述式b化合物的第一混合物
[0013][0014]
其中m为na或k,r1为c
1-c3烷基;
[0015]
加热所述第一混合物;
[0016]
b)通过向所述第一混合物中加入水、强碱和锌金属来制备第二混合物;
[0017]
c)加热所述第二混合物;和
[0018]
d)分离所述式a化合物。
[0019]
本技术也涉及由式d化合物制备式b化合物的方法
[0020][0021]
所述式b化合物可以在包括以下步骤的方法中制备:
[0022]
a)通过将水-有机溶剂两相体系、氨源、氰化物源和所述式d化合物组合在一起来制备第一混合物;
[0023]
b)从所述第一混合物中分离出作为有机溶剂中的溶液的含有式e化合物的第二混合物;
[0024][0025]
c)向所述第二混合物中加入无机酸水溶液,形成第三混合物;
[0026]
其中所述无机酸为hcl、hbr、h2so4、hno3或h3po4;
[0027]
d)从第三混合物中分离出第四混合物,所述第四混合物为含有式f化合物的含水混合物;
[0028][0029]
其中x为cl、br、hso4、no3或h2po4;
[0030]
e)向所述第四混合物中加入溴化剂,形成第五混合物,所述溴化剂选自包括以下物质的组:
[0031]
i)溴化物以及与氧化剂的溴化物混配物;和
[0032]
ii)与氧化剂的溴化物混配物;和
[0033]
f)从所述第五混合物中分离出所述式b化合物。
[0034]
本技术也涉及由式d化合物制备式g化合物的方法
[0035][0036]
所述式g化合物可以在包括以下步骤的方法中制备:
[0037]
a)通过将水、有机溶剂、氨源、氰化物源和所述式d化合物组合在一起来制备第一混合物;
[0038]
b)从所述第一混合物中分离出作为有机溶剂中的溶液的含有式e化合物的第二混合物;
[0039][0040]
c)向所述第二混合物中加入无机酸水溶液,形成第三混合物;
[0041]
其中所述无机酸为hcl、hbr、h2so4、hno3或h3po4;
[0042]
d)从第三混合物中分离第四混合物,所述第四混合物为含有式f化合物的含水混合物;
[0043][0044]
其中x为cl、br、hso4、no3或h2po4;
[0045]
e)向所述第四混合物中加入溴化剂,形成第五混合物,所述溴化剂包括溴化物与氧化剂;和
[0046]
f)从所述第五混合物中分离出所述式g化合物。
[0047]
本发明包括:
[0048]
1.由式b化合物制备式a化合物的方法
[0049][0050]
其中r1为c
1-c3烷基;
[0051][0052]
所述方法包括以下步骤:
[0053]
a)制备含有式c碱金属醇盐和所述式b化合物的第一混合物
[0054][0055]
其中m为na或k,r1为c
1-c3烷基;
[0056]
加热所述第一混合物;
[0057]
b)通过向所述第一混合物中加入水、强碱和锌金属来制备第二混合物;
[0058]
c)加热所述第二混合物;和
[0059]
d)分离所述式a化合物。
[0060]
2.项1的方法,其中m为na,r1为甲基。
[0061]
3.项1的方法,其中所述第一混合物进一步包含选自包括dmso、甲醇、环丁砜及其混合物的组的溶剂。
[0062]
4.项1的方法,其中所述强碱选自氢氧化钠或氢氧化钾。
[0063]
5.项1的方法,其进一步包括在加热所述第二混合物之前从所述第二混合物中除去锌盐或锌金属的步骤。
[0064]
6.由式d化合物制备式b化合物的方法
[0065][0066]
所述方法包括以下步骤:
[0067]
a)通过将水-有机溶剂两相体系、氨源、氰化物源和所述式d化合物组合在一起来制备第一混合物;
[0068]
b)从所述第一混合物中分离出作为有机溶剂中的溶液的含有式e化合物的第二混合物;
[0069][0070]
c)向所述第二混合物中加入无机酸水溶液,形成第三混合物,其中所述无机酸为hcl、hbr、h2so4、hno3或h3po4;
[0071]
d)从第三混合物中分离出第四混合物,所述第四混合物为含有式f化合物的含水混合物;
[0072][0073]
其中x为cl、br、hso4、no3或h2po4;
[0074]
e)向所述第四混合物中加入溴化剂,形成第五混合物,所述溴化剂包括溴化物、与氧化剂的溴化物混配物、以及它们的混合物;和
[0075]
f)从所述第五混合物中分离出所述式b化合物。
[0076]
7.项6的方法,其中所述有机溶剂选自包括以下物质的组:mtbe,乙酸乙酯,乙酸异丙酯,thf,2-methf,甲苯,二甲苯或二甲苯的混合物,以及它们的混合物。
[0077]
8.项6的方法,其中所述无机酸为氢溴酸。
[0078]
9.项6的方法,其中x为br。
[0079]
10.项6的方法,其中溴化物为氢溴酸、或选自nabr和kbr的溴化物盐与酸的组合。
[0080]
11.项6的方法,其中所述氧化剂选自过氧化氢和过氧化单硫酸钾。
[0081]
12.由式d化合物制备式g化合物的方法
[0082][0083]
所述方法包括以下步骤:
[0084]
a)通过将水-有机溶剂两相体系、氨源、氰化物源和所述式d化合物组合在一起来制备第一混合物;
[0085]
b)从所述第一混合物中分离出作为有机溶剂中的溶液的含有式e化合物的第二混合物;
[0086][0087]
c)向所述第二混合物中加入无机酸水溶液,形成第三混合物,其中所述无机酸为hcl、hbr、h2so4、hno3或h3po4;
[0088]
d)从第三混合物中分离第四混合物,所述第四混合物为含有式f化合物的含水混合物;
[0089][0090]
其中x为cl、br、hso4、no3或h2po4;
[0091]
e)向所述第四混合物中加入溴化剂,形成第五混合物,所述溴化剂包括溴化物与氧化剂;和
[0092]
f)从所述第五混合物中分离出所述式g化合物。
[0093]
13.项12的方法,其中所述有机溶剂选自包括以下物质的组:mtbe,乙酸乙酯,乙酸异丙酯,thf,2-methf,甲苯,二甲苯或二甲苯的混合物,以及它们的混合物。
[0094]
14.项12的方法,其中所述无机酸为氢溴酸。
[0095]
15.项12的方法,其中x为br。
[0096]
16.项12的方法,其中溴化物为氢溴酸、或选自nabr和kbr的溴化物盐与酸的组合。
[0097]
17.项12的方法,其中所述氧化剂选自过氧化氢和过氧化单硫酸钾。
具体实施方式
[0098]
本技术所用的术语“分离(isolate)”、“分离(isolating)”或“分离(isolation)”是指将所需产物从成品化学工艺混合物的其它组分中使用标准方法部分或完全除去或分离,所述标准方法例如但不限于,过滤、萃取、蒸馏、结晶、离心、研磨、液-液相分离或本领域普通技术人员已知的其它方法。分离的产物可具有《50%至》50%的纯度,并且可使用标准纯化方法纯化至更高的纯度水平。分离的产物也可以经纯化或不经纯化在后续工艺步骤中使用。
[0099]
在本技术所述的方法中,4-烷基-3-羟基吡啶甲腈由4,6-二溴-3-羟基吡啶甲腈经一系列化学步骤制备,所述化学步骤包括溴取代、腈水解和卤素还原。本技术描述利用更有效的“一锅法”由4,6-二溴-3-羟基吡啶甲腈制备4-烷氧基-3-羟基吡啶甲酸的改进方法。
[0100]
本技术也描述由糠醛制备4,6-二溴-3-羟基吡啶甲腈的改进方法。该方法利用原位生成溴的溴化物/氧化剂配对试剂来部分或完全取代溴。这种工艺改进降低处理元素溴的需求并且提高溴原子利用的效率。
[0101]
本技术描述由糠醛制备4,6-二溴-3-羟基吡啶甲腈时的原位生成溴相当于使用元素溴,并且令人惊讶的是,氧化剂的存在不会对strecker或重排反应产生负面影响。此外,也令人惊讶的是,氧化剂不会导致4,6-二溴-3-羟基吡啶甲腈的吡啶环或腈基降解或氧化。
[0102]
a.制备式a化合物
[0103]
描述使用更有效的“一锅法”由4,6-二溴-3-羟基吡啶甲腈(化合物b)制备式a的4-烷氧基-3-羟基吡啶甲酸的改进方法。该方法包括首先用醇钠处理式b化合物,然后用锌金属、强碱水溶液处理,并且任选地加入另外的强碱水溶液,最后用强酸水溶液酸化最终反应混合物以制备式a化合物(其中r1为c
1-c3烷基)。
[0104]
在该方法的一种实施方式中,式b化合物与甲醇钠的反应可在偶极非质子溶剂如dmso或环丁砜中(任选加入甲醇)进行,或在作为溶剂的甲醇中进行。使用至少2摩尔当量的甲醇钠(优选2.5-3摩尔当量),并且在约50℃至约80℃加热约1小时至约24小时,完成4-溴基团用甲醇盐的取代。然后可用水和强碱水溶液如氢氧化钾或氢氧化钠(2-3摩尔当量)稀释所得反应混合物,
[0105]
方案i
[0106][0107]
用约1至约3摩尔当量的锌金属(即,粒度《10μm的zn粉、粒度《150μm的zn粉或另一高表面积的zn固体)处理并且在约20℃至约70℃搅拌直到完成6-溴基团还原。然后可以加入另外的强碱水溶液(2-3摩尔当量),并且将所得混合物在约80℃至约95℃加热约4小时至约24小时。可通过酸化反应混合物并且采用标准分离和纯化技术分离所需式a化合物(其中r1为甲基)。
[0108]
在该方法的另一种实施方式中,在式b化合物与甲醇钠反应完成后,然后可将所得反应混合物用水、强碱水溶液(4-6摩尔当量)和锌金属稀释,然后保持在约20℃至约95℃的
温度达约2小时至约48小时。完成锌还原和碱水解反应后,可通过酸化反应混合物并且采用标准分离和纯化技术分离所需产物。
[0109]
在该方法的另一种实施方式中,在式b化合物与甲醇钠反应完成后,然后所得反应混合物可用水和强碱水溶液(4-6摩尔当量)稀释,并且所得混合物在约80℃至约95℃加热达约4小时至约24小时,以完成腈基的水解。然后所得混合物可用锌金属处理,然后保持在约20℃至约70℃的温度直到完成6-溴基团的还原。完成锌还原和碱水解反应后,可通过酸化反应混合物并且采用标准分离和纯化技术分离所需产物。
[0110]
在该方法的另一种实施方式中,在式b化合物与甲醇钠反应完成后,腈基的水解和6-溴基的还原可以如下同时进行:向反应容器加入水、强碱水溶液和锌金属(一次性或在一段时间内加入),并且将其在约80℃至约95℃加热达到完成腈基水解和6-溴基团还原所需要的时间。
[0111]
b.制备式b化合物
[0112]
如方案ii中所示,糠醛(式d)可以在使用化学步骤a、b和c的方法中转化为4,6-二溴-3-羟基吡啶甲腈(式b)。
[0113]
方案ii
[0114][0115]
式f的氰基(呋喃-2-基)甲铵卤化物盐在双相法(有机相-水相,两相溶剂体系)中如下制备:
[0116][0117]
首先使糠醛(式d)与至少一个当量的氨源和氰化物源(步骤a)在本领域已知的α-氨基腈strecker合成的反应中反应,所述strecker合成描述于organic syntheses,coll.vol.i第21页和coll.vol.iii第84和88页中,得到式e的氨基(呋喃-2-基)乙腈。适宜的氨源包括:铵盐,例如但不限于,乙酸铵、溴化铵、氯化铵、甲酸铵、硫酸铵和氰化铵;溶解在有机溶剂中的氨,例如在甲醇中的氨、在乙醇中的氨和在二氧六环中的氨;在水中的氨(即氢氧化铵);和液体的无水氨或气态氨。适宜的氰化物源包括:氰化物盐,例如但不限于,氰化钠、氰化钾和氰化铵;和可以以连续加入方式与氨一起加入到糠醛中的氰化氢。反应(步骤a)在由水和水不混溶性的溶剂组成的两相溶剂体系中进行,所述水不混溶性的溶剂选自:醚,如乙醚、甲基叔丁基醚(mtbe)、四氢呋喃(thf)和2-甲基四氢呋喃(2-methf);酯,如乙酸乙酯和乙酸异丙酯;烷烃,如己烷、环己烷、庚烷和辛烷;芳族化合物,例如苯甲醚、甲苯和二甲苯或二甲苯的混合物,以及它们的混合物。这种反应描述于国际申请2000049008第55页中。本技术反应通常在足以保持基本均匀反应物混合物的搅拌下进行。这种反应可在约15℃至约30℃之间进行约1小时至约50小时。
[0118]
在制备式e的氨基(呋喃-2-基)乙腈的反应完成后,含有式e的化合物的两相溶剂
体系的有机相通过标准相分离和萃取方法容易地与水相分离。然后将作为有机相中的溶液的式e的化合物通过用无机酸水溶液处理转化为式f的盐。适宜的无机酸包括但不限于,氢溴酸(hbr)、盐酸(hcl)、硝酸(hno3)、硫酸(h2so4)和磷酸(h3po4)。本技术反应可在约0℃至约25℃进行。在将含有式e的化合物的有机相和无机酸水溶液适宜混合后,含有式f的氰基(呋喃-2-基)甲铵卤化物盐的酸性水溶液通过标准相分离和萃取方法从有机相中分离出来,并且准备进行最终的溴化/重排反应(方案ii,步骤c),以制备式b化合物。
[0119]
在该方法的溴化/重排反应步骤中,用溴化剂如溴处理式f的氰基(呋喃-2-基)甲铵盐,得到式b的产物。式f的起始原料(其中x为br、cl、no3、hso4或h2po4)可以用适宜溴化剂处理。
[0120][0121]
可以使用约3至约6摩尔当量的溴。该反应优选使用约3-5摩尔当量的溴和式f化合物的溴化物盐(x=br)进行。使用过量的溴化剂(例如过量5摩尔%、10摩尔%或15摩尔%)通常是方便的,以确保反应进行完全。该反应优选在质子溶剂或反应介质如水、或水与水溶性有机溶剂的混合物中进行,所述有机溶剂例如甲醇、乙醇、四氢呋喃、二氧六环或乙腈。进行反应的温度通常在约10℃至约25℃之间。溴加入完成后,可使反应混合物缓慢升温至室温并且搅拌10-48小时,或反应可在约30-40℃加热以完成反应。任选地,通过向反应中加入碱,例如2-4摩尔当量的乙酸钠,可以缩短反应时间。反应完成后,通过采用标准分离和纯化技术回收所需产物。
[0122]
在本技术的一些实施方式中,式f化合物的溴化/重排可包括使用一种或多种选自以下的溴化剂:(1)溴,和(2)与氧化剂配对的溴化物。文献中已知溴化物例如hbr、kbr和nabr在与氧化剂例如过氧化氢、过氧化单硫酸钾(即)、dmso或叔丁基过氧化氢结合时,在适宜反应条件下,可以产生溴(这在本技术中称为溴的原位生成)。使用溴化物(其为盐,例如nabr或kbr)原位生成溴,也需要使用酸来形成溴)。酸可选自包括hbr、hcl、h2so4、hno3、h3po4、乙酸及其混合物的组。这种涉及原位生成溴的方法提供以下优点:限制或消除元素溴的使用,提高该方法的溴原子效率,并且减少溴化物废物流的形成和处置。
[0123]
在本技术的一些实施方式中,在由式f的化合物(x=br)制备式b的化合物的方法中,使用溴化物例如hbr、kbr或nabr与氧化剂如过氧化氢配对可以如下进行:在环境温度将过氧化氢(氧化剂)缓慢加入到式f的化合物和溴化物(即,kbr或nabr作为溴化物,溴化物需要使用酸进行原位溴形成),并且在加入过程中使温度保持低于约50℃。在该方法中在足够量的溴化物(2-5摩尔当量)和酸的存在下可以使用相对于式b的化合物为约3-5摩尔当量的过氧化氢。
[0124]
描述溴化物与氧化剂一起用于溴化化学的化学文献包括:a)“simple and practical halogenation of arenes,alkenes,and alkynes with hydrohalic acid/h2o2(or tbhp),”tetrahedron,55,(1999)1127-1142,b)“oxidative halogenation with“green”oxidants:oxygen and hydrogen peroxide,”angew.chem.int.ed.,2009,48,
8424,以及其中的参考文献。描述由溴化物盐或hbr与过氧化氢反应生成溴的专利包括u.s.5,266,295、u.s.4,029,732和u.s.2,772,302。
[0125]
c.制备式g的化合物
[0126]
本技术的另一种实施方式涉及由糠醛制备式g的化合物的方法。在该方法的第一部分中,使用本技术描述的双相法将糠醛转化为式f的氰基(呋喃-2-基)甲铵溴化物盐(x为br)。在该方法的下一步骤中,将式f的溴化物盐与另外的hbr水溶液(1.5当量)合并,然后与约3至约4摩尔当量的过氧化氢(相对于式f的溴化物盐)反应,得到3-羟基-吡啶-2-甲腈(式g)。
[0127][0128]
可以进行过氧化氢添加的温度在约0℃至约50℃之间。过氧化氢添加完成后,使反应混合物在室温搅拌约1小时至约24小时。反应完成后,通过采用标准分离和纯化技术回收所需产物。
[0129]
d.制备式h的化合物
[0130]
将式a的4-烷氧基-3-羟基吡啶甲酸转化为式h的3-乙酰氧基化合物可以如下实现:用一种或多种选自乙酸酐和乙酰氯的乙酰化试剂,选自吡啶、烷基取代的吡啶和三烷基胺的碱,或利用肖顿-鲍曼(schotten-baumann)反应条件,使式a的化合物乙酰化。
[0131][0132]
通过这些方法中的任一种得到的产物可以通过常规方法回收,常规方法例如蒸发、过滤或萃取,并且可以通过标准方法纯化,标准方法例如重结晶或色谱法。
[0133]
给出以下实施例以说明本技术。
[0134]
实施例
[0135]
实施例1a. 3-羟基-4-甲氧基吡啶甲酸
[0136][0137]
用50ml无水dmso和1ml meoh制备甲醇钠(25g,0.45mol)的浆料。向该浆料中加入4,6-二溴-3-羟基-2-吡啶-2-甲腈(50g,0.181mol)和约50ml无水dmso的溶液,该溶液在30分钟内加入。加入过程中反应保持在50-65℃。加入完成后,反应在》50℃搅拌另外1小时。通过1h nmr分析确定反应完成。使反应冷却至35℃,然后将100ml水、接着45%koh(40ml,468mmol)加入到反应溶液中。然后在15min间隔以5g的份数加入锌粉(15.4g,234mmol;《10微米的粒度),这导致温度升至约45℃。使反应在环境温度搅拌过夜。反应未完成,因此将反
应加热至50℃,然后加入另外的zn粉(4.8g,74mmol)。3小时后反应完成。将另外的koh(45%的水溶液,40ml,468mmol)加入到反应混合物中。然后将反应在94℃加热12小时以完成水解。将反应冷却至环境温度,然后过滤除去固体。将固体用约100ml水洗涤到反应溶液中。然后将合并的滤液和洗涤溶液的ph用12n hcl调节至0.4。使所得混合物搅拌约1小时以确保ph稳定,然后通过过滤收集固体。所得灰白色固体用丙酮洗涤。将该物质在50℃的真空烘箱中干燥,得到4-甲氧基-3-羟基吡啶甲酸,为微浅黄色粉末(19.22g,纯度96%时的收率为63.2%,相当于60.7%收率)。通过hplc测定,有机物纯度为99.75%。1h nmr(400mhz,dmso-d6)δ8.03(d,j=6.4hz,1h),7.39(d,j=6.4hz,1h),4.04(s,3h)。
[0138]
实施例1b. 3-羟基-4-甲氧基吡啶甲酸
[0139][0140]
在30分钟时间内将纯甲醇钠(14.7g,271mmol)加入到4,6-二溴-3-羟基吡啶甲腈(30.2g,109mmol)和环丁砜(120g)的溶液中,这导致温度升至50℃。然后在60℃将反应加热18小时。使反应溶液冷却至环境温度,然后向反应中加入150ml去离子水,接着加入50ml 45wt%koh(5.4当量,586mmol)。加入zn粉(113mmol,7.5g),然后将反应加热至40℃。2小时后,加入另外的zn粉(2.5g,38mmol),然后将反应加热至60℃达另外2小时。使反应在环境温度搅拌过夜。反应混合物的1h nmr分析表明脱溴完成。反应过滤以除去固体,将45%koh(50ml,596mmol)加入滤液中,然后将所得溶液加热至约90℃。使反应在约90℃搅拌5.5小时,这导致接近完全转化。使反应在约90℃搅拌过夜。将反应混合物冷却至《30℃,然后用40%硫酸将ph调节至0.8,这导致形成固体。通过过滤分离固体,然后干燥,得到固体,其收率大于100%。将该物质在0.5ph盐酸中搅浆过夜。然后通过过滤和干燥分离该物质,得到10.3克4-甲氧基-3-羟基吡啶甲酸,为灰白色粉末,通过hplc测定其纯度为94%(53%收率)。
[0141]
实施例1c. 3-羟基-4-甲氧基吡啶甲酸
[0142][0143]
向500ml三颈烧瓶中加入甲醇钠(25g,0.462mol)和25ml二甲基亚砜。将甲醇钠/dmso混合物置于惰性气体下并且机械搅拌以产生自由流动的浆料。在单独容器中制备4,6-二溴-3-羟基吡啶甲腈(50.3g,0.181mol,dbhp,96.2wt%纯度)在约25ml无水dmso中的溶液。在50分钟内通过注射泵将dbhp溶液加入到甲醇钠/dmso混合物中。加入过程中温度保持在60℃以下。加入完成后,使反应搅拌另外1小时。在此时间内,反应混合物固化。向固化的反应混合物中加入100ml水,然后加入50%koh(50ml,941mmol)。将所得混合物搅拌约1.5小时,以使固体破碎成稠的浆料。然后以约5克的份数、间隔约20分钟加入zn粉(14.8g,226mmol),这导致温度升至约40℃。在zn溶解过程中,反应稀化为易混合的浆料。使反应在环境温度搅拌过夜。然后将反应加热至95℃达24小时。将反应冷却至《20℃,然后将溶液的
ph用hcl水溶液(12n)调节至0.6,这导致产物沉淀。过滤分离固体,用约50ml水洗涤,然后用约25ml丙酮洗涤。使所得浅黄色粉末在通风橱中干燥,得到23.3g产物。通过1h nmr(相对于内标)测定,产物纯度为96%,相当于所需产物的收率为76%,基于起始原料和最终产物的纯度。1h nmr(400mhz,dmso-d6)δ8.03(d,j=6.4hz,1h),7.39(d,j=6.4hz,1h),4.04(s,3h)。
[0144]
实施例1d.氰基(呋喃-2-基)甲基溴化铵
[0145][0146]
向配备有搅拌棒的500ml烧瓶中加入33.65g乙酸铵(436mmol)、150ml乙酸乙酯,30ml去离子水和10g kcn(154mmol)。然后通过注射器将糠醛(14g,145mmol)加入到反应器中。反应器中的温度从约15℃增至24℃。使反应在环境温度搅拌过夜。乙酸乙酯相的1h nmr分析显示转化率为》95%完全转化。向反应器中加入75ml 20%碳酸钠水溶液,使得搅拌10分钟。除去碳酸钠溶液,然后将反应混合物用40ml饱和盐水洗涤。1h nmr(400mhz,dmso-d6)δ7.53(dd,j=2.0,1.0hz,1h),6.47(dd,j=3.4,1.1hz,1h),6.42(dd,j=3.3,1.7hz,1h),5.08(s,1h)。
[0147]
除去盐水相后,向反应中加入24.5ml在约130ml去离子水中稀释的48%hbr水溶液(1当量,145mmol)。反应混合15分钟。除去水层并且置于单独容器中。然后有机层用2
×
25ml去离子水洗涤。将每次洗涤物加入到具有初始hbr萃取相的保持容器中。得到总共210.5g水相,其含有约14.06wt%的氰基(呋喃-2-基)甲基溴化铵。
[0148]
实施例1e. 4,6-二溴-3-羟基吡啶甲腈
[0149][0150]
将52.5g含有7.38g(36mmol)氰基(呋喃-2-基)甲基溴化铵(14.06wt%,在水中)的水相置于配备有搅拌棒的250ml烧瓶中。然后将烧瓶置于冰浴中。冷却至《10℃后,然后在15分钟内将5.8g溴(36mmol)滴加到反应中,导致形成固体。搅拌1小时后,使反应温热至环境温度。过硫酸氢钾复合盐(27g,87.8mmol)分批加入到反应中,导致固体溶解和红棕色的液相,其在搅拌1小时后缓慢转变成圆形颗粒状物质。反应用饱和亚硫酸氢钠水溶液淬灭。然后通过过滤分离固体,用去离子水洗涤,然后干燥过夜,得到6.25g棕褐色粉末。1h nmr分析表明该产物由4,6-二溴-3-羟基吡啶甲腈(96.6mol%,60.3%收率)和6-溴-3-羟基吡啶甲腈(3.4mol%,2.2%收率)组成。1h nmr(400mhz,dmso-d6)δ8.27(s,1.00h),7.75(d,j=8.9hz,0.034h),7.44(d,j=8.9hz,0.034h)。hrms(m/z)正离子模式[m+1],c6h3br2n2o的计算值为276.8612;实测值为276.8611。
[0151]
实施例1f. 4,6-二溴-3-羟基吡啶甲腈
[0152][0153]
将52.5g含有7.38g(36mmol)氰基(呋喃-2-基)甲基溴化铵(14.06wt%,在水中)的水相置于配备有搅拌棒的250ml烧瓶中。然后将烧瓶置于冰浴中。冷却至《10℃后,然后在约15分钟内将5.8g溴(36mmol)滴加到反应中,导致形成固体。搅拌1小时后,在20-30分钟内通过注射器向反应中加入30%过氧化氢(9.4ml)。这导致固体溶解,然后在1-2小时内沉淀出细粉末。反应用饱和亚硫酸氢钠淬灭。然后通过过滤分离固体,用去离子水洗涤,然后干燥过夜,得到6.03g棕褐色粉末。1h nmr分析表明该产物由4,6-二溴-3-羟基吡啶甲腈(94.5mol%,57.3%收率)和6-溴-3-羟基吡啶甲腈(5.5mol%,3.2%收率)组成。1h nmr(400mhz,dmso-d6)δ8.27(s,1.00h),7.75(d,j=8.9hz,0.075h),7.44(d,j=8.9hz,0.075h).
13
c nmr(101mhz,dmso-d6)δ157.65,141.95,135.55,128.76,124.37,120.34,115.97。hrms(m/z)正离子模式[m+1],c6h3br2n2o
+
的计算值为276.8612;实测值为276.8609。
[0154]
实施例1g. 3-羟基吡啶甲腈
[0155][0156]
将52.5g含有7.38g(36mmol)氰基(呋喃-2-基)甲基溴化铵(14.06wt%,在水中)的水相置于配备有搅拌棒的250ml烧瓶中。在搅拌下将48%hbr(6.2ml,55mmol)加入烧瓶中。然后将烧瓶置于冰浴中。冷却至《5℃后,在20-30分钟内通过注射器向反应中加入约7ml的30%过氧化氢。这导致非常少的放热。使反应温热至环境温度,此时反应开始自热至约50℃。将反应冷却至20℃,然后加入7ml 30%过氧化物,这导致形成沉淀。使反应搅拌约20分钟,然后反应用饱和亚硫酸氢钠淬灭,结果温度升至约40℃。升温期间,固体溶解。然后将反应物置于冰浴中。搅拌约45分钟后,固体产生。过滤收集固体并用去离子水洗涤。分离3-羟基吡啶甲腈(1.63克),为棕褐色结晶固体(37.3%收率)。1h nmr(400mhz,dmso-d6)δ11.67(s,1h),8.19(dd,j=4.4,1.3hz,1h),7.56(dd,j=8.6,4.4hz,1h),7.47(dd,j=8.7,1.4hz,1h)。
13
c nmr(101mhz,dmso-d6)δ157.67,141.93,135.56,128.75,125.99,124.37,120.34,115.97。hrms(m/z)负离子模式[m-1],c6h4n2o的计算值为119.0246;实测值为119.0240。
[0157]
实施例1h. 4,6-二溴-3-羟基吡啶甲腈
[0158][0159]
将53g含有7.45g(37mmol)氰基(呋喃-2-基)甲基溴化铵(14.06wt%,在水中)的水相置于配备有搅拌棒的250ml烧瓶中。在搅拌下将48%hbr(8.2ml,73mmol)加入烧瓶中。将烧瓶置于冰浴中。冷却至《5℃后,在20-30分钟内通过注射器向反应中加入6至7ml的30%过氧化氢。这导致非常少的放热。使反应温热至环境温度,此时反应开始自热至约46-48℃并
且变成橙黄色(均匀)。反应冷却至20℃,然后在15-20分钟内通过注射器加入另外7ml的30%过氧化氢,这导致形成沉淀。使反应搅拌过夜。反应用亚硫酸氢钠淬灭,得到含有固体的浅黄色溶液。过氧化物测试条表明没有残留的过氧化物。过滤收集固体,用水洗涤,干燥,得到6.22克浅棕褐色粉末。1h nmr分析表明该产物由4,6-二溴-3-羟基吡啶甲腈(收率58.1%)和6-溴-3-羟基吡啶甲腈(收率3.8%)组成。1h nmr(400mhz,dmso-d6)δ8.27(s,1.00h),7.75(d,j=8.9hz,0.064h),7.44(d,j=8.9hz,0.064h)。
[0160]
实施例1i. 4,6-二溴-3-羟基吡啶甲腈
[0161][0162]
使用39.21g糠醛、20g氰化钠、96g乙酸铵在300ml乙酸乙酯和260ml水中制备氰基(呋喃-2-基)甲基氯化铵的储备溶液。形成α-氨基腈后,向混合物中加入75ml饱和碳酸钠,使得混合20-30分钟。除去水相,然后用2
×
50ml饱和盐水洗涤有机相。将在约260ml去离子水中稀释的34ml 12n hcl水溶液(1当量,408mmol)加入到有机相中。将所得混合物混合(》500rpm)15分钟。沉降后,除去含有氰基(呋喃-2-基)甲基氯化铵的水层并且置于塑料保持容器中以形成储备溶液。然后有机层用44ml去离子水萃取,接着用46ml去离子水萃取。将每一含水提取物置于保持容器中,得到约460g水相。将水相稀释至总共480克,其含有约64.70g(13.5wt%)的氰基(呋喃-2-基)甲基氯化铵。
[0163]
将60g含有约8.1g(51mmol氰基(呋喃-2-基)甲基氯化铵)的储备溶液置于配有搅拌棒的250ml圆底烧瓶中。向烧瓶中加入约4.2ml(50.4mmol)12n hcl和10.4g(101mmol)nabr。在50分钟内向烧瓶中滴加30%过氧化氢(20g,176mmol)。加入(加入约7.5g过氧化物)期间的25分钟内,反应自热至56℃,此时反应冷却至约36℃。40分钟后,固体开始形成。使反应搅拌另外6小时。过滤收集固体,用去离子水洗涤,然后干燥。得到6.28g自由流动的浅棕褐色粉末,通过1h nmr测定,其为4,6-二溴-3-羟基吡啶甲腈(39.1wt%收率)、6-溴-3-羟基吡啶甲腈(3.0wt%收率)、6-氯-3-羟基-吡啶甲腈(6.23wt%收率)和4/6-氯/溴-3-羟基吡啶甲腈异构体(0.4wt%收率)的混合物。观测到总收率为51%。所需产物的1h nmr(400mhz,dmso-d6):δ8.27(s,1.00h),δ8.18(s,0.11h),δ7.75(d,j=8.9hz,0.64h),7.65(d,j=8.9hz,0.01h),7.53(d,j=8.9hz,0.01h),7.44(d,j=8.9hz,0.064h)。
[0164]
实施例1j. 4,6-二溴-3-羟基吡啶甲腈
[0165][0166]
向配备有搅拌棒的500ml烧瓶中加入36g乙酸铵(467mmol),200ml乙酸乙酯和7.5g nacn(153mmol)。使用75ml水将残留的氰化钠洗涤到烧瓶中并且从漏斗中洗出。然后通过注射器向反应器中快速加入糠醛(12.7ml,14.7g,153mmol)。反应器内的温度从约15℃升至24℃。使反应在环境温度(18℃)搅拌过夜。关闭搅拌以使两个液相分离。然后取样有机相进行1h nmr分析,确定反应仅完成约80%。然后将反应在25℃(使用水浴)搅拌另外6小时。通过1h nmr显示反应完成约90%。向反应器中加入75ml 20%碳酸钠水溶液并且搅拌30分钟,
然后将混合物静置20-30分钟。除去水相,然后用2
×
50ml饱和盐水洗涤含有糠醛的α-氨基腈在乙酸乙酯中的有机相。
[0167]
将10n硫酸(15ml,1当量,153mmol)在约225ml去离子水中稀释。用稀硫酸溶液以约1/3的比例萃取含有糠醛的α-氨基腈的乙酸乙酯溶液。将每次萃取物置于配有搅拌棒的500ml圆底烧瓶中。用另外5ml去离子水萃取有机溶液。向合并的含水酸提取物中加入47g溴化钠(459mmol),然后在2小时内加入过氧化氢(30%,360mmol),导致温度从19℃升至约50℃。使反应搅拌过夜。1h nmr分析表明该反应是6-溴-3-羟基吡啶甲腈和4,6-二溴-3-羟基吡啶甲腈的1:1混合物。将另外15ml 10n硫酸和13.5g的30%过氧化物(107mmol)加入到反应溶液中并且将反应加热至45℃。2小时后,1h nmr分析表明,反应完成。过滤收集固体,用水洗涤,干燥,得到21.9g浅棕褐色粉末。1h nmr分析表明该粉末由4,6-二溴-3-羟基吡啶甲腈(49.8%收率)和6-溴-3-羟基吡啶甲腈(2.4%收率)组成。
[0168]
实施例1k. 3-(乙酰氧基)-4-甲氧基吡啶甲酸
[0169][0170]
在环境温度下,将3-羟基-4-甲氧基吡啶甲酸(5.0g,29.6mmol)在50ml吡啶和50ml乙酸酐中搅浆。1小时后,形成黄色溶液,然后搅拌过夜。将溶液在45℃(2mmhg)蒸发,得到6.28g褐色固体(99%收率,mp=132
–
134℃)。1h nmr(400mhz,dmso-d6)δ13.32(s,1h),8.43(d,j=5.5hz,1h),7.40(d,j=5.5hz,1h),3.91(s,3h),2.27(s,3h)。
13
c nmr(101mhz,dmso-d6)δ167.95,164.81,158.34,147.87,142.77,136.18,110.87,56.59,20.27。hrms(m/z)c9h9no5的计算值为211.0478,实测值为211.0481([m]
+
)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1